

Abstract
Fibrotic disease is characterized by the pathological accumulation of extracellular matrix (ECM) proteins. Surprisingly, very little is known about the synthesis and degradation rates of the many proteins and proteoglycans that constitute healthy or pathological extracellular matrix. A comprehensive understanding of altered ECM protein synthesis and degradation during the onset and progression of fibrotic disease would be immensely valuable. We have developed a dynamic proteomics platform that quantifies the fractional synthesis rates of large numbers of proteins via stable isotope labeling and LC/MS-based mass isotopomer analysis. Here, we present the first broad analysis of ECM protein kinetics during the onset of experimental pulmonary fibrosis. Mice were labeled with heavy water for up to 21 days following the induction of lung fibrosis with bleomycin. Lung tissue was subjected to sequential protein extraction to fractionate cellular, guanidine-soluble ECM proteins and residual insoluble ECM proteins. Fractional synthesis rates were calculated for 34 ECM proteins or protein subunits, including collagens, proteoglycans, and microfibrillar proteins. Overall, fractional synthesis rates of guanidine-soluble ECM proteins were faster than those of insoluble ECM proteins, suggesting that the insoluble fraction reflected older, more mature matrix components. This was confirmed through the quantitation of pyridinoline cross-links in each protein fraction. In fibrotic lung tissue, there was a significant increase in the fractional synthesis of unique sets of matrix proteins during early (pre-1 week) and late (post-1 week) fibrotic response. Furthermore, we isolated fast turnover subpopulations of several ECM proteins (e.g. type I collagen) based on guanidine solubility, allowing for accelerated detection of increased synthesis of typically slow-turnover protein populations. This establishes the presence of multiple kinetic pools of pulmonary collagen in vivo with altered turnover rates during evolving fibrosis. These data demonstrate the utility of dynamic proteomics in analyzing changes in ECM protein turnover associated with the onset and progression of fibrotic disease.
The extracellular matrix (ECM)1 comprises an intricate network of cell-secreted collagens, proteoglycans, and glycoproteins providing structural and mechanical support to every tissue. The dynamic interplay between cells and ECM also directs cell proliferation, migration, differentiation, and apoptosis associated with normal tissue development, homeostasis, and repair (1, 2). Tissue repair following acute injury is typically characterized by the recruitment of inflammatory cells, enzymatic degradation of ECM immediately adjacent to the damaged tissue site, and subsequent infiltration of fibroblasts depositing new ECM. However, in the case of chronic tissue injury and inflammation, abnormal signaling pathways can stimulate uncontrolled ECM protein deposition, ultimately resulting in fibrosis and organ failure (3–6). In fact, fibrotic diseases including idiopathic pulmonary fibrosis, liver cirrhosis, systemic sclerosis, and cardiovascular disease have been estimated to account for over 45% of deaths in the developed world (1).
Despite the wide prevalence of fibrotic diseases, there is currently a paucity of anti-fibrotic drug treatments and diagnostic tests (7, 8). Median survival rates for idiopathic pulmonary fibrosis, for example, range from only two to five years following diagnosis (9, 10). Failure in the development of successful anti-fibrotic treatments can in part be attributed to a poor understanding of the active and dynamic role played by the ECM during various stages of fibrotic disease. ECM components influence myofibroblast differentiation not only through their modulation of fibrogenic growth factor activity (e.g. TGF-β), but also through mechanotransductive pathways whereby cells interpret altered ECM mechanical properties (3, 5, 11–13). The search for novel target pathways in the development of anti-fibrotic therapies would benefit from a better understanding of dynamic ECM synthesis and degradation associated with the various stages of fibrotic disease.
The combination of stable isotope labeling and proteomic analysis provides a new approach for interrogating dynamic changes in ECM protein synthesis associated with fibrotic disease. We have developed a platform termed “dynamic proteomics,” whereby protein synthesis rates from tissue samples are measured following the administration of stable isotope tracers (e.g. 2H, 15N) (14). Label incorporation into newly synthesized proteins is assessed via LC/MS analysis of mass isotopomer distributions in peptides derived from parent proteins through enzymatic degradation, providing a means to quantify the fractional synthesis rate (FSR) of individual proteins over the labeling period. Unlike traditional static proteomic techniques, this strategy provides valuable information regarding which proteins are actively synthesized or degraded during any specific stage of the disease process. Moreover, as measurements of label incorporation do not fluctuate based on the amount or yield of protein isolated (14–16), dynamic proteomic strategies also offer additional robustness relative to traditional quantitative proteomic techniques.
The detection of ECM components in highly cellular tissues such as liver and lung poses an additional stumbling block in the proteomic analysis of fibrotic ECM. The identification of less abundant matrix components is limited by the overwhelming number of cellular proteins present in standard homogenized tissue samples. Standard global protein fractionation techniques (e.g. gel electrophoresis) are inefficient at enriching targeted subsets of proteins. Tissue decellularization techniques commonly utilized in regenerative medicine offer a novel approach toward the enrichment of ECM proteins prior to proteomic analysis (17). Tissue samples are incubated under mechanical agitation in the presence of weak detergents that solubilize cell membranes, releasing cellular protein components into solution while keeping the surrounding structural ECM intact. This technique has recently been applied in the compositional proteomic analysis of cardiovascular, lung, and colon tissues, leading to the identification of ECM-related proteins previously not associated with those tissues (11, 18–20).
We present here the first study to combine dynamic proteomics with tissue decellularization in order to analyze altered ECM protein synthesis associated with pulmonary fibrosis. Bleomycin and sham-dosed mice were labeled for up to three weeks with heavy water (2H2O), and lung tissue was subsequently collected and fractionated into cellular and extracellular components. Further fractionation of ECM based on guanidine solubility resulted in the identification of protein fractions with kinetically distinct characteristics composed of a variety of collagens, basement membrane proteoglycans, and microfibrillar proteins. Label incorporation into ECM proteins in sham-dosed control lungs was generally faster in the guanidine-soluble fraction, suggesting that the insoluble pool reflected more stable, slower-turnover matrix components. In bleomycin-dosed lungs, however, there was a significant increase in the synthesis of both guanidine-soluble and insoluble ECM proteins. These labeling and fractionation methods should be easily adaptable to a variety of animal and human tissue types and could provide a new approach toward actively monitoring the dynamic changes in ECM synthesis and composition associated with fibrotic disease.
EXPERIMENTAL PROCEDURES
Animal Protocols
10-week-old C57Bl/6 mice (Jackson, Sacramento, CA) underwent 2H2O labeling according to a protocol similar to that previously described (21). Briefly, animals received a bolus intraperitoneal injection of 2H2O in 0.9% NaCl to bring total body water enrichment to ∼5%, followed by 8% 2H2O drinking water to maintain body water enrichment at 5% for the remainder of the study. Shortly following initial 2H2O administration, mice were dosed intratracheally with 1.5 units/kg of bleomycin (Sigma, St. Louis, MO) or saline as sham treatment similar to that previously described (22). Sham-dosed mice were euthanized at 6 and 21 days (n = 3), and bleomycin-dosed mice were euthanized at 5 (n = 3) and 17 or 21 days (n = 1, 2). Premature euthanization of some mice (day 5 or day 17) was performed because of excessive weight loss and morbidity relative to control animals associated with bleomycin exposure. Plasma was collected via cardiac puncture. Bronchial lavage was performed with 0.9% NaCl. Lung tissue was then perfused with 0.9% NaCl, collected, snap frozen in liquid nitrogen, and stored at −80¦°C. Details regarding individual animal weights and labeling durations are provided in Table I. Approximate labeling times of 1 and 3 weeks are reported hereinafter to simplify interpretation of the data. All procedures were Institutional Animal Care and Use Committee approved.
Table I. Duration of D2O labeling following bleomycin/saline delivery, initial and final body weights, and final lung weight for each mouse analyzed.
| Animal | Days of label (post-intubation) | Final animal weight (g) | Final lung weight (mg) |
|---|---|---|---|
| Control 1.1 | 6 | 19.7 | 258 |
| Control 1.2 | 6 | 18.6 | 231.9 |
| Control 1.3 | 6 | 19 | 338 |
| Bleomycin 1.1 | 5 | 15 | 447.2 |
| Bleomycin 1.2 | 5 | 15.8 | 371.5 |
| Bleomycin 1.3 | 5 | 14.8 | 321.5 |
| Control 2.1 | 21 | 20.5 | 359.7 |
| Control 2.2 | 21 | 19.4 | 262.9 |
| Control 2.3 | 21 | 19.7 | 251.3 |
| Bleomycin 2.1 | 17 | 16.7 | 368.6 |
| Bleomycin 2.2 | 21 | 19.6 | 385.2 |
| Bleomycin 2.3 | 21 | 20.9 | 385.2 |
Lung Tissue Preparation
Sequential extraction of lung tissue was performed to fractionate cellular and extracellular proteins, similar to previous work (23). 50 mg of lung tissue was minced with a razorblade and placed in 2-ml screw-cap vials. Tissues were rinsed four times with cold PBS for 5 min on a benchtop rotator to remove residual blood proteins. Tissues were then suspended in 0.5 m NaCl in 10 mm Tris-HCl, pH 7.5, at a 10:1 volume:mass ratio and vortexed at low speed for 2 h to extract NaCl-soluble loosely bound proteins. The supernatant was transferred to a low-binding microcentrifuge tube (Corning, Tewksbury, MA), spun at 16,000 × g for 30 min to clear any insoluble proteins, and stored at −80¦°C. Remaining tissues were rinsed with H2O, resuspended in 0.08% SDS at a 10:1 volume:weight ratio, and vortexed at low speed overnight (16 h) to extract cellular proteins. The supernatant was then removed, cleared of insoluble proteins, and stored as described above. Remaining tissue was once again rinsed with H2O; resuspended in 4 m guanidine HCl, 50 mm sodium acetate, pH 5.8, at a 10:1 volume:mass ratio; and vortexed at high speed for 48 h. The supernatant was then removed, cleared of insoluble proteins, and stored as described above. Residual guanidine-insoluble tissue was rinsed three times with H2O and stored at −80¦°C. All extraction buffers and rinses were spiked with protease inhibitor mixture at manufacturer-recommended concentrations (Millipore, Billerica, MA). Soluble proteins from extracted fractions were precipitated by overnight incubation at −20¦°C in ethanol (5:1 ethanol:extraction buffer) followed by centrifugation at 16,000 × g for 45 min. Pelleted proteins were rinsed twice with 90% ethanol, allowed to air dry, and resuspended in 8 m urea prior to trypsin digestion. Insoluble proteins were digested directly from residual guanidine-insoluble tissue fragments.
Protein concentrations in extractable fractions were determined via NanoDrop A280 measurement (Thermo, Waltham, MA). Up to 80 μg of each fractionated protein sample was denatured using ProteaseMax surfactant (0.1%; Promega, Madison, WI) and 4 m urea in 25 mm ammonium bicarbonate (pH 8). Proteins were reduced with TCEP (5 mm) for 20 min at room temperature with vortexing and then incubated with iodoacetamide (10 mm) in the dark for 20 min to chemically modify reduced cysteines. Proteins were then digested with trypsin (Promega) at 37¦°C overnight using a 1:25 trypsin:protein mass ratio. Guanidine-insoluble protein fractions were processed in an identical manner using a volume of trypsin sufficient for 80 μg of protein. The following day, formic acid was added to a total concentration of 5%, and samples were centrifuged at 14,000 × g for 30 min. The supernatant was transferred to a fresh tube, desalted with a C18 spec tip (Varian, Palo Alto, CA), dried via vacuum centrifugation, and resuspended in 0.1% formic acid/3% acetonitrile prior to LC/MS analysis.
Whole lung tissue homogenate was prepared using a Fast Prep-24 (MP Biomedical, Burlingame, CA) bead mill. 50 mg of lung tissue was suspended in H2O at a 10:1 volume:mass ratio with protease inhibitors and two 2.3-mm chrome steel beads (BioSpec, Bartlesville, OK) in a 2-ml screw-cap tube. Samples were homogenized at high speed three times for 30 s with 5-min intervals on ice and stored at −80¦°C. Proteins were precipitated with acetone at a 5:1 acetone:homogenate ratio by incubation at −20¦°C for 20 min followed by centrifugation at 2000 × g for 5 min at 4¦°C prior to hydrolysis and GC-MS analysis.
Plasma 2H2O Measurement
2H2O enrichment from 100 μl of mouse plasma was determined using a previously described method (24). Briefly, body water was evaporated from plasma via overnight incubation at 80¦°C. Samples were then mixed in 10 m NaOH and acetone and underwent a second overnight incubation. This material was extracted in hexane and dried with Na2SO4 prior to GC-MS analysis alongside a standard curve of samples prepared at known 2H2O concentrations.
LC-MS Peptide Analysis and Kinetic Calculations
Trypsin-digested peptides were analyzed on an Agilent 6520 quadrupole time-of-flight mass spectrometer with a 1260 Chip Cube nano-electrospray ionization source (Agilent Technologies, Santa Clara, CA). Peptides were separated chromatographically using a Polaris HR chip (Agilent #G4240–62030) consisting of a 360-nl enrichment column and a 0.075 × 150 mm analytical column, each packed with Polaris C18-A stationary phase with a 3-μm particle size. Mobile phases were (A) 5% v/v acetonitrile and 0.1% formic acid in deionized water and (B) 95% acetonitrile and 0.1% formic acid in deionized water. Peptides were eluted at a flow rate of 350 nl/min during a 27-min nano-LC gradient (2% B at 0 min, 5% B at 1 min, 30% B at 18 min, 50% B at 22 min, 90% B at 22.1–33 min, 2% B at 33.1 min; stop time: 38 min). Each sample was analyzed twice, once for protein/peptide identification in data-dependent MS/MS mode and once for peptide isotope analysis in MS-only mode. Acquisition parameters were as follows: MS/MS acquisition rate = 6 Hz MS and 4 Hz MS/MS with up to 12 precursors per cycle; MS acquisition rate = 0.9 Hz; ionization mode = positive electrospray; capillary voltage = −1980 V; drying gas flow = 4 l/min; drying gas temperature = 290¦°C; fragmentor = 170 V; skimmer = 65 V; maximum precursor per cycle = 20; scan range = 100–1700 m/z (MS), 50–1700 m/z (MS/MS); isolation width (MS/MS) = medium (∼4 m/z); collision energy (V) = −4.8 + 3.6*(precursor m/z/100); active exclusion enabled (exclude after one spectrum, release after 0.12 min); charge state preference = 2, 3, >3 only, sorted by abundance; total ion chromatogram target = 25,000; reference mass = 922.009798 m/z.
Acquired MS/MS spectra were extracted and searched using Spectrum Mill Proteomics Workbench software (version B.04.00, Agilent Technologies) and a UniProtKB/Swiss-Prot mouse protein database (16,473 proteins, release 2012 02). Data files were extracted with the following parameters: fixed modification = carbamidomethylation of cysteine; scans with the same precursor mass merged by spectral similarity within tolerances (retention time ± 10 s, mass ± 1.4 m/z); precursor charge maximum z = 6; precursor minimum MS1 S/n = 10; and 12C precursor m/z assigned during extraction. Extracted files were searched with the following parameters: enzyme = trypsin; Mus musculus; fixed modification = carbamidomethylation of cysteine; variable modifications = oxidized methionine + pyroglutamic acid + hydroxylation of proline; maximum number of missed cleavages = 2; minimum matched peak intensity = 30%; precursor mass tolerance = 10 ppm; product mass tolerance = 30 ppm; minimum number of detected peaks = 4; maximum precursor charge = 3. Search results were validated at the peptide and protein levels with a global false discovery rate of 1%. Details regarding specific proteins identified and unique peptide coverage are presented in the supplemental material.
Proteins with scores greater than 11.0 were reported, and a list of peptides with scores greater than 6 and scored peak intensities greater than 50% was exported from Spectrum Mill and condensed to a non-redundant peptide formula database using Excel. This database, containing peptide elemental composition, mass, and retention time, was used to extract MS spectra (M0–M3) from corresponding MS-only acquisition files with the Find-by-Formula algorithm in Mass Hunter Qualitative Analysis software (version B.05.00, Agilent Technologies). MS spectra were extracted with the following parameters: extracted ion chromatogram integration by Agile integrator; peak height > 10,000 counts; include spectra with average scans > 12% of peak height; no MS peak spectrum background; unbiased isotope model; isotope peak spacing tolerance = 0.0025 m/z plus 12.0 ppm; mass and retention time matches required; mass match tolerance = ±12 ppm; retention time match tolerance = ±0.8 min; charge states z = +2 to +4; chromatogram extraction = ±12 ppm (symmetric); extracted ion chromatogram extraction limit around expected retention time = ±1.2 min.
Details of FSR calculations were described previously (14). Briefly, in-house software was developed to calculate the peptide elemental composition and curve fit parameters for predicting isotope enrichments of peptides in newly synthesized proteins based on precursor body water enrichment (p) and the number (n) of amino acid C-H positions per peptide actively incorporating H and 2H from body water. Incorporation of 2H into tryptic peptides decreases the relative proportion of M0 within the overall isotope envelope spanning M0–M3. Fractional synthesis was calculated as the ratio of excess %M0 (EM0) for each peptide to the maximal absolute EM0 possible at the measured body water enrichment. Data handling was performed using Microsoft Excel templates, with input of precursor body water enrichment for each subject, to yield FSR data at the protein level.
Data from individual biological samples were filtered to exclude protein measurements with fewer than two peptide spectra measurements per protein. FSR data at individual time points (1 or 3 weeks) are reported as a cumulative value (percentage of protein newly synthesized over the entirety of the labeling period). The fold change in mean protein FSR between groups (bleomycin:control) was determined for both early (0 to 1 week) and late (1 to 3 weeks) fibrotic response by calculating the slope increase of FSR between collected data points. Protein FSR on day 0 was assumed to be 0%.
GC-MS OHPro Analysis
GC-MS analysis of OHPro FSR was carried out as previously described (21). Briefly, lung tissue protein fractions and whole homogenate proteins were hydrolyzed in 6N HCl at 110¦°C for 18 h. Extracted protein fractions were spiked with known amounts of 2H3-labeled OHPro to provide an internal standard for quantitation. The amine group was deactivated with a solution of pentafluorabenzyl bromide, acetonitrile, water, and phosphate buffer. In order to silylate the hydroxyl moiety of OHPro, samples were incubated with a solution of acetonitrile, N-methyl-N-[tert-butyldimethyl-silyl]trifluoroacetamide, and methylimidizole. This material was extracted in petroleum ether and dried with Na2SO4. Derivatized OHPro was analyzed via GC-MS using selected ion monitoring of 424, 425, and 427 m/z ions in negative chemical ionization mode. Incorporation of 2H into OHPro was calculated as excess %M1 (EM1). Fractional collagen synthesis was calculated as the ratio of EM1 to the maximal EM1 possible at the measured body water enrichment. The concentration of OHPro was determined using the 2H3-OHPro internal standard and a standard curve analyzed with each batch of samples. Total lung collagen was determined using total lung tissue weights recorded at the time of collection.
Pyridinoline Cross-link Quantitation
Pyridinoline cross-links were quantitated by means of ELISA using the MicroVue Serum PYD Assay (Quidel, San Diego, CA) per the manufacturer's instructions. Lung tissue protein fractions were hydrolyzed as described previously for GC-MS analysis and diluted within the working concentration range of the assay similarly to what was previously described (25). Samples were adjusted to neutral pH with NaOH prior to analysis.
Statistical Analyses
Means and standard deviations (error bars) of fractional protein synthesis between groups (n = 3) were compared via Student's t test at each time point. A Holm-Sidak correction for multiple comparisons was performed for all ECM proteins detected within each protein fraction. Analysis of variance was used for assessing statistically significant differences among three or more groups. Statistical significance was defined as a p value < 0.05.
RESULTS
Verification of Sequential Protein Extraction
Dynamic proteomic analysis of mouse lung ECM protein fractional synthesis was performed following fibrotic induction with bleomycin or sham treatment. Lung tissue proteins were fractionated to enrich for ECM and then trypsinized and analyzed via LC-MS to measure shifts in peptide mass isotopomer distributions (Fig. 1). Measurements of ECM protein enrichment from lung tissue were carried out through the sequential extraction of proteins into four fractions, NaCl-soluble, SDS-soluble, guanidine HCl-soluble, and insoluble, the latter two of which were enriched for ECM proteins (e.g. collagens, proteoglycans, etc.) as determined from LC-MS peptide spectral identification (Fig. 2). No appreciable enrichment for ECM proteins was detected in the NaCl- or SDS-soluble fractions.
Fig. 1.
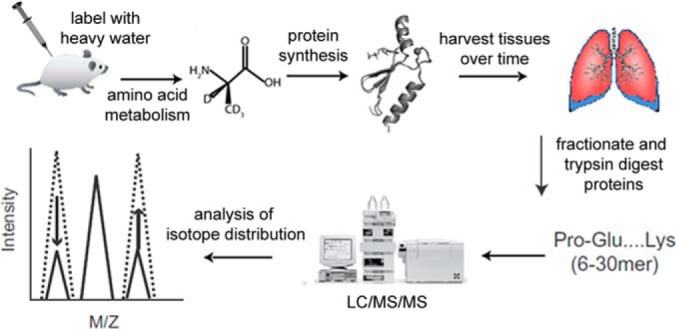
Flowchart of dynamic proteomic analysis. Following the administration of bleomycin or vehicle, mice are continuously labeled with 2H2O, which is incorporated into newly synthesized proteins over time. Proteins from harvested tissues are trypsinized into peptides and analyzed via LC-MS to measure isotopic shifting reflective of the fraction of each protein that was synthesized during the labeling period.
Fig. 2.
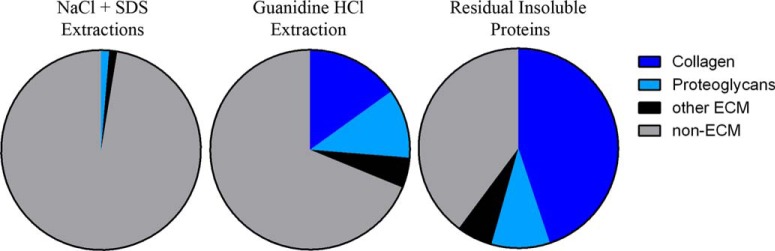
ECM enrichment following sequential protein extraction. Spectrum mill identification of MS/MS peptide spectra from NaCl/SDS-soluble, guanidine-soluble, and residual insoluble pulmonary protein fractions. Peptides are divided into collagens, proteoglycans, other ECM, and non-ECM.
Kinetics of Guanidine-soluble ECM Proteins
Guanidine-soluble protein extraction successfully enriched for a variety of pulmonary proteoglycans, as well as additional ECM proteins including fibronectin, collagen I, and collagen VI (Table II). Fractional synthesis of basement-membrane-associated proteoglycans ranged from roughly 10% to 20% and 30% to 50% newly synthesized molecules present in control lungs after 1 and 3 weeks of label, respectively. Fractional synthesis of the same proteoglycans in bleomycin-dosed lungs was significantly higher in most cases, with the majority approaching 60% to 80% labeled at 3 weeks. Guanidine-soluble collagens and collagen-associated small leucine-rich proteoglycans also attained significantly greater label incorporation following bleomycin exposure. Fractional synthesis of guanidine-soluble collagens (types I and VI) increased from ∼10% and 20% in control lungs to 20% and 50% in bleomycin-dosed lungs at 1 and 3 weeks, respectively. FSRs for biglycan and decorin, two small leucine-rich proteoglycans associated with collagen fibril assembly and growth factor signaling, were noted to be particularly rapid (>60% labeled in control lungs at 1 week). Label incorporation into fibronectin was also expeditious, reaching greater than 75% in both control and bleomycin-dosed lungs prior to 1 week. Protein-glutamine γ-glutamyltransferase 2 (a.k.a. tissue transglutaminase), an enzyme involved in protein cross-linking, also showed increased fractional synthesis at both time points observed after bleomycin administration.
Table II. Percentage of newly synthesized guanidine-soluble ECM proteins extracted from control and bleomycin-induced fibrotic lung tissue after 1 and 3 weeks of label. Values represent mean ± S.D. (n = 3) where protein data were available from three distinct biological samples.
| Protein | UniProt accession number | Average F control, 1 week (%) | Average F bleomycin, 1 week (%) | Average F control, 3 weeks (%) | Average F bleomycin, 3 weeks (%) |
|---|---|---|---|---|---|
| Agrina | A2ASQ1 | 11.1 ± 0.5 | 14.0 ± 2.7 | 30.7 ± 0.3 | 64.4 ± 6.9 |
| Biglycanb | P28653 | 65.8 ± 4.2 | 86.7 ± 5.1 | 85.1 ± 2.2 | 95.9 ± 2.1 |
| Collagen α-1(I) chainb | P11087 | 10.3 ± 1.7 | 21.4 ± 4.9 | 17.9 ± 2.4 | 52.7 ± 3.2 |
| Collagen α-2(I) chaina | Q01149 | 10.3 ± 1.5 | 17.9 ± 3.2 | 17.6 ± 2.6 | 53.8 ± 2.3 |
| Collagen α-1(V) chainc | O88207 | 9.9 ± 2.4 | 25.3 ± 8.0 | 47.0 | 62.7 |
| Collagen α-2(V) chainc | Q3U962 | 7.6 ± 0.1 | 27.2 ± 6.4 | 20.4 ± 4.3 | 58.7 |
| Collagen α-1(VI) chainb | Q04857 | 6.7 ± 1.0 | 18.3 ± 1.8 | 14.5 ± 0.1 | 58.9 ± 11.4 |
| Collagen α-2(VI) chaina | Q02788 | 9.3 | 19.1 ± 1.7 | 15.7 ± 1.6 | 62.1 ± 10.7 |
| Decorin | P28654 | 89.6 | N/A | N/A | 98.4 |
| Dermatopontinb | Q9QZZ6 | 7.2 ± 1.0 | 22.3 ± 6.2 | 16.4 ± 1.4 | 64.3 ± 5.9 |
| Fibronectinb | P11276 | 77.4 ± 2.9 | 96.2 ± 3.0 | 76.4 ± 2.5 | 94.7 ± 2.3 |
| Laminin subunit α-4a | P97927 | 20.7 ± 8.1 | 24.6 ± 4.8 | 44.9 ± 1.4 | 69.2 ± 4.2 |
| Laminin subunit α-5b | Q61001 | 8.6 ± 1.9 | 21.8 ± 1.0 | 23.5 ± 3.2 | 51.9 ± 4.0 |
| Laminin subunit β-1b | P02469 | 13.9 ± 2.6 | 31.5 ± 6.9 | 45.6 ± 7.3 | 81.1 ± 8.7 |
| Laminin subunit β-2a | Q61292 | 16.8 ± 3.7 | 25.5 ± 4.7 | 39.1 ± 1.1 | 70.9 ± 4.5 |
| Laminin subunit γ-1a | P02468 | 14.0 ± 0.7 | 23.3 ± 2.0 | 42.2 ± 1.3 | 67.4 ± 3.8 |
| Microfibril-associated glycoprotein 4a | Q9D1H9 | 8.1 ± 0.6 | 11.7 ± 3.6 | 22.7 ± 3.1 | 61.0 ± 3.3 |
| Nidogen-1a | P10493 | 16.3 ± 1.3 | 25.7 ± 5.0 | 44.2 ± 1.5 | 74.2 ± 4.8 |
| Nidogen-2a | O88322 | 9.4 ± 1.1 | 13.0 ± 4.7 | 28.6 ± 1.0 | 49.3 ± 10.1 |
| Perlecanb | Q05793 | 22.5 ± 1.2 | 33.3 ± 7.3 | 51.5 ± 1.5 | 79.9 ± 1.9 |
| Prolargin | Q9JK53 | 55.5 | 78.5 ± 6.8 | 76.6 ± 10.2 | 99.1 |
| Protein–glutamine γ-glutamyltransferase 2b | P21981 | 44.7 ± 2.2 | 63.3 ± 6.3 | 86.2 ± 2.3 | 97.5 ± 2.4 |
a p < 0.05 at 3 weeks only.
b p < 0.05 at both time points.
c p < 0.05 at 1 week only.
Kinetics of Insoluble ECM Proteins
Insoluble pulmonary protein fractions were enriched for a variety of collagens and microfibrillar proteins (Table III). Fractional synthesis of fibrillar collagens (types I, III, and V), those most associated with fibrotic scar tissue, was not significantly increased in bleomycin-dosed lungs after 1 week of label. However, fibrillar collagen fractional synthesis was remarkably elevated by 3 weeks, reaching a 6-fold higher percentage of label relative to control lungs. Insoluble type VI collagen fractional synthesis was significantly higher in bleomycin-dosed lungs at both time points, whereas type IV collagen fractional synthesis was significantly increased only at 3 weeks. Fractional synthesis of elastin, EMILIN-1, fibrillin-1, and fibulin-5, proteins associated with elastic microfibril formation, was also significantly higher in bleomycin-dosed lungs, with elastin reaching a greater than 8-fold increase in FSR at 3 weeks. Basement membrane proteoglycans laminin and perlecan were also detected in the insoluble protein pool, but their fractional synthesis was only elevated in fibrotic lungs following 3 weeks of label. These results confirm a time-dependent increase in insoluble protein deposition in the bleomycin lung model, with the majority occurring more than 1 week post-bleomycin exposure.
Table III. Percentage of newly synthesized guanidine-insoluble ECM proteins present in control and bleomycin-induced fibrotic lung tissue after 1 and 3 weeks of label. Values represent mean ± S.D. (n = 3) where protein data were available from three distinct biological samples.
| Protein | Accession number | Average F control, 1 week (%) | Average F bleomycin, 1 week (%) | Average F control, 3 weeks (%) | Average F bleomycin, 3 weeks (%) |
|---|---|---|---|---|---|
| Collagen α-1(I) chaina | P11087 | 5.0 ± 0.9 | 12.0 ± 5.1 | 7.4 ± 1.2 | 57.3 ± 8.5 |
| Collagen α-2(I) chaina | Q01149 | 5.2 ± 1.0 | 11.9 ± 4.1 | 8.4 ± 1.8 | 58.2 ± 8.7 |
| Collagen α-1(III) chaina | P08121 | 6.2 ± 0.8 | 11.8 ± 2.9 | 9.0 ± 1.5 | 51.8 ± 7.1 |
| Collagen α-1(IV) chaina | P02463 | 8.7 ± 1.4 | 13.1 ± 1.0 | 19.4 ± 1.0 | 57.6 ± 7.9 |
| Collagen α-2(IV) chaina | P08122 | 8.8 ± 1.0 | 14.9 ± 1.1 | 20.4 ± 0.8 | 59.1 ± 9.7 |
| Collagen α-1(V) chaina | O88207 | 5.8 ± 1.7 | 13.0 ± 2.0 | 11.8 ± 2.1 | 57.0 ± 8.4 |
| Collagen α-1(VI) chainb | Q04857 | 12.5 ± 2.9 | 29.8 ± 5.4 | 26.4 ± 4.1 | 78.8 ± 6.6 |
| Collagen α-2(VI) chainb | Q02788 | 12.5 ± 2.8 | 28.6 ± 4.9 | 26.1 ± 3.5 | 74.2 ± 7.4 |
| Elastina | P54320 | 4.7 ± 1.1 | 11.9 ± 7.0 | 8.5 ± 1.2 | 74.4 ± 11.6 |
| EMILIN-1b | Q99K41 | 27.2 ± 0.5 | 54.7 ± 5.6 | 58.1 ± 3.7 | 84.7 ± 0.3 |
| Fibrillin-1b | Q61554 | 11.3 ± 1.2 | 25.9 ± 6.3 | 22.5 ± 2.3 | 83.6 ± 4.2 |
| Fibulin-5a | Q9WVH9 | 7.7 ± 1.6 | 7.0 ± 1.8 | 14.7 ± 0.7 | 45.3 ± 0.6 |
| Laminin subunit α-3a | Q61789 | 9.7 ± 0.6 | 9.9 ± 2.9 | 22.6 ± 0.9 | 54.1 ± 13.6 |
| Laminin subunit α-5a | Q61001 | 8.2 ± 0.6 | 9.8 ± 2.2 | 13.8 ± 0.6 | 35.5 ± 6.5 |
| Laminin subunit β-2a | Q61292 | 6.8 ± 0.4 | 7.7 ± 2.0 | 13.2 ± 0.5 | 33.3 ± 6.1 |
| Laminin subunit β-3a | Q61087 | 12.6 ± 0.1 | 8.6 ± 1.7 | 22.0 ± 2.7 | 45.6 ± 5.0 |
| Laminin subunit γ-1a | P02468 | 7.4 ± 0.4 | 8.5 ± 1.9 | 13.4 ± 0.5 | 35.4 ± 5.4 |
| Microfibrillar-associated protein 2a | P55002 | 10.0 ± 2.4 | 13.5 | 23.9 ± 1.3 | 64.4 ± 7.8 |
| Nephronectina | Q91V88 | 10.3 ± 0.6 | 8.9 ± 2.7 | 22.4 ± 1.7 | 34.7 ± 3.1 |
| Periostina | Q62009 | 34.7 ± 4.2 | 36.5 ± 11.4 | 68.4 ± 2.1 | 92.1 ± 3.2 |
| Perlecana | Q05793 | 13.1 ± 0.2 | 15.1 ± 4.1 | 29.1 ± 1.4 | 60.1 ± 2.7 |
a p < 0.05 at 3 weeks only.
b p < 0.05 at both time points.
Kinetics of Individual ECM Proteins Fractionated by Guanidine Solubility
We identified multiple ECM proteins present in both guanidine-soluble and insoluble protein fractions, including collagen I, collagen VI, perlecan, and laminin. For the majority of these proteins, including laminin subunit β-2, perlecan, and collagen α-1(I), fractional synthesis in control lungs was significantly higher in the guanidine-soluble fraction than in the insoluble fraction (Figs. 3A–3C). Although bleomycin administration did not appear to affect this trend for the two proteoglycans, the ratio of labeled to unlabeled collagen I across the two protein fractions was altered. Interestingly, guanidine-insoluble collagen VI fractional synthesis was higher than that of the soluble form, a trend that was maintained following the onset of fibrosis (Fig. 3D). Solubility-related changes in fractional synthesis were most pronounced for extracellular proteins compared with other classes of proteins, as demonstrated by very little change in α-smooth muscle actin kinetics across protein fractions (Fig. 5E).
Fig. 3.

ECM proteins fractionated into two subpopulations by guanidine solubility display distinct kinetics. Comparison of newly synthesized guanidine-soluble and insoluble laminin β-2 (A), perlecan (B), collagen α-1(I) (C), collagen α-1(VI) (D), and α-smooth muscle actin (E) present in control and bleomycin-induced fibrotic lung tissue. Values are means ± S.D. (n = 3) with statistical comparison between protein fractions at each time point (*p < 0.05).
Fig. 5.

PYD cross-link quantitation. Concentration of pyridinoline cross-links present in guanidine-soluble and insoluble pulmonary protein fractions from control animals (n = 6), early fibrotic animals (1 week post-bleomycin; n = 3), and late fibrotic animals (3 weeks post-bleomycin; n = 3). Cross-link concentration was determined via ELISA and GC-MS quantitation of OHPro. Values are means ± S.D. with statistical comparison between protein fractions (*p < 0.05).
Early versus Late Fibrotic ECM Kinetics
Pulmonary administration of bleomycin has previously been shown to result in an early inflammatory phase (pre-1 week), followed by a later fibrotic phase (post-1 week) (26, 27). To better understand how ECM protein synthesis is altered during these distinctive stages of fibrotic disease, we calculated the fold-change in ECM protein FSR between bleomycin-dosed and control lungs for these time periods (Fig. 4). Global ECM protein fractional synthesis appeared to be elevated in bleomycin-dosed lung tissue during both the early inflammatory and late fibrotic phase, and a small subset of proteins were particularly elevated during the late fibrotic phase. In the guanidine-soluble protein pool, labeling with collagens I and VI appeared to be most accelerated in the late fibrotic phase of disease, along with dermatopontin and MFAP-4 (Fig. 4A). These latter proteins play roles in TGF-β signaling pathways and cell-matrix interactions, respectively (28, 29). An analysis of the insoluble ECM protein pool identified fibrillar collagens (types I, III, and V) and microfibrillar proteins (elastin, fibulin-5, and fibrillin-1) as most elevated in fractional synthesis during the late fibrotic phase of disease (Fig. 4B). It is important to note that this method of analysis is less accurate for fast-turnover proteins, which are close to fully labeled at 1 week (e.g. biglycan, fibronectin, EMILIN-1), so that if any differences between groups were present at 3 weeks, they would not be apparent.
Fig. 4.

Early- and late-stage ECM kinetics in response to bleomycin. Fold change (bleo:control) in guanidine-soluble (A) and insoluble (B) ECM protein fractional synthesis following induction of fibrosis with bleomycin. Data represent group means and are divided into early (pre-1 week) and late (post-1 week) fibrotic response sorted by magnitude of fold change in late-responding proteins. Results for late response (1 to 3 weeks) were calculated using group differences in fractional synthesis at 1 and 3 weeks (as described in the text).
GC-MS Analysis of Pulmonary OHPro Fractional Synthesis
To further characterize sequentially extracted collagen subsets, we utilized methods similar to those previously published for determining total OHPro mass and FSR in tissues via GC-MS (21, 30). OHPro was present in each pulmonary tissue protein fraction in different quantities (Table IV). The mass of OHPro present in the NaCl and SDS-soluble protein pools was minimal, comprising roughly 0.3% of total OHPro detected across all protein fractions. OHPro measured in the guanidine-soluble protein fraction accounted for roughly 2.5% to 5% of total collagen, and insoluble collagens made up the remaining 95% to 97.5%. Although the OHPro mass was elevated in the NaCl, SDS, and insoluble protein fractions following fibrotic induction with bleomycin, guanidine-soluble OHPro levels were unchanged.
Table IV. Quantitation of total OHPro present in lung protein extracts 1 and 3 weeks post-bleomycin. Total lung OHPro quantity from control animals (n = 6), early fibrotic animals (1 week post-bleomycin; n = 3), and late fibrotic animals (3 weeks post-bleomycin; n = 3). Values are means ± S.D. The percentage of total OHPro in each fraction was calculated for each experimental group (controls, bleomycin 1 week, bleomycin 3 weeks).
| Experimental group | Lung tissue fraction | OHPro per lung (ng) | OHPro per group (%) |
|---|---|---|---|
| Controls | NaCl soluble | 218 ± 47 | 0.05 |
| Controls | SDS soluble | 636 ± 252 | 0.15 |
| Controls | Guanidine soluble | 17,242 ± 6569 | 4.14 |
| Controls | Insoluble | 398,370 ± 179,903 | 95.65 |
| Bleomycin 1 week | NaCl soluble | 376 ± 107 | 0.08 |
| Bleomycin 1 week | SDS soluble | 1180 ± 208 | 0.26 |
| Bleomycin 1 week | Guanidine soluble | 18,299 ± 4140 | 4.03 |
| Bleomycin 1 week | Insoluble | 434,746 ± 61,429 | 95.63 |
| Bleomycin 3 weeks | NaCl soluble | 792 ± 253 | 0.11 |
| Bleomycin 3 weeks | SDS soluble | 1178 ± 233 | 0.17 |
| Bleomycin 3 weeks | Guanidine soluble | 18,055 ± 3727 | 2.60 |
| Bleomycin 3 weeks | Insoluble | 674,629 ± 185,995 | 97.12 |
Quantification of pyridinilone cross-link density in the guanidine-soluble and insoluble protein pools revealed significantly elevated concentrations in the insoluble pool of control lungs, indicative of enhanced collagen stability and maturity (Fig. 5). Although no longer significantly different, pyridinoline cross-link density did not appear to be altered after 3 weeks.
Similar to the collagen data observed in our dynamic proteomic analyses, the fractional synthesis rate of OHPro was significantly increased following the induction of fibrosis (Fig. 6A). Rapid label incorporation occurred in the NaCl and SDS-soluble OHPro pools, indicating that these fractions were largely populated by recently synthesized collagen proteins. Administration of bleomycin elevated label incorporation in these pools to nearly 100% at 1 week. OHPro fractional synthesis was also significantly higher in the guanidine-soluble and insoluble protein fractions. Importantly, label incorporation was similar to that observed in fibrillar collagens via LC-MS analysis. A comparison of total lung OHPro fractional synthesis (GC-MS) and insoluble collagen α-1(I) fractional synthesis (LC-MS) demonstrated close agreement between the two kinetic assays (Fig. 6B). The combination of OHPro mass and fractional synthesis data calculated from our GC-MS analysis also allowed for absolute quantitation of the newly synthesized OHPro present within each protein fraction (Fig. 6C). Note that these data are presented in log scale because of the dynamic range of collagen present in the various protein fractions. Newly synthesized guanidine-soluble and insoluble OHPro quantities were roughly 3-fold and 15-fold higher in bleomycin-dosed lung tissue than in control tissue at 3 weeks, respectively. Although NaCl and SDS-soluble OHPro masses were elevated in bleomycin-dosed mice, 100% label incorporation (i.e. plateau labeling) prevented an accurate assessment of absolute synthesis rates in those fractions.
Fig. 6.

OHPro and collagen kinetics. A, fraction of newly synthesized OHPro present in protein extracts from control and bleomycin-induced fibrotic lung tissue. B, linear regression analysis of insoluble collagen α-1(I) turnover (LC-MS) and total OHPro turnover (GC-MS). C, absolute OHPro synthesis in pulmonary protein extracts from control and bleomycin-induced fibrotic lung tissue (note log scale). Values are means ± S.D. (n = 3) with statistical comparison between control and treatment groups at each time point (*p < 0.05).
DISCUSSION
A combination of dynamic proteomics and tissue decellularization was utilized to quantify changes in ECM fractional synthesis associated with the onset and progression of experimental fibrotic disease in vivo in the mouse. FSRs for dozens of ECM proteins were determined by monitoring stable isotope incorporation into newly synthesized proteins in a common model of pulmonary fibrosis. Conventional proteomic strategies targeting fibrosis-associated proteins are typically limited to semi-quantitative snapshots of ECM content, providing little to no insight into protein dynamics. Our analysis of healthy mouse lung tissue measured ECM protein FSRs ranging from less than 10% per week (e.g. type I collagen, elastin) to greater than 75% per week (e.g. fibronectin), demonstrating the complex dynamic state of pulmonary ECM. Following bleomycin exposure. ECM protein fractional synthesis was significantly altered, with some proteins affected more than others during early and late disease response. As fibrotic disease is characterized by perturbations in normal ECM dynamics resulting in ECM accumulation, we posit that the measurement of protein fractional synthesis provides a unique perspective on ECM accumulation and turnover in the development of fibrotic disease.
The overwhelming majority of ECM proteins were detected in the guanidine-soluble and insoluble pulmonary tissue protein fractions. Overall, guanidine-soluble ECM protein FSRs were higher than insoluble FSRs in sham control mice. The elevated pyridinoline cross-link density detected in the insoluble protein fraction provides one explanation for differential protein extractability. This supports FSR data indicating slower overall ECM protein turnover in the insoluble protein fraction, as cross-linking promotes collagen fibril stability. Interestingly, several individual proteins identified in both fractions had significantly different FSRs, allowing for a direct comparison of guanidine-soluble and insoluble protein pool kinetics. Label incorporation occurred faster in the guanidine-soluble forms of collagen I, perlecan, and laminin than it did for the same proteins in the insoluble form in control lungs. This indicates that guanidine extraction of acellular lung tissue favors the enrichment of a subpopulation of more recently synthesized, less mature ECM proteins. Collagen VI demonstrated the opposite phenomenon, with the insoluble pool turning over at a faster rate than its guanidine-soluble counterpart. This heterogeneity in differential FSRs across guanidine-soluble and insoluble protein fractions might result from the preferential interaction of newly synthesized protein populations with other, more mature protein populations, or vice versa, and deserves further exploration.
Measurement of increased collagen content is currently the gold standard for assessing the severity of fibrotic tissue disease. We therefore focused much of our analytic effort on the characterization of collagen fractional synthesis across different protein fractions. Dynamic proteomic analysis revealed a dramatic increase in fibrillar collagen turnover (types I, III, and V) following bleomycin administration, in both the guanidine-soluble and the insoluble protein pools. Whereas label incorporation occurred more slowly in insoluble collagens than in guanidine-soluble collagens in control mice, bleomycin administration made label incorporation virtually indistinguishable between the two pools after 3 weeks. This reflects a dramatic accumulation of typically stable, slowly turning over collagen, most of which appeared to occur between 1 and 3 weeks post-induction of pulmonary fibrosis. Although bleomycin also increased the FSR of basement membrane proteoglycans (laminin, perlecan) in both fractions, the proportion of newly synthesized protein in each fraction was similar.
GC-MS analysis of total OHPro quantity and turnover provided additional insight into collagen flux within the various protein fractions. The relatively small but fast turnover pool of OHPro isolated in the NaCl and SDS-soluble protein fractions is indicative of newly synthesized collagens. Increased OHPro quantity and FSR within these fractions following bleomycin administration likely reflects an increase in new collagen synthesis. Guanidine-soluble OHPro fractional synthesis closely matched that of type I collagen as determined via LC-MS analysis following bleomycin administration, but no change was detected in OHPro quantity in this fraction. A higher FSR with no change in pool size reflects the presence of a steady state in which increased guanidine-soluble collagen synthesis is balanced with degradation or the conversion of newly synthesized protein molecules to an insoluble form. Accumulation of insoluble collagen was confirmed by an increased FSR and a roughly 70% increase in insoluble OHPro content at 3 weeks post-bleomycin. Elevated concentrations of pyridinoline cross-links present in the insoluble collagen fraction provide one means for collagen transformation between guanidine-soluble and insoluble states. Additional forms of collagen cross-linking might also contribute, as we also detected increased fractional synthesis of tissue transglutaminase in fibrotic tissues (31).
Along with collagens, elastic microfibrils are highly prevalent in lung tissue, contributing to pulmonary viscoelastic properties (5). We observed significantly elevated fractional synthesis of microfibril-related proteins including elastin, fibrillin-1, EMILIN-1, and fibulin-5 following administration of bleomycin, particularly during the later phase of disease response (post 1 week). Previous studies showed an increase in elastic fiber content associated with fibrotic disease (5, 32, 33). It is therefore likely that increased labeling of microfibrillar proteins comes as a result of increased synthesis and accumulation rather than an increase in the degradation of existing unlabeled proteins. These data indicate that like fibrillar collagen FSRs, elastic microfibril-related protein FSRs also might serve as effective markers of fibrotic disease activity.
Basement membrane proteoglycan FSRs were also altered by bleomycin administration. Guanidine-soluble proteoglycans had higher FSRs than insoluble proteoglycans in bleomycin-dosed tissue during both early and later disease response. Insoluble proteoglycan turnover, in contrast, was altered only during the later fibrotic response (1 to 3 weeks). Interestingly, collagen IV, though detectable only in the insoluble protein fraction, appeared to more closely resemble the fractional synthesis profile of guanidine-soluble basement membrane proteoglycans, potentially reflective of an interaction between these protein populations.
Other proteins of interest included small leucine-rich proteoglycans, which were observed to have a wide range of turnover rates. Biglycan and decorin, two commonly studied small leucine-rich proteoglycans associated with collagen fibril formation and TGF-β superfamily growth factor activity (34, 35), were nearly fully labeled in control lungs at 1 week. Although this experimental design factor diminished the absolute difference that we were able to detect in labeling between experimental groups, statistical differences in biglycan fractional synthesis were still observed. These differences may result from a combination of increased protein pool size and the presence of a small pool with a very slow turnover rate. Similar results were observed for fibronectin, an abundant ECM glycoprotein previously shown to increase in quantity shortly following bleomycin administration (36). Future experiments utilizing shorter labeling periods would be useful for further study of fast-turnover ECM proteins, which might represent robust dynamic markers of fibrotic disease.
Dermatopontin, another proteoglycan associated with TGF-β activity through its interaction with decorin (37), fell well within the range of our labeling period. Dermatopontin turnover was higher in bleomycin-dosed lungs than in control tissues at both time points, indicative of a role in the fibrotic tissue response. Other ECM proteins including MFAP-2, MFAP-4, nephronectin, and periostin demonstrated very little change between bleomycin-dosed and control groups at 1week but large changes at 3 weeks. Such differences in individual ECM protein FSRs over time might allow for the identification of specific dynamic protein markers of different stages of fibrotic disease.
The applications for ECM-focused dynamic proteomics in the diagnosis and treatment of fibrotic diseases are potentially important. From a basic research perspective, these techniques are useful in profiling ECM protein flux associated with the onset and developmental stages of fibrotic disease. Identification of dynamic biomarkers could provide novel therapeutic targets, as well as allow for more accurate diagnosis of disease progression or anti-fibrotic drug efficacy. Comparisons of global ECM protein dynamics in various animal models of fibrosis with those observed in human disease might also provide valuable information regarding the validity of those animal models (i.e. reverse translation). This might be particularly relevant in the study of pulmonary fibrosis, where there is currently debate over the relevance of the bleomycin model to human idiopathic pulmonary fibrosis (27, 38, 39). As stable isotopes including D2O are routinely used in human subjects, the methods described herein are safely translatable to biopsied human tissue. Dynamic biomarkers of pulmonary fibrosis might also be obtainable in biofluids such as bronchial lavage fluid or plasma, potentially acting as surrogate markers of disease. This strategy is supported by multiple studies quantifying ECM breakdown products in plasma that appear to correlate with fibrotic disease (40–43).
It is important to note that allowing for the hydroxylation of proline as a post-translational modification during LC-MS/MS peptide identification was a vital step in our analysis of collagen FSR, as >90% of extracellular collagen I peptides detected in this study included OHPro residues. We also considered the effect of proline hydroxylation on our calculation of collagen turnover, but we detected no change in collagen peptide FSR related to the presence of one or more OHPro residues (data not shown). Although proline hydroxylation eliminates one 2H-labeling site in the de novo proline synthesis pathway, the impact of this difference on peptide FSR is minimized by two factors: the relatively greater abundance of alternative sources of proline (e.g. diet or protein degradation products), and the limited proportion of OHPro relative to other amino acids present in any given collagen peptide (21).
One shortcoming of this study was our inability to perfectly match the labeling times of animal groups at early and late collection points. Because of weight loss and morbidity associated with bleomycin administration, early sacrifice of some animals was required. However, as we report here increased ECM protein synthesis rates as a result of pulmonary exposure to bleomycin, shorter labeling periods in animals exposed to bleomycin do not account for these findings. In addition, we chose not to represent FSR data as a daily rate by fitting to a one-phase exponential association because of the high, presumably plateaued FSRs of many ECM proteins at both time points.
Another technical challenge lay in the difficulty of interpreting ECM protein FSR data during the onset of fibrotic disease because of the large changes in total ECM protein quantity. For example, it has been reported that the total ECM quantity may increase as much as 6-fold following the onset of liver fibrosis (44). Such drastic changes in pool size can make it difficult to interpret corresponding changes in protein FSR, as the ratio of synthesis to degradation shifts away from a steady state. In the case of collagen, the quantitation of total OHPro provided one solution, allowing us to calculate absolute collagen synthesis over the labeling period. Additional quantitative proteomics-based and non-proteomics-based techniques would also assist in understanding quantitative changes in particular proteins of interest. Future studies administering isotope label only at the later stages of disease might also ameliorate this problem, by distinguishing fractional synthesis associated with disease onset from that associated with the chronic fibrotic state. Although we do not report turnover data associated with cellular proteins here, such data will also likely be valuable in understanding disease progression. For example, smooth muscle actin, a marker of myofibroblast activation that we found to be present across multiple protein fractions, showed an increased FSR in bleomycin-dosed tissues.
Fibrotic diseases, characterized by a chronic imbalance in ECM turnover favoring elevated matrix deposition, present a significant worldwide medical problem with little currently available in the way of effective diagnostic or therapeutic strategies. Here, we demonstrate a technique combining dynamic proteomics and tissue decellularization biochemical procedures to quantify the fractional synthesis of a broad array of ECM proteins associated with fibrotic disease development. Fractionation of matrix proteins based on solubility resulted in the identification of physically separable ECM protein subpopulations with distinctive kinetic behaviors in both healthy and fibrotic pulmonary tissues. Moreover, we observed striking increases in fibrillar collagen synthesis 1 to 3 weeks post-bleomycin exposure, consistent with a pathogenic accumulation of mature cross-linked ECM. These techniques have implications in the development of improved diagnostics and ultimately treatments for fibrotic disease via improved understanding of matrix dynamics during the various stages of tissue fibrogenesis.
Supplementary Material
Acknowledgments
We thank J. Price, T. Angel, T. Riiff, and C. Khambatta for discussions regarding data analysis and presentation and/or critical reading of the manuscript. All authors are current employees of KineMed Inc.
Footnotes
Author contributions: M.L.D., M.K.H., S.M.T., and C.L.E. designed research; M.L.D., M.G., S.F., and F.L. performed research; W.E.H. contributed new reagents or analytic tools; M.L.D. and K.L. analyzed data; M.L.D. and W.E.H. wrote the paper; K.L., M.K.H., S.M.T., and C.L.E. edited the paper.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- ECM
- extracellular matrix
- FSR
- fractional synthesis rate
- GC-MS
- gas chromatography–mass spectrometry
- LC-MS
- liquid chromatography–mass spectrometry
- OHPro
- hydroxyproline.
REFERENCES
- 1. Cox T. R., Erler J. T. (2011) Remodeling and homeostasis of the extracellular matrix: implications for fibrotic diseases and cancer. Dis. Model. Mech. 4, 165–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Daley W. P., Peters S. B., Larsen M. (2008) Extracellular matrix dynamics in development and regenerative medicine. J. Cell Sci. 121, 255–264 [DOI] [PubMed] [Google Scholar]
- 3. Kisseleva T., Brenner D. A. (2008) Mechanisms of fibrogenesis. Exp. Biol. Med. 233, 109–122 [DOI] [PubMed] [Google Scholar]
- 4. Lu P., Takai K., Weaver V. M., Werb Z. (2011) Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 3:a005058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Suki B., Bates J. H. (2008) Extracellular matrix mechanics in lung parenchymal diseases. Resp. Physiol. Neurobiol. 163, 33–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wight T. N., Potter-Perigo S. (2011) The extracellular matrix: an active or passive player in fibrosis? Am. J. Physiol. Gastrointest. Liver Physiol. 301, G950–G955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wynn T. A., Ramalingam T. R. (2012) Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat. Med. 18, 1028–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cowan M. L., Rahman T. M., Krishna S. (2010) Proteomic approaches in the search for biomarkers of liver fibrosis. Trends Mol. Med. 16, 171–183 [DOI] [PubMed] [Google Scholar]
- 9. Noble P. W., Barkauskas C. E., Jiang D. (2012) Pulmonary fibrosis: patterns and perpetrators. J. Clin. Invest. 122, 2756–2762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ueha S., Shand F. H., Matsushima K. (2012) Cellular and molecular mechanisms of chronic inflammation-associated organ fibrosis. Frontiers Immunol. 3, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Booth A. J., Hadley R., Cornett A. M., Dreffs A. A., Matthes S. A., Tsui J. L., Weiss K., Horowitz J. C., Fiore V. F., Barker T. H., Moore B. B., Martinez F. J., Niklason L. E., White E. S. (2012) Acellular normal and fibrotic human lung matrices as a culture system for in vitro investigation. Am. J. Resp. Crit. Care Med. 186, 866–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Smits N. C., Shworak N. W., Dekhuijzen P. N., van Kuppevelt T. H. (2010) Heparan sulfates in the lung: structure, diversity, and role in pulmonary emphysema. Anat. Rec. 293, 955–967 [DOI] [PubMed] [Google Scholar]
- 13. Kim S. H., Turnbull J., Guimond S. (2011) Extracellular matrix and cell signalling: the dynamic cooperation of integrin, proteoglycan and growth factor receptor. J. Endocrinol. 209, 139–151 [DOI] [PubMed] [Google Scholar]
- 14. Price J. C., Holmes W. E., Li K. W., Floreani N. A., Neese R. A., Turner S. M., Hellerstein M. K. (2012) Measurement of human plasma proteome dynamics with (2)H(2)O and liquid chromatography tandem mass spectrometry. Anal. Biochem. 420, 73–83 [DOI] [PubMed] [Google Scholar]
- 15. Hellerstein M. K. (2004) New stable isotope-mass spectrometric techniques for measuring fluxes through intact metabolic pathways in mammalian systems: introduction of moving pictures into functional genomics and biochemical phenotyping. Metab. Eng. 6, 85–100 [DOI] [PubMed] [Google Scholar]
- 16. Hellerstein M. K., Neese R. A. (1999) Mass isotopomer distribution analysis at eight years: theoretical, analytic, and experimental considerations. Am. J. Physiol. 276, E1146–E1170 [DOI] [PubMed] [Google Scholar]
- 17. Crapo P. M., Gilbert T. W., Badylak S. F. (2011) An overview of tissue and whole organ decellularization processes. Biomaterials 32, 3233–3243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Barallobre-Barreiro J., Didangelos A., Schoendube F. A., Drozdov I., Yin X., Fernandez-Caggiano M., Willeit P., Puntmann V. O., Aldama-Lopez G., Shah A. M., Domenech N., Mayr M. (2012) Proteomics analysis of cardiac extracellular matrix remodeling in a porcine model of ischemia/reperfusion injury. Circulation 125, 789–802 [DOI] [PubMed] [Google Scholar]
- 19. Didangelos A., Yin X., Mandal K., Baumert M., Jahangiri M., Mayr M. (2010) Proteomics characterization of extracellular space components in the human aorta. Mol. Cell. Proteomics 9, 2048–2062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Naba A., Clauser K. R., Hoersch S., Liu H., Carr S. A., Hynes R. O. (2012) The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol. Cell. Proteomics 11, M111.014647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gardner J. L., Turner S. M., Bautista A., Lindwall G., Awada M., Hellerstein M. K. (2007) Measurement of liver collagen synthesis by heavy water labeling: effects of profibrotic toxicants and antifibrotic interventions. Am. J. Physiol. Gastrointest. Liver Physiol. 292, G1695–G1705 [DOI] [PubMed] [Google Scholar]
- 22. Atabai K., Jame S., Azhar N., Kuo A., Lam M., McKleroy W., Dehart G., Rahman S., Xia D. D., Melton A. C., Wolters P., Emson C. L., Turner S. M., Werb Z., Sheppard D. (2009) Mfge8 diminishes the severity of tissue fibrosis in mice by binding and targeting collagen for uptake by macrophages. J. Clin. Invest. 119, 3713–3722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Didangelos A., Yin X., Mayr M. (2012) Method for protein subfractionation of cardiovascular tissues before DIGE analysis. Methods Mol. Biol. 854, 287–297 [DOI] [PubMed] [Google Scholar]
- 24. McCabe B. J., Bederman I. R., Croniger C., Millward C., Norment C., Previs S. F. (2006) Reproducibility of gas chromatography-mass spectrometry measurements of 2H labeling of water: application for measuring body composition in mice. Anal. Biochem. 350, 171–176 [DOI] [PubMed] [Google Scholar]
- 25. Boutten B., Brazier M., Morche N., Morel A., Vendeuvre J. L. (2000) Effects of animal and muscle characteristics on collagen and consequences for ham production. Meat Sci. 55, 233–238 [DOI] [PubMed] [Google Scholar]
- 26. Chaudhary N. I., Schnapp A., Park J. E. (2006) Pharmacologic differentiation of inflammation and fibrosis in the rat bleomycin model. Am. J. Resp. Crit. Care Med. 173, 769–776 [DOI] [PubMed] [Google Scholar]
- 27. Peng R., Sridhar S., Tyagi G., Phillips J. E., Garrido R., Harris P., Burns L., Renteria L., Woods J., Chen L., Allard J., Ravindran P., Bitter H., Liang Z., Hogaboam C. M., Kitson C., Budd D. C., Fine J. S., Bauer C. M., Stevenson C. S. (2013) Bleomycin induces molecular changes directly relevant to idiopathic pulmonary fibrosis: a model for “active” disease. PLoS One 8, e59348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Okamoto O., Fujiwara S. (2006) Dermatopontin, a novel player in the biology of the extracellular matrix. Connective Tissue Res. 47, 177–189 [DOI] [PubMed] [Google Scholar]
- 29. Molleken C., Sitek B., Henkel C., Poschmann G., Sipos B., Wiese S., Warscheid B., Broelsch C., Reiser M., Friedman S. L., Tornoe I., Schlosser A., Kloppel G., Schmiegel W., Meyer H. E., Holmskov U., Stuhler K. (2009) Detection of novel biomarkers of liver cirrhosis by proteomic analysis. Hepatology 49, 1257–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Blaauboer M. E., Emson C. L., Verschuren L., van Erk M., Turner S. M., Everts V., Hanemaaijer R., Stoop R. (2013) Novel combination of collagen dynamics analysis and transcriptional profiling reveals fibrosis-relevant genes and pathways. Matrix Biol. 32, 424–431 [DOI] [PubMed] [Google Scholar]
- 31. Popov Y., Sverdlov D. Y., Sharma A. K., Bhaskar K. R., Li S., Freitag T. L., Lee J., Dieterich W., Melino G., Schuppan D. (2011) Tissue transglutaminase does not affect fibrotic matrix stability or regression of liver fibrosis in mice. Gastroenterology 140, 1642–1652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Baranova A., Lal P., Birerdinc A., Younossi Z. M. (2011) Non-invasive markers for hepatic fibrosis. BMC Gastroenterol. 11, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lorena D., Darby I. A., Reinhardt D. P., Sapin V., Rosenbaum J., Desmouliere A. (2004) Fibrillin-1 expression in normal and fibrotic rat liver and in cultured hepatic fibroblastic cells: modulation by mechanical stress and role in cell adhesion. Lab. Invest. 84, 203–212 [DOI] [PubMed] [Google Scholar]
- 34. Kolb M., Margetts P. J., Sime P. J., Gauldie J. (2001) Proteoglycans decorin and biglycan differentially modulate TGF-beta-mediated fibrotic responses in the lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 280, L1327–L1334 [DOI] [PubMed] [Google Scholar]
- 35. Kalamajski S., Oldberg A. (2010) The role of small leucine-rich proteoglycans in collagen fibrillogenesis. Matrix Biol. 29, 248–253 [DOI] [PubMed] [Google Scholar]
- 36. Hernnas J., Nettelbladt O., Bjermer L., Sarnstrand B., Malmstrom A., Hallgren R. (1992) Alveolar accumulation of fibronectin and hyaluronan precedes bleomycin-induced pulmonary fibrosis in the rat. Eur. Resp. J. 5, 404–410 [PubMed] [Google Scholar]
- 37. Okamoto O., Fujiwara S., Abe M., Sato Y. (1999) Dermatopontin interacts with transforming growth factor beta and enhances its biological activity. Biochem. J. 337, 537–541 [PMC free article] [PubMed] [Google Scholar]
- 38. Gauldie J., Kolb M. (2008) Animal models of pulmonary fibrosis: how far from effective reality? Am. J. Physiol. Lung Cell. Mol. Physiol. 294, L151. [DOI] [PubMed] [Google Scholar]
- 39. Moeller A., Ask K., Warburton D., Gauldie J., Kolb M. (2008) The bleomycin animal model: a useful tool to investigate treatment options for idiopathic pulmonary fibrosis? Int. J. Biochem. Cell Biol. 40, 362–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Veidal S. S., Bay-Jensen A. C., Tougas G., Karsdal M. A., Vainer B. (2010) Serum markers of liver fibrosis: combining the BIPED classification and the neo-epitope approach in the development of new biomarkers. Dis. Markers 28, 15–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Veidal S. S., Karsdal M. A., Nawrocki A., Larsen M. R., Dai Y., Zheng Q., Hagglund P., Vainer B., Skjot-Arkil H., Leeming D. J. (2011) Assessment of proteolytic degradation of the basement membrane: a fragment of type IV collagen as a biochemical marker for liver fibrosis. Fibrogenesis Tissue Repair 4, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Veidal S. S., Karsdal M. A., Vassiliadis E., Nawrocki A., Larsen M. R., Nguyen Q. H., Hagglund P., Luo Y., Zheng Q., Vainer B., Leeming D. J. (2011) MMP mediated degradation of type VI collagen is highly associated with liver fibrosis—identification and validation of a novel biochemical marker assay. PLoS One 6, e24753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Veidal S. S., Vassiliadis E., Bay-Jensen A. C., Tougas G., Vainer B., Karsdal M. A. (2010) Procollagen type I N-terminal propeptide (PINP) is a marker for fibrogenesis in bile duct ligation-induced fibrosis in rats. Fibrogenesis Tissue Repair 3, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Arthur M. J. (2000) Fibrogenesis II. Metalloproteinases and their inhibitors in liver fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 279, G245–G249 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.