

Abstract
Cilia and flagella are complex organelles made of hundreds of proteins of highly variable structures and functions. Here we report the purification of intact flagella from the procyclic stage of Trypanosoma brucei using mechanical shearing. Structural preservation was confirmed by transmission electron microscopy that showed that flagella still contained typical elements such as the membrane, the axoneme, the paraflagellar rod, and the intraflagellar transport particles. It also revealed that flagella severed below the basal body, and were not contaminated by other cytoskeletal structures such as the flagellar pocket collar or the adhesion zone filament. Mass spectrometry analysis identified a total of 751 proteins with high confidence, including 88% of known flagellar components. Comparison with the cell debris fraction revealed that more than half of the flagellum markers were enriched in flagella and this enrichment criterion was taken into account to identify 212 proteins not previously reported to be associated to flagella. Nine of these were experimentally validated including a 14-3-3 protein not yet reported to be associated to flagella and eight novel proteins termed FLAM (FLAgellar Member). Remarkably, they localized to five different subdomains of the flagellum. For example, FLAM6 is restricted to the proximal half of the axoneme, no matter its length. In contrast, FLAM8 is progressively accumulating at the distal tip of growing flagella and half of it still needs to be added after cell division. A combination of RNA interference and Fluorescence Recovery After Photobleaching approaches demonstrated very different dynamics from one protein to the other, but also according to the stage of construction and the age of the flagellum. Structural proteins are added to the distal tip of the elongating flagellum and exhibit slow turnover whereas membrane proteins such as the arginine kinase show rapid turnover without a detectible polarity.
Cilia and flagella are prominent organelles of many eukaryotic cells. The names “cilia” and “flagella” are often related to historical reasons but they correspond to the same entity: a cylindrical organelle surrounded by a membrane and composed of an axoneme, a set of nine doublet microtubules originating from the basal body. Motile cilia usually contain a central pair of single microtubules and various substructures involved in the generation or the control of flagellar or ciliary beating, such as dynein arms, radial spokes, or central pair projections. This structural organization is remarkably well conserved across evolution, being encountered from protists to mammals (1). The conservation is also found at the molecular level as observed by comparative genomics between species with or without cilia and flagella (2, 3). Nevertheless, proteomic analysis revealed that in addition to the common core, many components unique to each group of eukaryotes are also present (4–8).
The cilium represents a separate compartment from the cell body and does not contain any ribosomes or vesicles of any kind. The base of cilia and flagella contains projections that link each microtubule triplet of the basal body to the flagellum membrane (9). This region has been proposed to act as a barrier restricting entry of cytoplasmic proteins and ensuring retention of flagellum matrix elements (10). The transition zone is found in-between this area and the axoneme and contains several complexes of proteins (many of whom are mutated in the case of ciliopathies, genetic diseases affecting cilia function and/or formation) that contribute to the definition of the ciliary compartment (11, 12). Recent data showed that dextrans of low molecular weight are free to diffuse in the ciliary compartment as well as in the nucleus, whereas molecules of higher size (30 kDa or above) could not access these organelles. This led to the finding that a structure equivalent to the nucleopore complex is localized at the basal body area and could control access to the ciliary compartment (13). Finally, a septin barrier appears to be present close to the basis of the cilium and could control the trafficking of specific ciliary membrane proteins (14). The existence of a specific compartment comprising a large number of skeletal, matrix, and membrane proteins raises the issue of its internal organization. Key questions include the distribution of proteins, the mechanisms involved in specific distribution and the turnover during the life of the organelle.
We selected to address these basic phenomena in the protist Trypanosoma brucei, well known as the etiological agent of sleeping sickness in Africa, but that is also an amenable model for cilia studies (15). It possesses a single flagellum that contains a typical 9 + 2 axoneme emerging from a depression of the cell surface called the flagellar pocket. This structure can be related to the ciliary pocket found at the base of different types of cilia in mammalian cells (16, 17). The axoneme is flanked by a lattice-like structure called the paraflagellar rod (PFR)1 that is present as soon as the flagellum emerges from the pocket and runs to its distal end (18). The PFR contains at least 30 different proteins (19) and has been proposed to contribute to cell motility because its ablation results in cell paralysis in T. brucei (20) and in the related parasite Leishmania mexicana (21). The flagellum is attached to the cell body for most of its length, with the PFR lying close to the cell body side where a specific cytoskeletal structure termed the flagellum attachment zone (FAZ) is found (22). It is made of a unique filament composed of trypanosome-specific proteins (23, 24) and of four specialized microtubules flanked by the smooth endoplasmic reticulum (25). The flagellum plays key cellular functions as it drives cell motility (4, 26, 27), controls cell morphogenesis (28) and is responsible for parasite attachment during invasion of the salivary glands in the tsetse fly (29). Moreover, it could perform sensory functions and contribute to detection of the environment during the parasite life cycle (30). Recent data revealed the essential role of flagellum beating during fly invasion (31) but surprisingly reduction of forward motility did not affect infectivity in a mouse model (32).
Purification of intact flagella from trypanosomes is a challenging task because of the adhesion to the cell body. Detergent and high-salt treatment have been used to efficiently purify the skeletal fraction of the flagellum that contains the axoneme, the PFR, and the basal body but that also includes the kinetoplast (mitochondrial genome), the FAZ, and the flagellar pocket collar (4, 33, 34). However, membrane and matrix components are totally lost during this procedure. For example, none of the intraflagellar transport (IFT) proteins that normally traffic in the flagellum matrix along peripheral microtubules (35) could be detected in samples purified by this procedure (4). We therefore decided to purify intact flagella by using a mutant strain called FLA1RNAi where expression of an mRNA encoding a protein essential for flagellum attachment to the cell body (36) can be conditionally knocked-down by RNAi (37). FLA1RNAi cells exhibit detached flagella from the main cell body, with the exception of the anchoring point at the basal body (37). By mechanical shearing, we found out that flagella could be severed from the cell body while preserving their membrane and their matrix elements. After purification, flagellar fractions were exhaustively characterized at the level of light and electron microscopy and their content was determined by mass spectrometry that confirmed the presence of the majority of known flagellar markers and revealed novel flagellar components. Three previously characterized proteins (the arginine kinase and two 14-3-3 proteins) and 10 hypothetical proteins were investigated in detail. Out of these 13 candidate proteins, 10 turned out to be associated to the flagellum whereas the others could not be detected experimentally. The novel ones were termed FLAM, for Flagellum Members. Remarkably, these proteins showed very specific location patterns within the flagellum including the membrane, the distal tip of the axoneme or the first proximal half of the axoneme, and displayed unexpected variations in their turnover rate. Overall, we revealed the existence of multiple subdomains within the flagellum with very specific dynamics, further demonstrating the highly sophisticated organization of the organelle.
EXPERIMENTAL PROCEDURES
Trypanosome Cell Line and Culture
All cells used for this work were derivatives of T. brucei strain 427 (procyclic stage) and were cultured in SDM79 medium supplemented with hemin and 10% fetal calf serum. The FLA1RNAi cell line was generated by transfection of the p2T7FLA1 plasmid (37) in the 29–13 cell line (38).
Purification of Flagella
The FLA1RNAi cell line was grown in the presence of tetracycline for 3–4 days. After microscope verification of the phenotype and of cell viability, cells were spun down at 420 × g for 20 min, then the pellet was washed with buffer A (25 mm tricine, pH7, 1% BSA, 0.1 mm CaCl2, 0.2 mm EDTA, 5 mm MgCl2, and 12 mm mercaptoethanol) containing 0.32 m sucrose and the suspension was again centrifuged at 420 × g for 10 min. Finally, the pellet was carefully resuspended at a concentration of 3 × 108 cells/ml in buffer A containing 0.32 m sucrose. The suspension was transferred to 50 ml Falcon tubes (2.5 ml maximum per tube) and was vortexed continuously for 5 min, followed by centrifugation at 420 × g for 10 min (Sorval HS-4 rotor). The supernatant was recovered and recentrifuged twice while the pellet was discarded. The last supernatant was centrifuged at maximum speed in a table top centrifuge for 5 min at 4 °C. The flagellar pellet was resuspended in 25 μl of sterile PBS and either frozen or further treated to obtain ultra-pure flagella by dilution in 500 μl of buffer A containing 0.32 m sucrose and loading on top of a discontinuous gradient of sucrose made of 2.5 ml layers of 1.49/1.66/1.84/2.02 M sucrose in a Beckman plastic tube. The tube was then centrifuged in a Beckman SW-41Ti (8E4245) at 130,000 × g (28,000 rpm) for 4–15 h. The flagella equilibrate between 1.49 and 1.66 m sucrose and this interphase (recognized by its cloudy aspect) was carefully removed and spun down at maximum speed in a microfuge for 30 min. The pellet was washed once in buffer A for 15 min and finally resuspended in 25 μl of sterile PBS.
Flagella Shaving
The amount of protein per flagella preparation was evaluated using a Quick Start Bradford Dye Agent (Bio-Rad, Hercules, CA). For shaving (39), trypsin (Trypsin Gold Mass Spec Grade, Promega, Madison, WI) was added to each flagella preparation suspended in PBS with an enzyme to protein ratio of 1:50 and the mixture was incubated for 1h at 37 °C. It was then centrifuged for 5 min at maximum speed in a microfuge at 4 °C. The supernatant was transferred to a fresh tube and the pellet collected in 25 μl of sterile PBS. The composition of both samples was determined using nanoLC-MS/MS.
Trypsin Digestion
Supernatant and pellet were dried down in a vacuum concentrator and reconstituted respectively in 100 mm Tris HCl, pH 8.5, 8 m urea and in 100 mm ammonium bicarbonate. Samples were reduced (5 mm TCEP, 30 min at room temperature) and alkylated (10 mm iodoacetamide, 30 min at room temperature in the dark). For the supernatant after flagella shaving, Lys-C was added in a 1:40 (w/w) ratio and incubated for 5 h at 37 °C. Samples were diluted four times with 100 mm Tris HCl, pH 8.5. Trypsin (Trypsin Gold Mass Spec Grade, Promega) was added in a 1:100 (w/w) ratio and the incubation was carried out overnight at 37 °C. The next day, the same amount of enzyme was added and samples were further incubated for 5 h at 37 °C.
For the pellet after flagella shaving, samples were dried down in a vacuum concentrator after the reduction/alkylation steps and reconstituted in methanol/100 mm ammonium bicarbonate (60:40). 2 min vortex mixer and 1 min sonication under cold conditions were repeated five times (40, 41). Trypsin was added in a 1:100 (w/w) ratio and the incubation was carried out overnight at 37 °C. The next day, the same amount of enzyme was added and samples were further incubated for 5 h at 37 °C. Digestions were stopped by adding formic acid to 5% final concentration. Digest lysates were desalted by OMIX pipette Tips C18 solid phase extraction (Agilent Technologies, Santa Clara, CA), dried down and reconstituted in water/acetonitrile/formic acid (98:2:0.1).
Mass Spectrometry
Tryptic peptides were analyzed by nanoLC-MS/MS using an Ultimate 3000 system (Dionex, Amsterdam, The Netherlands) coupled to an LTQ-Orbitrap Velos (Thermo Fisher Scientific, Bremen). Each sample was loaded on a C18 precolumn (300 μm inner diameter × 5 mm; Dionex) at 30 μl/min in 2% acetonitrile, 0.1% formic acid. After 3 min desalting, the precolumn was switched online with the 15 cm capillary column (75 μm diameter, PepMap C18 3 μm) equilibrated in 96% solvent A (2% acetonitrile, 0.1% formic acid) and 4% solvent B (80% acetonitrile, 0.08% formic acid). Peptides were eluted using a 4% to 60% gradient of solvent B for 90 min at 300 nL/min flow rate. The LTQ-Orbitrap Velos was operated in data-dependent acquisition mode with the Xcalibur software (Thermo Fisher Scientific, Bremen). Survey scan MS were acquired in the Orbitrap in the 300–2000 m/z range with the resolution set to a value of 60,000 at m/z = 400. The 20 most intense ions per survey scan were selected for CID fragmentation, and the resulting fragments were analyzed in the linear trap (LTQ). Dynamic exclusion was employed within 90 s and repeated during 30 s to prevent repetitive selection of the same peptide.
Control samples (0 to 24 h induction) and flagella-enriched fractions (48 to 72 h induction) were analyzed in 3 and 5 biological replicates respectively.
Data Analysis
Raw data were analyzed using MaxQuant (MQ) software (V 1.3.0.5) with Andromeda as the search engine (42). A MudPit analysis was done for each replicate (pellet and supernatant). The search was done against a database containing the Trypanosoma brucei proteins (TbruceiTreu927AnnotatedProteins_TriTrypDB-4.1.fasta - 9826 sequences). Andromeda searches were performed choosing trypsin as specific enzyme with a maximum number of two missed cleavages. Possible modifications included carbamidomethylation (Cys, fixed), pyroglutamylation (Nterminus/Gln/Glu, variable), and oxidation (Met, variable). The mass tolerance in MS was set to 20 ppm for the first search then 6 ppm for the main search and 0.5 Da for the MS/MS. Maximum peptide charge was set to seven and seven amino acids were required as minimum peptide length. The “match between runs” feature was used with a maximal retention time window of 2 min. Proteins were considered as identified when found with at least two peptides in a minimum of three experiments.
The “proteinGroup.txt” file of MaxQuant was further used to extract LFQ (Label Free Quantitation) intensity values and calculate, for each protein, the ratio between the control (cell debris fraction) and the experiments (flagella fraction). For each group (debris and flagella fraction) an average of all LFQ intensities was used (supplemental Table S1, S2 and S3). Proteins were considered as enriched in the flagella fraction when the ratio was larger than 1.5.
Immunoblot Analysis
Samples were boiled in Laemmli buffer (2× stock: 0.5 m Tris pH 6.8 containing 20% Glycerol, 4% DTT, 4% SDS, Bromo-phenol blue) before SDS-PAGE separation, loading 2 μg of total cell protein per lane. The Criterion system (Bio-Rad) was used for electrophoresis: Criterion XT precast gels with XT Mops running buffer (20 x stock solution) in the Criterion electrophoresis cell (at 200 V constant). Proteins were transferred to PVDF membranes (Hybond-P from Amersham Biosciences) in the Criterion blotter (Bio-Rad) for 45 min at 100 V constant in TG buffer (10x stock: 0.25 mm Tris pH 8.3, 1.92 mm glycine). The membrane was blocked overnight with 5% skimmed milk in PBS and incubated with primary antibodies diluted in 1% milk and 0.1% Tween20 in PBS for 1 h. Membrane washes were performed with 0.2% Tween20 in PBS. Species specific secondary antibodies coupled to HRP (GE Healthcare) were diluted 1/20,000 in 1% milk and 0.1% Tween20 in PBS and incubated with the membranes for 1 h. Final detection was carried out by using an ECL kit according to manufacturer's instructions (Amersham Biosciences) and exposure of Hyperfilm-ECL (Amersham Biosciences). Signal intensity was analyzed using ImageJ and calibrated against a range of dilutions of loaded proteins.
Electron Microscopy
Cell fixation, embedding, and sectioning for transmission electron microscopy of whole cells or of flagella preparations was carried out as described previously (26). For scanning electron microscopy, cells were washed in PBS, fixed with 2.5% glutaraldehyde and treated as reported previously (43).
Expression of YFP Fusion Proteins and RNAi Silencing
Endogenous tagging of FLAM genes was achieved with plasmids that contain a fragment of the target gene sequence flanked by YFP in 5′ or 3′ positions. Before transfection, the plasmid was linearized in the target gene sequence, allowing homologous recombination with the target allele. As a result, the expression of the fluorescent fusion protein is under the control of one of the untranslated regions from the gene locus and one coming from the inserted plasmid. Different vectors for endogenous tagging were used that are described in (44) and sequences are found in (http://web.me.com/mc115/mclab/resources.html). The p3329 plasmid allows tagging with eYFP at the C-terminal end of the protein and requires selection with puromycin (1 μg/ml). It was used for all FLAM genes with the exception of FLAM4 and FLAM7 where N-terminal tagging with eYFP was chosen because of the presence of possible conserved domains in C-terminal positions. This was achieved using the p2675 vector with puromycin selection (see supplemental Table S5 for more information). For RNAi silencing, gene fragments were cloned in the pZJM vector (45) where they are flanked by T7 promoters facing each other, allowing tetracycline-inducible dsRNA expression in the cell line 29–13 that expresses a T7 RNA-Polymerase and tetracycline repressor (38). The sequences for dsRNA expression (supplemental Table S6) were selected using the RNAit algorithm (46). All the target sequence fragments were chemically synthesized by GeneCust Europe (Dudelange, Luxembourg) and subcloned into the appropriate vectors. For the generation of cell lines expressing dsRNA and the corresponding YFP fusion protein, 29–13 cells were nucleofected with both plasmids simultaneously and selection was carried out using phleomycin (2.5 μg/ml) and puromycin (1 μg/ml). For RNAi against arginine kinase (AK), a suitable targeting sequence of 406 bp (spanning bp 226 to 631, when the numbering was set to zero at the AK1 sequence start) was selected with RNAit in order to target transcripts from all three very similar AK genes. The fragment was amplified using the primers: forward 5′ AAGCTTACTGTGTTTGCCGACCTCTT ′3 (HindIII site underlined) and reverse 5′ CTCGAGAGATACCACGACCAGTTGGC ′3 (XhoI site underlined). Before transfection into trypanosomes, all pZJM plasmids were linearized at the unique NotI site in the rDNA intergenic targeting region. For pZJMAK1–3, linearization was performed with ClaI, as the fragment contains a NotI recognition sequence. The enzymes used to linearize the plasmids designed for FLAM endogenous tagging can be found at supplemental Table S5.
Trypanosomes were transfected with the linearized plasmid constructs by Nucleofector® technology (Lonza, Italy) as described (47). Transgenic cell lines were selected in medium supplemented with the respective antibiotics. For induction of RNAi, tetracycline (Sigma) was added to the medium at each dilution step at a final concentration of 1 μg/ml. In order to obtain clonal cell lines, the selected population was serially diluted and the dilutions spread to 96-well plates to obtain ¼ of the wells with 50 cells/well, ¼ of wells with 5 cells/well and half with in theory 0.5 cells/well. For this procedure, to facilitate growth of procyclic cells at low densities they were diluted in conditioned medium (medium that had been used to culture procyclic forms up to a density of 107 cells/ml, then has been centrifuged and the supernatant filtered through a filter with pores of 0.2 μm diameter from Millipore).
Immunogold Labeling
Trypanosomes were washed twice in SDM79 without serum by a 350 × g centrifugation of 5 min at 20 °C. The parasite membrane was stripped by a 10-min Triton X 100 treatment at 4 °C in PBS (48). Then, cytoskeletons were fixed in a mixture of 2% paraformaldehyde and 0.1% glutaraldehyde for 10 min. After 3 washes of 5 min and a neutralization step (addition of 50 mm NH4Cl for 20 min), samples were blocked for 20 min in PBS containing 0.1% bovine serum albumin (BSA) and incubated for 45 min at room temperature with a specific anti-GFP antibody (Invitrogen: A6455) diluted 1 in 50 in PBS containing 0.1% BSA. After two washes with PBS-0.1% BSA, a secondary anti-rabbit antibody coupled to 20 nm gold particles was diluted 1 in 30 in PBS-0.1% BSA and incubated for 15 min. Finally, parasites were washed 3 times in PBS and post-fixed in 2.5% glutaraldehyde in PBS for 30 min. The fixed samples were treated with 1% (w/v) OsO4, dehydrated, critical point dried (CPD 7501, Polaron), and coated with carbon powder (Ion Beam Coater 681, Gatan, CA, USA). Samples were visualized with the scanning electron microscope JEOL JSM 6700F (Japan).
Indirect Immunofluorescence Assay (IFA)
Cultured parasites were washed twice in SDM79 medium without serum and spread directly on poly-l-lysine coated slides (Menzel-Gläser, Braunschweig) before fixation. For methanol fixation, parasites were air dried and fixed in methanol at -20 °C for 30 s to 5 min followed by a rehydration step for 15 min in PBS. For PFA fixation, parasites were left to settle on slides, rinsed in PBS before being incubated for 30 min at room temperature with a 4% PFA solution in PBS at pH 7. After a permeabilization step with 0.1% Nonidet P-40 (Fluka) in PBS, samples were blocked for 1 h with 1% BSA in PBS. To obtain demembranated parasites, the cells were left to settle on poly-l-lysine coated slides for 10 min, rinsed in PBS and treated for 7 s with 1% Nonidet P-40 in PEM buffer (0.1 m PIPES pH 6.9, 2 mm EGTA, 1 mm MgSO4). After thorough washes, the samples were fixed in 4% PFA in PBS for 30 min and washed again.
For immunodetection, slides were incubated with the appropriate dilution of the first antibody in 0.1% BSA in PBS for 1 h. MAb25 that recognizes the axonemal protein TbSAXO1 (49) was used as a marker of the axoneme (50), L8C4 that picks up the PFR2 protein served as a PFR marker. After three 5 min-washes, species and subclass-specific secondary antibodies coupled to the appropriate fluorochrome (Alexa 488, Cy3, or Cy5, Jackson ImmunoResearch, West Grove, PA) were diluted 1/400 in PBS containing 0.1% BSA and were applied for 1 h. After washing as above, cells were stained with a 1 μg/ml solution of the DNA-dye DAPI (Roche) and mounted with the ProLong antifade reagent (Invitrogen, Carlsbad, CA). Slides were either stored at −20 °C or immediately analyzed, either with a DMR microscope (Leica) and images captured with a CoolSnap HQ camera (Roper Scientific, Trenton, NJ) or with a DMI4000 microscope (Leica) and images acquired with a Retiga-SRV camera (Q-Imaging, Surrey, BC, Canada). Pictures were analyzed using the IPLab Spectrum 3.9 software (Scanalytics and BD Biosciences) and were merged and superimposed using Adobe Photoshop (CS2). Measurements of cell parameters were performed with the ImageJ 1.38× software (NIH).
Quantification of the FLAM8 Signals
FLAM8::YFP expressing cells were fixed in methanol and stained by IFA with the anti-GFP (green) and mAb25 (red), as well as DAPI as described above. Z-stacks were acquired for each channel (100 planes in Z-steps of 100 nm). Stacks were deconvolved using the Huygens Essential software with the theoretical point spread function calculated for the used objective. To make sure that weak signals were not erased by this procedure, the non-deconvolved raw data stacks were analyzed in parallel. Using the ImageJ software, the stack corresponding to the Mab25 image was assembled to one plane using the Z-project tool. Cells with 2 flagella were selected and length of both flagella was measured. The DAPI content was exploited to define cells as 1Kd1N (K for kinetoplast, N for nucleus, d for dividing, recognizable as a single large kinetoplast), 1Kb1N (b for bone-shaped kinetoplast clearly incised in the middle), 2K1N, and 2K2N. On the green channel (FLAM8::YFP detected by the anti-GFP), a Z-projection was made without averaging, so that the pixel with the maximum intensity remains as such. In order to determine the range of fluorescence intensity for each cell, the tool HiLo (in the lookup tables) was used to code the dimmest pixel blue and the brightest red. Using the adjust brightness and contrast, the background on the overall cell was set to being all blue and as the tip of the old flagellum was the brightest signal in the cell, it was set to just underneath being saturated. This determines the threshold of fluorescence intensity and can set the viewing options in-between the minimum and maximum value. This procedure was done for each cell separately to determine its specific background and brightest signal. After setting the threshold, the area that is in-between can be measured to get the output of the area, the mean gray value, the minimum and maximum gray value, and the integrated density (the product of area and mean gray value). A total of 25 microscopic fields containing more or less cells in division were analyzed, containing a total of 46 cells at various stages of the cell cycle. The measurements made with ImageJ were exported to Excel to calculate the ratio of the new flagellar length versus the old one, the difference of maximum to minimum signal for old and new flagellar tip providing the ratio of new versus old, and similarly the ratio of the integrated density of new versus old.
Fluorescent Recovery After Photobleaching (FRAP) Analysis
The expression of FLAM8::YFP was first observed directly with a DMI4000 Leica microscope using a mercury bulb for excitation to verify correct protein expression and localization. For FRAP analysis, a Zeiss inverted microscope (Axiovert 200) equipped with an oil immersion objective (magnification x63 with a 1.4 numerical aperture) and a spinning disk confocal head (CSU22, Yokogawa) was used (51). Images were acquired using Volocity software with an EMCCD camera (C-9100, Hamamatsu) operating in streaming mode. A sample (a 100 μl drop) was taken directly from the culture grown at 6 to 8.106 cells/ml and trapped between slide and coverslip. The samples were kept at 27 °C using a fast response mini-stage temperature controller allowing rapid changes in temperatures. Time-lapse sequences were acquired to analyze GFP signal recovery after photo-bleaching. A total of 12 image sequences were acquired in three independent FRAP experiments. Movies were taken using a time lapse of 30 s. Exposure time was 0.8 s per frame (binning was 1 × 1 pixels). Fluorescence intensity was measured in the region of interest using the ImageJ software. To normalize data, background was subtracted and overall photo-bleaching over time was normalized using another equivalent source of GFP (the fluorescent spot at the distal tip of a flagellum in a cell that had not been bleached). Because two obviously distinct profiles were obtained, films were separated in two categories for final presentation: those exhibiting recovery and those that did not.
RESULTS
Purification and Characterization of Intact Trypanosome Flagella
To purify intact flagella from T. brucei, the FLA1RNAi cell line (corresponding to the procyclic stage that normally proliferates in the midgut of the tsetse fly) was used. It expresses double-stranded RNA corresponding to the FLA1 gene that encodes a protein necessary for flagellum adhesion to the cell body under the control of tetracycline-inducible promoters (37). In our hands, close to 100% of the cells exhibited a flagellum detached from the cell body 2–4 days after addition of tetracycline (supplemental Fig. S1A, S1B). Longer induction periods resulted in multiple cell cycle defects as previously reported (37). Therefore, experiments were carried out with cells that were grown in the presence of tetracycline for a maximum of 3 days. To mechanically shear flagella from the cell body, different approaches were attempted: Dounce homogenizer, syringe passage, sonication, or vortex. The latter one turned out to be the most efficient in releasing intact flagella (supplemental Fig. S1C). Centrifugation was used to precipitate cell bodies and remnants and the supernatant was layered first on top of a 0.32 m sucrose solution, then on top of a 0.61 m sucrose solution. Fractions were collected and analyzed by light microscopy, revealing a high enrichment in flagella (supplemental Fig. S1D). Flagellar samples were then processed for scanning electron microscopy analysis that confirmed the absence of major contamination, although cell remnants could occasionally be detected (supplemental Fig. S1E, S1F). Higher magnification suggested that most flagella were intact, although some membrane damage was occasionally observed (supplemental Fig. S1G, S1H).
Immunofluorescence assay (IFA) analysis using the monoclonal antibodies MAb25 and L8C4 as typical markers of the axoneme (49) and the PFR (52) confirmed flagellar identity (supplemental Fig. S2 and data not shown). To evaluate whether these flagella still possess their membrane, they were probed by IFA with antibodies against the IFT components IFT172 (35) (supplemental Fig. S2) and IFT22/RABL5 (53) (data not shown). IFT proteins are transported as large complexes along the axoneme microtubules underneath the flagellum membrane and are lost upon membrane removal (35, 53). Both antibodies produced a significant signal on most flagella, showing that these are likely to be intact. DAPI staining turned out to be almost negative, suggesting that flagella were severed from the cell body without the kinetoplast (normally physically linked to the proximal portion of the basal body (54)). However, light microscopy is neither sensitive enough to examine the actual structure of the flagella nor to detect potential small contaminants.
Further investigation of two purified flagella preparations was carried out by transmission electron microscopy (Fig. 1). This revealed that the membrane was still present in more than 80% of the flagellar sections (Fig. 1A, 1B). Sections in the basal body area confirmed that flagella got rooted out from the cell body without the kinetoplast (Fig. 1C–1E). The membrane appeared to seal at the basal body, hence resulting in the recovery of complete flagella without contamination from the kinetoplast. Some exquisite sections through the proximal portion of the flagellum were observed, including the basal body made of triplet microtubules, the transition zone, the axoneme and the emergence of the PFR, only present once the flagellum exits from the flagellar pocket (Fig. 1C). Cross-sections through these different areas confirmed the normal structure of typical elements such as the transition zone made of doublet microtubules and its “Y” fibers (Fig. 1F), the 9 + 2 axoneme with dynein arms and radial spokes (Fig. 1G, 1H) and the PFR (Fig. 1H). IFT particles were commonly detected (Fig. 1H) and positioned along specific microtubules doublets 3–4 and 7–8 as previously reported (35). No evidence for contamination by cytoskeleton-associated structures such as the flagellar pocket collar (34) or the flagellum attachment zone (23) could be observed. Finally, despite the significant enrichment in flagella, remnants of cell bodies were sometimes detected (arrow on Fig. 1B). Overall, these data demonstrate the high enrichment of mostly intact flagella in terms of structure, with little contamination from other cellular elements.
Fig. 1.

Morphological characterization of purified T. brucei flagella. Flagella were purified from procyclic FLA1RNAi cells grown in the presence of tetracycline for 72h, fixed, and sectioned before analysis by transmission electron microscopy. A, Low magnification view of a preparation of purified flagella. The membrane is preserved in ∼80% of the sectioned flagella. B, Higher magnification of a separate preparation of flagella. Although flagella are highly abundant, small cell debris (containing membrane and subpellicular microtubules) are sometimes encountered (arrow). Flagellar membrane remnants are also present. C, Section through the base of a purified flagellum reveals the typical aspect of the basal body (BB), the transition zone (TZ), the axoneme (AXO). and the emergence of the paraflagellar rod (PFR). D, E, Sections through the basal body area of purified flagella showing the absence of the kinetoplast. F–H, Sections through the transition zone (F), the axoneme (G) or the axoneme and the PFR (H) of purified flagella showed that these structures appear intact. IFT particles can be detected on cross-sections of flagella (white arrows) and occupy the usual position relative to axoneme microtubules. Scale bars, A–B, 2 μm; D–H, 100 nm.
Determination of the Molecular Composition of Purified Intact Flagella
The molecular composition of the purified flagella from induced FLA1RNAi cells was investigated by Western blotting experiments using antibodies for recognized flagellar marker proteins (Fig. 2). A ∼15-fold enrichment was observed for two well characterized components of the flagellar skeleton: the axoneme protein TbSAXO1 (49) (Fig. 2A) and one of the main PFR proteins PFR2 (52) (Fig. 2B, top). Importantly, an almost similar enrichment was observed for the calflagins, a family of calcium-binding proteins associated to the flagellum membrane (55) (Fig. 2B). Variable enrichment levels were seen for IFT proteins, ranging from ∼3-fold for IFT172 to ∼15-fold for RABL5/IFT22 (Fig. 2B), perhaps reflecting a different distribution of these proteins between the cytoplasm and the flagellar compartment (35, 53). In contrast, the endoplasmic reticulum marker BiP was depleted from the flagellar fractions but was still detected, although in much lower proportions compared with flagellar proteins (Fig. 2A, 2B). These results add on the structural data and demonstrate the quality of the flagella purification method.
Fig. 2.
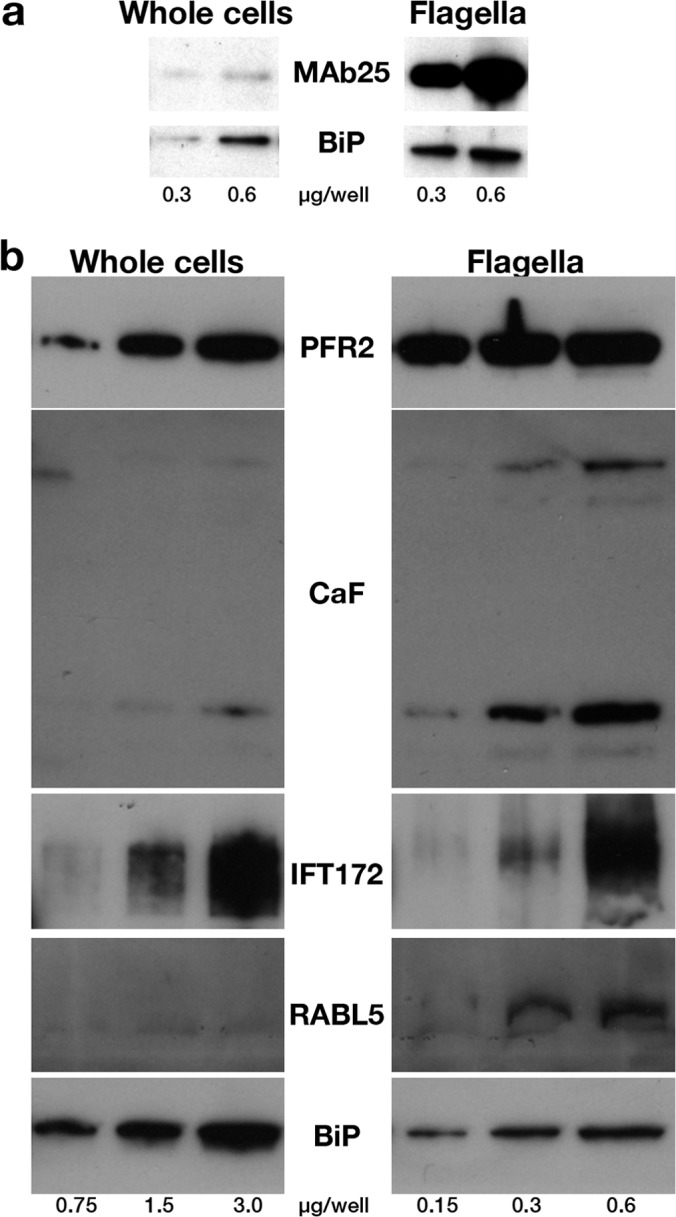
Marker proteins are enriched in flagellar preparations. A, Western blotting analysis of increasing concentrations of total cells or purified flagella (indicated in μg of proteins per well below the figure) probed with the axonemal marker MAb25 or with the endoplasmic reticulum component BiP. A 15-fold enrichment of the MAb25 signal is observed relative to that obtained for BiP. B, Western blotting analysis of increasing concentrations of total cells or a separate preparation of purified flagella (indicated in μg of proteins per lane below the figure) probed with markers of the PFR (PFR2), the membrane (calflagins) or of IFT particles (IFT172, RABL5/IFT22). Flagellar proteins are enriched three- to 15-fold compared with the endoplasmic reticulum marker BiP.
A semi-quantitative LC-MS approach was chosen to identify the proteins enriched in the flagellar fraction. For this quantitative analysis, five different samples of flagellar fractions obtained after 48 or 72 h culture of induced FLA1RNAi cells (in which flagella are totally detached from the body) were compared with three control samples consisting of fractions obtained after 0 or 24 h culture of FLA1RNAi cells (in which flagella are not yet detached). After centrifugation, the control samples mainly contained cell debris.
To ensure better access to membrane proteins on intact flagella, as well as to complex skeletal structures on flagella deprived of membrane (that represent 20% of the sample), an initial trypsin digest was applied to the preparations, in a so-called “shaving” procedure (39), followed by centrifugation. All fractions (supernatant and pellet) were analyzed by LC-MS/MS using an LTQ Orbitrap Velos.
This analysis led to the identification of 751 proteins (supplemental Table S1). These included 255 proteins out of the 380 proteins retrieved in the proteome of the flagellar skeleton published by Broadhead et al. (4). We then sought for the presence of typical flagellar markers (56). A list of 122 markers was established based on either direct experimental evidence from T. brucei or a closely related organism (other Trypanosoma subspecies or Leishmania) or similarity to well-recognized flagellar components that are highly conserved across evolution such as dynein arm, radial spoke or IFT components (supplemental Table S2). A total of 108 of these proteins were detected in our flagellar proteome, representing ∼88.5% of the expected flagellar markers. A breakdown analysis revealed the high abundance of skeletal proteins (81/89) including axoneme (46/48), PFR (31/35), and basal body components (4/6) (Table S2); but also of membrane and matrix components (27/33). None of the known components of the flagellar adhesion zone were detected, in agreement with the absence of visible contamination by this structure.
These results are very encouraging but nevertheless, electron microscopy analysis revealed the low but consistent occurrence of small cell body remnants in the flagellar preparations. This could explain the presence of a significant number of proteins known to be abundant in the cell body such as translation factors, ribosomal proteins, glycosomal enzymes, mitochondrial components, or histones. To evaluate the contribution of this material to the list of identified proteins, three control experiments were carried out using FLA1RNAi cells that had been induced for 0–24 h, when flagella are not yet detached from the cell body. These cells were treated the same way as samples induced for 48–72 h and fractions usually containing flagella were collected from the gradient. However, microscope observation revealed that these fractions contained few flagella but mostly cell debris.
A quantitative analysis using LFQ values obtained from MaxQuant was used to determine the ratio of each protein between the flagella and the cell debris fraction. In case a protein was not encountered at all in the cell debris fraction, this ratio becomes infinite (363 proteins). The threshold to consider that a protein was enriched in the flagellum fraction was set to a ratio of 1.5. This approach revealed that 67 out of the 108 flagellar markers identified in the flagellum proteome were actually enriched (62%). Enrichment scores were high for most skeletal elements (outer and inner dynein arms, radial spokes, central pair, PFR, and basal body), in agreement with the fact that these elements have little or no soluble cytoplasmic pool in trypanosomes (20). As expected, scores were relatively neutral for proteins known to be also present in the cell body such as tubulins or IFT proteins (12 out of 21 were enriched in flagella, in agreement with the large cytoplasmic pool (57)) (Table S2). In contrast, only six out of the 20 ribosomal proteins were enriched in the flagellar preparations (supplemental Table S1).
This repartition criterion was used as a guide to screen the list of identified proteins to select the ones that were likely to be enriched in flagella. Please note that this stringency criterion does not prove that a protein is missing from the flagellum, but actually indicates that it is less likely to be found in the flagellum than in the cell body. Supplemental Table S3 groups proteins that had not been found in the proteome of the flagellar skeletons (4), that were detected in all five experiments and that showed a relative score flagella::cell debris above 1.5, yielding a list of 212 candidate proteins.
While this article was in the final stages of preparation, the purification of intact flagella from the bloodstream stage of T. brucei was reported (8). In this case, cells were surface biotinylated, then flagella were purified, lysed in detergent, and the soluble fraction containing membrane and matrix proteins was applied to a streptavidin column. The bound fraction was defined as the flagellum surface proteome (FSP) whereas the unbound corresponded to the flagellum matrix proteome (FMP) (8). These results were compared with the protein list obtained here from procyclic T. brucei (supplemental Table S3). Only 16 proteins were shared with the bloodstream surface proteome, likely reflecting the wide difference in the environment in which procyclic and bloodstream trypanosomes develop (58). More expectedly, 73 proteins (34.4%) identified here were also detected in the FMP of bloodstream cells (supplemental Table S3). Finally, a total of 16 proteins (7.5%) were picked-up in another proteomic study of intact flagella obtained from wild-type procyclic trypanosomes (59).
Two approaches were selected to validate this proteome. First, we used available antibodies to known proteins such as two 14-3-3 proteins (60) that turned out to be enriched in the flagellar fraction. Because the repartition criterion is not definitive in the case of proteins present in both the cell body and the flagellum, we also investigated the arginine kinase, a protein known to exhibit such a profile (61) and that produced a relatively neutral score (1.27) but with a high number of peptides (supplemental Table S1). Second, in order to find new members present in the flagellum, we selected 10 proteins listed in supplemental Table S3 that were indicated as hypothetical for further biological validation. Eight of them were selected based on the enrichment criterion described above and because of their high abundance, and two hypothetical proteins that did not meet our criterion but showed a fairly large number of identified peptides (respectively 12 and 15) in the flagellar fraction were also added.
Novel Proteins Localize to Distinct Subdomains of the Flagellum
To experimentally validate the presence of these proteins in the flagellum, we first used existing antibodies against proteins previously studied in a different context. Antibodies turned out to be available against the arginine kinase (AK) and two 14-3-3 proteins. AK is an enzyme of the phosphagen biosynthetic pathway and is encoded by three very closely related genes in T. brucei (62). The three encoded proteins share a highly conserved central domain with only tiny extensions providing protein specificity for cytoplasmic, glycosomal or flagellar localization (61). An anti-AK antiserum was raised in rabbits against the Trypanosoma cruzi protein where it stains the cytosol (62) but was reported to cross-react with its T. brucei counterparts by immunoblotting (63). The antibody was employed for immunofluorescence assay (IFA) either on paraformaldehyde (PFA) or on methanol-fixed trypanosomes and in both cases produced a clear flagellum signal (supplemental Fig. S3). Upon PFA fixation, some cytoplasmic signal was detected but more strikingly, a double line delimiting the flagellum was also observed, strongly indicative of a flagellar membrane location (supplemental Fig. S3A). Accordingly, methanol fixation that is known to damage membranes resulted in a weaker signal (supplemental Fig. S3B). Formal demonstration of membrane association was provided by detergent extraction that led to an almost complete loss of the AK signal (supplemental Fig. S3C). This was observed on all cells no matter the stage of the cell cycle and the length of the flagellum. The flagellar location of AK was further confirmed upon PFA fixation by triple staining with the axoneme marker antibody MAb25 (Fig. 3B) or the anti-PFR2 L8C4 antibody (Fig. 3C). The AK signal was forming a tube-like structure that encompassed the axoneme and the PFR staining (see magnified images at Fig. 3A–C). AK was detected from the flagellum base where it corresponded to the MAb25 signal to the flagellum tip (Fig. 3B), in contrast to the PFR staining that is only present from the exit of the flagellar pocket (Fig. 3C). Finally, triple staining with the anti-AK, MAb25, and a monoclonal antibody against the intraflagellar transport protein IFT172 on methanol-fixed cells showed very close co-localization between all three markers (Fig. 3D–F). These results confirm that at least one protein product issued of the 3 AK genes is localized to the flagellum membrane.
Fig. 3.
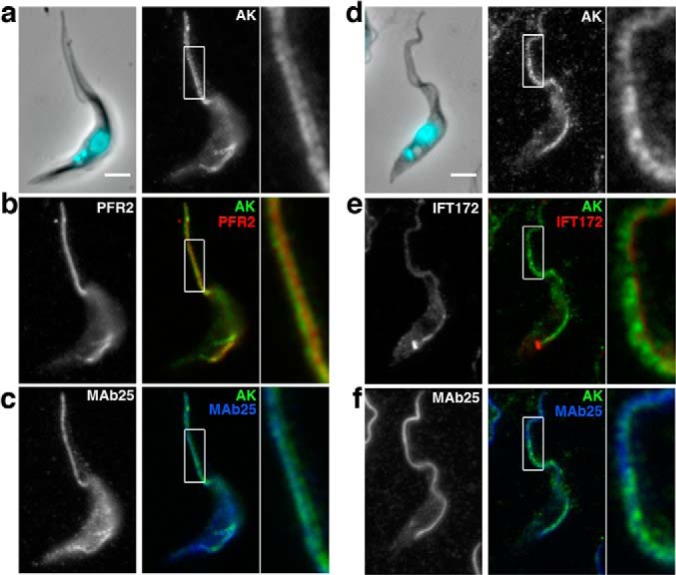
Arginine kinase is localized to the flagellum membrane. A–C, IFA of PFA-fixed control trypanosomes stained with an antiserum against the arginine kinase of T. cruzi (white in a, green in B, C), with the PFR marker L8C4 (B, red) and with the axonemal marker MAb25 (C, blue) reveals a membrane location for arginine kinase. D–F, IFA of methanol-fixed trypanosomes stained with the anti-arginine kinase (white in D, green in E, F), with the anti-IFT172 monoclonal antibody (E, red), and with the axonemal marker MAb25 (F, blue). Scale bar, 3 μm. White boxes indicate the area of magnification shown on the right of each panel.
14-3-3 proteins are a family of molecules able to bind to proteins containing phosphoserine or phosphothreonine and have been involved in multiple functions and in diverse organisms. Functional studies in bloodstream T. brucei suggest a role in cell motility (60). Rat antibodies against the two 14-3-3 (60) proteins identified in our list were used by IFA to evaluate a possible flagellar location. Whereas in our hands the anti-14-3-3-II antibody did not produce any reproducible signal by IFA on procyclic trypanosomes, the anti-14-3-3-I antibody revealed a flagellum localization upon methanol fixation (supplemental Fig. S4A). This signal survived detergent extraction (supplemental Fig. S4B) and colocalized with that obtained for MAb25, indicative of an axoneme location. This association to the flagellum could explain the cytokinesis phenotype reported upon RNAi silencing in the bloodstream stage of the parasite (60) where alterations of flagellum motility (4, 26, 27) or integrity (64) can severely impair cell division without affecting the nucleus cycle.
While examining supplemental Table S3, we noticed that several of the most abundant proteins were annotated as “hypothetical” without indication about their possible function and location. We selected eight of them that to our knowledge had not yet been investigated in any organism. Two proteins that were not enriched but were fairly abundant in the flagellar proteome were also selected (Tb927.8.940 and Tb927.7.5340). A diagram of the primary structure of these proteins showing conserved domains is presented at supplemental Fig. S5. Remarkably, out of the eight domains identified by Pfam searches, five (TPR, tetratricopeptide repeat; LRR, leucin-rich repeat; WD40, 40 amino acid motif terminating in a tryptophane-aspartate; cNMP BD, cyclic nucleotide monophosphate binding domain, and IQ calmodulin binding domain) are over-represented in flagellar proteins (3). To evaluate a possible flagellum association, a series of plasmids were produced and transfected in wild-type procyclic trypanosomes for expression as YFP fusion proteins (supplemental Table S5) upon endogenous tagging. This approach is very efficient in trypanosomes and has the advantage to rely on the endogenous context of the locus for transcription and of at least one half of the regulatory sequences either at the 5′ or at the 3′ end of the fusion gene (44). Remarkably, all eight fusion proteins that were initially considered to be enriched in the flagella fraction turned out to be indeed localized in the flagellum, with an amazing diversity in distribution (Fig. 4 and supplemental Fig. S6). These proteins were therefore termed FLAM1–8, for Flagellum Members. The two other fusion proteins were not detected and hence their localization could not be established.
Fig. 4.

Novel proteins show distinct localizations within the flagellum. A–D, IFA of methanol-fixed trypanosomes expressing the indicated YFP fusion proteins from the endogenous locus stained with an anti-GFP antiserum (green) and with the axonemal marker MAb25 (red) reveals location to the PFR (A, FLAM1), to the PFR region limited to the adhesion domain to the cell body (B, FLAM3), to the proximal axoneme region (C, FLAM6, notice the yellow superposition profile with MAb25) or to the distal end of the flagellum (D, FLAM8). The white arrow in b indicates a partially detached flagellum, showing the association of FLAM3 with the flagellum and not with the attachment zone in the cell body. Scale bar, 5 μm.
The YFP::FLAM fusion proteins exhibited four distinct profiles (Fig. 4 and supplemental Fig. S6). The most common one was an association with the PFR as seen for FLAM1 (Fig. 4A) but also for FLAM2, FLAM4, FLAM5, and FLAM7 (supplemental Fig. S6). This is revealed by perfect co-localization with the PFR2 marker L8C4 whose signal is not detected before the flagellum exits from the flagellar pocket and runs till its distal tip. In contrast, it shows a side-by-side arrangement with the axoneme marker MAb25 and is not detected within the flagellar pocket (see magnified panels at supplemental Fig. S6). A second pattern was observed for FLAM3 that is also associated to the PFR but in contrast to the others, does not reach the distal end of the flagellum (Fig. 4B & supplemental Fig. S6). This could correspond to the flagellum attachment zone that runs alongside the flagellum on the cell body side (52). However, when flagella were occasionally partially detached from the cell body as a result of methanol dehydration, the FLAM3 signal was present on the flagellum side, supporting a flagellar location (arrow on Fig. 4B). The FLAM6 protein is found on the axoneme, from within the flagellar pocket area but extends only to the first half of the flagellum (Fig. 4C and supplemental Fig. S6). Finally, FLAM8 is found only at the distal tip of the flagellum, apparently beyond the axoneme signal (Fig. 4D and supplemental Fig. S6). Detergent extraction revealed that all FLAM proteins were still detected on cytoskeletal fractions, maintaining their specific association to the PFR (FLAM1, 2, 4, 5 & 7), the axoneme (FLAM6), or its distal tip (FLAM8), whereas FLAM3 was restricted to the FAZ region in both the flagellum and the cell body side (supplemental Fig. S7).
Dynamics of Unique Flagellar Proteins
This unexpected diversity of locations within the flagellum prompted us to investigate the way(s) by which these specific distributions were established and maintained. We first focused on FLAM6 and FLAM8 that showed the most restrictive location. The FLAM6 fusion protein is detected only in the proximal portion of the axoneme. This could be explained if the protein was only produced during the early phase of flagellum elongation that takes place via assembly at the distal end (65). Alternatively, FLAM6 might be restricted to the proximal half of the flagellum no matter its length. Trypanosomes in culture grow asynchronously and assemble a new flagellum during the cell cycle while maintaining the existing one (25), offering the advantage to compare growing and mature flagella in the same cell. The basal body of the flagellum is physically linked to the kinetoplast (DNA of the single large mitochondrion of the cell) and duplicates first, closely followed by the kinetoplast (33). DAPI staining therefore provides an easy way to position a cell in its cell cycle as the kinetoplast duplicates and segregates before nuclear mitosis, providing three intermediate situations during elongation of the new flagellum: cells with (1) one kinetoplast undergoing duplication but that have not yet segregated and one nucleus (Fig. 5B); (2) two kinetoplasts and one nucleus (Fig. 5C); and (3) two kinetoplasts and two nuclei (Fig. 5D). We measured the length of the FLAM6 signal in both old and new flagella and compared it with the length of the axoneme marked by MAb25. In mature flagella, the FLAM6 staining was limited to 55.7 ± 7.5% of the whole flagellum length (n = 42). In growing flagella, the length of the FLAM6 signal was correlated with that of the MAb25 signal, displaying a direct linear relationship (Fig. 5E). On average, FLAM6 was present on the first 45 ± 9% of the length of the axoneme (Fig. 5E). The FLAM6 protein was detected as soon as the MAb25 signal indicated the presence of two flagella, even when the new flagellum was short (Fig. 5B). Nevertheless, the FLAM6 signal never reached the tip of the flagellum. FLAM6 is therefore restricted to the first half of the flagellum no matter its length.
Fig. 5.
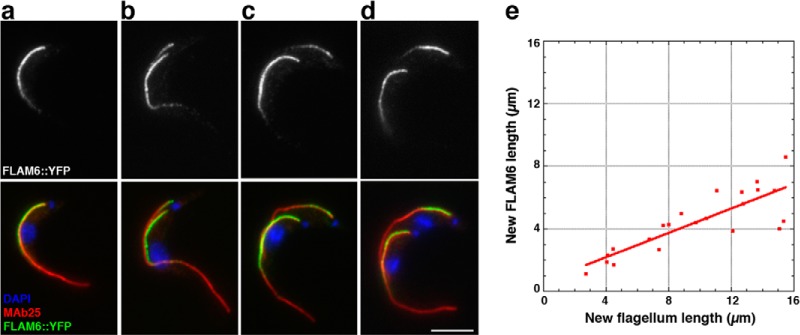
FLAM6 is restricted to the proximal portion of the axoneme at every stage of flagellum formation. A–D, IFA of methanol-fixed trypanosomes expressing the FLAM6::YFP fusion protein stained with an anti-GFP antiserum (green) and with the axonemal marker MAb25 (red) at various stages of flagellum elongation. FLAM6 is restricted to about half of the axoneme at all stages of formation of the new flagellum. E, The length of the FLAM6 signal was measured in 42 cells assembling a new flagellum and plotted versus the actual length of the new flagellum measured from the MAb25 signal. A clear linear relationship is observed, showing that FLAM6 is always restricted to ∼45% of the new flagellum length. Scale bar, 5 μm.
In FLAM8::YFP expressing cells, double IFA with anti-GFP and MAb25 showed that the signals were very close to each other but did not overlap (Fig. 4D and supplemental Fig. S6). This impression was confirmed by immunogold staining carried out on detergent-extracted trypanosome cytoskeletons (Fig 6A–C′). This revealed an exquisite concentration of FLAM8 only at the distal tip of the peripheral microtubule doublets (Fig. 6A–6C, 6A′–6C′). Examination of FLAM8 presence and abundance in growing flagella revealed that the protein was only detectable at relatively advanced stages of flagellum elongation (Fig. 6E–E′, 6F–6F′). The signal intensity for FLAM8:YFP was much weaker in the new flagellum compared with the old one, even in cells close to division (Fig. 6G–6G′). We therefore performed quantitative analysis of the FLAM8 signal at the distal tip of the growing flagellum and compared it with the one at the end of the mature flagellum in the same cell (Fig. 6H, 6I). The length of the old and the new axoneme was measured using the MAb25 staining. Two analyses were carried out, one based on the integrated density of the FLAM8 spot (Fig. 6H) and one relying on the difference between the maximum and the minimum signal for each spot (Fig. 6I). In both cases, a progressive build-up of FLAM8 takes place at the distal tip during flagellum assembly. Strikingly, FLAM8::YFP fluorescence intensity in the new flagellum only reached 25–45% of the signal measured in the old flagellum in cells about to divide, i.e. at a point where the flagellum had reached 90% of its mature length (Fig. 6H, 6I). This implies that exhaustive FLAM8 accumulation continues after cell division before the cell re-enters the cell cycle.
Fig. 6.

FLAM8 is localized at the tip of axonemal microtubules and accumulates during flagellum construction. A–C, Immunogold analysis of trypanosomes expressing the FLAM8::YFP fusion protein stripped with detergent and stained with an anti-GFP antiserum. The signal is concentrated at the tip of microtubule doublets. A–C shows low magnification (bar is 1 μm) and squares indicate areas that were magnified in A′–C′ (bar is 100 nm). D–G, IFA of methanol-fixed trypanosomes expressing the FLAM8::YFP fusion protein stained with an anti-GFP antiserum (green) and with the axonemal marker MAb25 (red) at various stages of flagellum elongation. The FLAM8 signal (D–G) was quantified and progressively accumulates at the distal tip of the new flagellum (yellow arrow) but its intensity is always lower compared with that at the distal tip of the old flagellum. Scale bar is 5 μm. H, I, The length of the new flagellum and the FLAM8::YFP signal intensity (H) or the difference between maximum and minimum intensity (I) were measured in 45 cells at different stages of the cell cycle and normalized by the equivalent parameter in the old flagellum of the same cell. The red and blue bars represent the flagellum length and the FLAM8 signal respectively.
FLAM8 could be part of a stable structure that accumulates material over time or could be turning over and be actively maintained at the distal tip, for example by the action of intraflagellar transport (35). This was addressed by performing FRAP (fluorescence recovery after photobleaching) analysis in cells bearing a single flagellum. Bleaching the FLAM8 signal was efficiently achieved in a single laser pulse and recovery was monitored upon image acquisition every 30 s. Two rather different situations were encountered: in half of the cells (6/12), recovery was either limited or even barely detectible whereas in the remaining cells (6/12) significant recovery was observed (Fig. 7). Films were therefore grouped in two categories: those with little or no recovery (less than 20% of the original value, Fig. 7A) (supplemental Movie S1) and those where recovery reached at least 25% of the original signal (Fig. 7B) (supplemental Movie S2). In the first situation (Fig. 7A), a brief recovery is observed at the first time point, possibly because of reversibility of YFP (66), but no further increase in signal could be observed over up to 10 min. In the second situation, fluorescence intensity increased steadily for the first 5 min before reaching a plateau, indicating a relatively rapid turnover. These data could be explained by a different age of the flagellum (see discussion).
Fig. 7.

Two different recovery profiles of FLAM8 signal after photobleaching. FRAP analysis of trypanosomes expressing the FLAM8::YFP fusion protein (see supplemental Movie S1 and S2). The distal tip signal was bleached with a brief laser pulse and recovery was monitored upon acquisition of an image every 30 s. Two profiles were observed, with six cells showing no detectable recovery (A) and six cells showing significant recovery (B). The bottom panels show magnification of the distal tip of the flagellum at the indicated time point.
To further understand the dynamics of the novel flagellar proteins, we used inducible RNAi to monitor their turnover in mature and growing flagella. The difference in age between the old and the new flagellum is a powerful molecular clock and was used to determine the site of incorporation of the flagellar proteins once their expression had been blocked by RNAi (67, 68). In trypanosomes, inducible RNAi can be generated by tetracycline-controlled expression of double-stranded RNA corresponding to a fragment of the gene of interest (45). For arginine kinase, the AK1–3RNAi cell line was produced targeting a segment common to all three AK genes. Cells were “induced” in the presence of tetracycline to trigger the production of dsRNA and kinetics of RNAi silencing was evaluated by IFA using the anti-AK antibody (Fig. 8A–8C). Noninduced cells still expressed arginine kinase that was detected in the membrane of both old and new flagella (Fig. 8A–8A′), as observed previously for wild-type cells. However, a rapid reduction in the amount of arginine kinase was detected in both old and new flagella as early as 4 h after the addition of tetracycline (Fig. 8B–8B′). The signal became virtually undetectable in both flagella 8–24 h after induction depending on the clones analyzed (Fig. 8C–C′). These data confirm both the specificity of the anti-AK antibody and the efficiency of the RNAi silencing. Comparison of signal intensity failed to reveal a significant difference between the old and the new flagellum and no gradients could be observed in contrast to what had been observed for structural proteins (67, 68).
Fig. 8.

Flagellar proteins show different turnover following RNAi silencing. Trypanosomes were transformed to express double-stranded RNA targeting AK1–3 (A–C), FLAM1 (D–F) or FLAM6 (G–I) leading to RNAi silencing. Cells were grown in the absence (non-induced, A–A′, D–D′, G–G′), or in the presence of tetracycline for 8 (B–B′), 24 (C–C′, E–E′, F–F′), or 48h (H–H′, I–I′) and analyzed by IFA following staining with the anti-arginine kinase antibody (white, A–C) in AK1–3RNAi cells; or with an anti-GFP in FLAM1RNAi cells expressing FLAM1::YFP (white, D–F) and in FLAM6RNAi cells expressing FLAM6::YFP (white, G–I). In these two cell lines, MAb25 was used to mark the axoneme (red) in double IFA with the anti-GFP (green, D′–I′). Only cells with two flagella are presented, showing equivalent bright staining between old and new flagella in non-induced samples. Whereas the arginine kinase signal goes down rapidly in both old and new flagella, the FLAM1::YFP and the FLAM6::YFP proteins disappear in the new flagellum but are still detected in the old flagellum (F–F′, I–I′). In intermediate situations, the signal is present at the proximal part of the flagellum but quickly becomes undetectable toward the end of the flagellum (yellow arrows on panels E–E′, H–H′), indicating polar assembly at the distal tip for both FLAM1 and FLAM6. Scale bar is 5 μm.
We then turned our attention to the FLAM proteins. Individual cell lines were generated by co-transfection of the recipient 29–13 cell line with the constructs allowing for constitutive expression of the YFP fusion proteins from their endogenous locus (supplemental Table S5) and the one designated for inducible expression of the corresponding dsRNA (supplemental Table S6). Unfortunately, leakiness of the inducible system led to the production of dsRNA even in non-induced controls, hindering the analysis of several cell lines where the YFP signal became too weak to be unambiguously detected (not shown). However, a clear-cut difference could be observed between non-induced and induced FLAM1RNAi cells expressing FLAM1::YFP and FLAM6RNAi cells expressing FLAM6::YFP (Fig. 8D–8I). First, RNAi against FLAM1 resulted in the absence of FLAM1 signal in the new flagellum but not in the old one 48 h after addition of tetracycline (Fig. 8F–8F′). This indicates that FLAM1 depletion prevented its incorporation in the new flagellum but did not affect significantly its presence in the old flagellum. Examination of earlier induction times identified intermediate situations where some FLAM1::YFP fusion protein was still detected in the new flagellum. However, this was not uniformly distributed but was always restricted to the proximal portion (Fig. 8E–8E′, yellow arrow), suggestive of distal addition of FLAM1 in the elongating structure. Analysis of induced FLAM6RNAi cells expressing FLAM6::YFP showed a similar profile, with the absence of the FLAM6 protein in the new flagellum but not in the old one (Fig. 8I–8I′) and with a lack of incorporation toward the distal end of the normally positive portion (Fig. 8H–8H′, yellow arrow), also supporting assembly by the distal tip, even for an axonemal protein showing limited distribution along the length of the axoneme. Overall, these analyses of flagellar protein distribution after RNAi silencing revealed very distinct protein dynamics and further highlighted the complexity of the flagellum, which contains several subcompartments whose components exhibit unique dynamics.
DISCUSSION
Quality of Purified Flagella
This work reports the first purification and characterization of intact flagella from procyclic trypanosomes. The quality of the purified flagella was confirmed at the structural level by scanning and transmission electron microscopy and at the molecular level by western blotting and mass spectrometry analysis. Up to 80% of the purified flagella were surrounded by an intact membrane and the axoneme, the PFR, and the IFT particles looked intact in most sections. This was confirmed by the significant enrichment of five flagellar markers measured by Western blotting and by the presence of close to 90% of all recognized flagellar markers. In contrast to the proteome of the skeletal flagellar fraction that did not identify a single membrane and matrix component, more than 80% of these were present in our study, including most of the IFT proteins and motors.
Three other proteomic studies of T. brucei flagella have been reported: two relied on extraction of flagellar skeletons from procyclic cells with detergent and salt (4, 59) whereas the third one used the FLA1RNAi mutant as reported here, but this time at the bloodstream stage and mechanical shearing of the flagella was achieved by multiple passages through a G-28 needle (8). Unfortunately, transmission electron microscopy analysis of the purified flagella has not been reported in any of these studies, making it difficult to evaluate the purity and the quality of the flagella. Nevertheless, the detergent and salt method is known to disrupt the membrane and to result in contamination by the flagellar pocket collar, the flagellum attachment zone (25) and another cytoskeletal structure known as the bilobe (69), as confirmed by the presence of various molecular components of these structures in the reported proteomes (4, 59). In the case of procyclic FLA1RNAi cells, our data show that flagella severed at the level of the connection between the kinetoplast and the basal body, presumably followed by rapid sealing of the membrane around the basal body area. This is different to what is observed in algae or ciliates where acidic shock or dibucain treatment leads to flagella/cilia severing at the level of the transition zone (70), excluding basal body proteins. Moreover, this severing was neither associated to disruption of the flagellar pocket collar nor of the FAZ. This was confirmed both at the structural and molecular level (no or very low contamination by any of the known components of these structures).
By using stringent criteria, we identified 212 candidates for novel flagellar proteins in procyclic T. brucei. Added to the skeletal fraction that contains at least 331 proteins (4), it would make a total of 543 proteins, a value quite close to what has been reported for the purification of intact Chlamydomonas flagella (5) or other flagellar/ciliary proteomes (71). Few proteins were shared with the surface proteome of bloodstream trypanosomes (8), which makes sense knowing the very different environments in which bloodstream and procyclic trypanosomes live and the exhaustive changes in cell surface composition that take place during differentiation from one stage to the other (72). In contrast, more than one third of the proteins present in the matrix proteome of bloodstream flagella were also found in the purified procyclic flagella, including several proteins potentially involved in signaling (8). Here, we focused on proteins that had not yet been studied and showed that 9 out of the 10 proteins investigated (FLAM1–8 and 14-3-3-I) were indeed associated to the flagellum. Moreover, the last one (14-3-3-II) could not be visualized experimentally and so might also be located to the flagellum. This reveals the quality of the purification and the stringency of the criteria applied because 100% of the selected proteins that we could detect experimentally were found in the flagellum.
Diversity of Sublocalization within the Flagellum and Implication for Targeting
Amazingly, the 10 novel proteins exhibited six different localization profiles within the flagellum, raising the issue of targeting and possibly retention at the restricted domain. FLAM3 is present on the PFR but is missing from the final 2 μm of the flagellum where it is not attached to the cell body. Its localization seems restricted to the PFR side that faces the cell body. It could correspond to the filaments that appear to link the proximal domain of the PFR to the part of the flagellum membrane that faces the FAZ on the cell body, as seen by transmission electron microscopy. These fibers survive ablation of most of the PFR structure in the absence of PFR2 where the flagellum remains normally attached to the cell body (20) but are missing in the kinesin KIF9B mutant that exhibits flagellum detachment (73). Functional analysis revealed that FLAM3 knockdown resulted to flagellum detachment in a similar manner as depletion of FLA1 (74). FLAM3 is therefore a key component of the attachment system and could provide a platform allowing the trans-membrane FLA1 binding protein (75) to partner with FLA1 for proper flagellum adhesion (74). FLAM3 could also interact with the recently identified putative calcium channel that shows restricted location to a similar area (8).
To our knowledge, the existence of a gradient along the trypanosome flagellum has only been shown for tyrosinated tubulin that is restricted to the distal tip of the elongating new flagellum before the final tyrosine residue is removed by a carboxypeptidase (76). In human cells, two axonemal dynein heavy chains exhibit restricted location either to the distal part of cilia in the respiratory epithelium for DNAH9 or at the proximal part of the sperm flagellum for DNAH5 (77). In Chlamydomonas, the dynein heavy chain DHC11 (belonging to the inner dynein arm) is restricted to a short proximal section of the flagellum. However, the protein covers the whole length of short flagella undergoing assembly (78), in contrast to what was observed here for FLAM6. DHC5, another dynein heavy chain occupies only the distal segment of the flagellum and it has been proposed that it competes with DHC11 for attachment to microtubules. The different distribution could result from shifted timing of protein expression during flagellum growth (78). Heterogeneity of inner dynein arm along the length of the flagellum has also been reported at the structural level (79). Here we have shown that FLAM6 displays restricted location at the proximal portion of the flagellum as soon as the flagellum starts to elongate. Such a profile could be obtained by different means. First, FLAM6 could be synthesized exclusively during the initial phase of flagellum construction. In these conditions, it would be added normally as other flagellar components but once its production stops, flagellum elongation continues and because assembly of microtubules takes place at the distal tip (76, 80), the distal portion of the flagellum would be assembled in the absence of FLAM6 and hence deprived of it. This control could be achieved at the mRNA level, because several sets of mRNA show a specific peak of production at different windows of the cell cycle, with genes encoding for basal body or IFT proteins peaking in late G1 whereas transcripts encoding axonemal proteins peak in S-phase, prior to PFR mRNA (81, 82). FLAM6 peaks at S-phase, indicating it behaves as the majority of axonemal proteins. An alternative model then would consist in considering that FLAM6 protein synthesis or assembly on the axoneme is twice slower than the rate of flagellum construction. A distinct possibility is that FLAM6 would be able to “sense” flagellar length and to restrict itself to the first half of the flagellum, no matter its length. This could be achieved by a competition with a protein with the inverse distribution, leading to mutual exclusion.
In addition to FLAM8, three other proteins have been identified at the distal tip of the trypanosome flagellum, suggesting the existence of a very specific microdomain: a calpain-like protein TbCALP1.3 (83) that was also found in our proteomic study (supplemental Table S2), the kinesin motor Kif13-2 (84) and an unknown antigen detected by a monoclonal antibody raised against bull sperm (85). The latter one and FLAM8 are resistant to detergent indicating that a specific structure could be present at the tip of the axoneme. This could be involved in microtubule assembly, stability or dynamics, as proposed in Chlamydomonas where HSP70 and EB1 have been localized to the distal tip of the flagellum (86, 87). It could also play a role in the control of flagellum beating, where main waves are initiated from the tip of the flagellum (88). Finally, it could also serve as a way to retain sensing molecules as found at the terminal end of sensory cilia (89).
Protein Turnover in the Trypanosome Flagellum
FLAM8 is progressively added to the new flagellum during its assembly but its concentration only reaches around one third of the amount present at the tip of the old flagellum when the cell is about to divide, meaning that material is still added after cell division. This could be brought by IFT that is active in mature flagella (35). FRAP analysis of the FLAM8 signal in cells with a single flagellum revealed two very different profiles: little or no recovery, or sustained recovery reaching up to 40% in a few minutes. This contrasts with what was observed upon bleaching fluorescent IFT proteins where rapid (∼20s) and significant (45%) recovery was measured in all cells analyzed (57) and with structural components of the axoneme that show no recovery even several hours after photobleaching (L.V. and P.B., unpublished data). It is tempting to speculate that the two profiles observed here could correspond respectively to cells that inherited the old flagellum where the FLAM8 signal has reached saturation and is turning over very slowly and to those that inherited the new flagellum and that still need to acquire 50% of the total pool of FLAM8 at the distal tip.
Inducible RNAi silencing is a powerful tool to examine protein turnover in the trypanosome flagellum (67, 68). It confirmed that structural elements exhibit little or very slow turnover in the mature flagellum for FLAM1 and FLAM6, exactly as had been observed for other PFR and axoneme components (26, 65, 67, 68). It also showed that FLAM1 is added at the distal tip of the growing flagellum and FLAM6 to the distal part of its own segment (but not the end of the flagellum). Results were strikingly different for the membrane protein arginine kinase whose silencing leads to a rapid drop of signal in both flagella, suggesting either: (1) a rapid turnover with similar kinetics between the two flagella; or (2) the possible exchange of arginine kinase molecules between the two flagella, leading to concomitant reduction in both.
Overall, these data show the high degree of sophistication of the trypanosome flagellum, within the organelle but also according to its age. This could be significantly relevant for the life cycle of the parasite where differential targeting of membrane protein between the old and the new flagellum have been observed at specific differentiation stage during infection of the tsetse fly (90).
Supplementary Material
Acknowledgments
We thank Pascal Lenormand for expert assistance with initial mass spectrometry analysis, Claudio Pereira for providing the anti-AK antibody, as well as Linda Kohl and Cher Pheng Ooi for critical reading of the manuscript. I.S. thanks Markus Engstler for his continuous support and Tim Krüger for sharing his expertise in microscopy.
Footnotes
Author contributions: I.S., D.J., A.N., and P.B. designed research; I.S., D.J., L.V., N.R., J.B., T.B., D.H., S.P., J.S., M.D., J.R., and N.C. performed research; J.R. contributed new reagents or analytic tools; I.S., D.J., L.V., M.D., V.H., J.C., and P.B. analyzed data; P.B. wrote the paper; J.C. corrected the paper.
* This work in the Trypanosome Cell Biology Unit was supported by the Institut Pasteur, the CNRS and ANR grants (ANR-06-MRAR-014 and ANR-08-MIE-027). Funding for the Orbitrap acquisition was secured through a DIM Malinf grant from the region Ile-de-France. I.S. was funded by a FNR (Fonds National de la Recherche du Luxembourg) fellowship; D.J. and L.V. were funded by Roux post-doctoral fellowships; J.B. was funded by MNRT, FRM and Pasteur-Weizmann doctoral fellowships; D.H. was funded by MNRT and FRM doctoral fellowships and J.S.-R. is funded by FRM and Institut Carnot post-doctoral fellowships.
 This article contains supplemental Figs S1 to S7, Tables S1 to S7 and Movies S1 and S2.
This article contains supplemental Figs S1 to S7, Tables S1 to S7 and Movies S1 and S2.
Original mass spectrometry data have been submitted to the ProteomeXchange data base with the accession number PXD000051.
1 The abbreviations used are:
- PFR
- paraflagellar rod
- IFA
- immunofluorescence assay
- FLAM
- flagellum member
- PFA
- paraformaldehyde
- IFT
- intraflagellar transport.
REFERENCES
- 1. Kohl L., Bastin P. (2005) The flagellum of trypanosomes. Int. Rev. Cytol. 244, 227–285 [DOI] [PubMed] [Google Scholar]
- 2. Avidor-Reiss T., Maer A. M., Koundakjian E., Polyanovsky A., Keil T., Subramaniam S., Zuker C. S. (2004) Decoding cilia function: defining specialized genes required for compartmentalized cilia biogenesis. Cell 117, 527–539 [DOI] [PubMed] [Google Scholar]
- 3. Li J. B., Gerdes J. M., Haycraft C. J., Fan Y., Teslovich T. M., May-Simera H., Li H., Blacque O. E., Li L., Leitch C. C., Lewis R. A., Green J. S., Parfrey P. S., Leroux M. R., Davidson W. S., Beales P. L., Guay-Woodford L. M., Yoder B. K., Stormo G. D., Katsanis N., Dutcher S. K. (2004) Comparative genomics identifies a flagellar and basal body proteome that includes the BBS5 human disease gene. Cell 117, 541–552 [DOI] [PubMed] [Google Scholar]
- 4. Broadhead R., Dawe H. R., Farr H., Griffiths S., Hart S. R., Portman N., Shaw M. K., Ginger M. L., Gaskell S. J., McKean P. G., Gull K. (2006) Flagellar motility is required for the viability of the bloodstream trypanosome. Nature 440, 224–227 [DOI] [PubMed] [Google Scholar]
- 5. Pazour G. J., Agrin N., Leszyk J., Witman G. B. (2005) Proteomic analysis of a eukaryotic cilium. J. Cell Biol. 170, 103–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Smith J. C., Northey J. G., Garg J., Pearlman R. E., Siu K. W. (2005) Robust method for proteome analysis by MS/MS using an entire translated genome: demonstration on the ciliome of Tetrahymena thermophila. J. Proteome Res. 4, 909–919 [DOI] [PubMed] [Google Scholar]
- 7. Ostrowski L. E., Blackburn K., Radde K. M., Moyer M. B., Schlatzer D. M., Moseley A., Boucher R. C. (2002) A proteomic analysis of human cilia: identification of novel components. Mol. Cell. Proteomics : MCP 1, 451–465 [DOI] [PubMed] [Google Scholar]
- 8. Oberholzer M., Langousis G., Nguyen H. T., Saada E. A., Shimogawa M. M., Jonsson Z. O., Nguyen S. M., Wohlschlegel J. A., Hill K. L. (2011) Independent analysis of the flagellum surface and matrix proteomes provides insight into flagellum signaling in mammalian-infectious Trypanosoma brucei. Mol. Cell. Proteomics : MCP 10, M111 010538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Czarnecki P. G., Shah J. V. (2012) The ciliary transition zone: from morphology and molecules to medicine. Trends Cell Biol. 22, 201–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Deane J. A., Cole D. G., Seeley E. S., Diener D. R., Rosenbaum J. L. (2001) Localization of intraflagellar transport protein IFT52 identifies basal body transitional fibers as the docking site for IFT particles. Curr. Biol. 11, 1586–1590 [DOI] [PubMed] [Google Scholar]
- 11. Craige B., Tsao C. C., Diener D. R., Hou Y., Lechtreck K. F., Rosenbaum J. L., Witman G. B. (2010) CEP290 tethers flagellar transition zone microtubules to the membrane and regulates flagellar protein content. J. Cell Biol. 190, 927–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chih B., Liu P., Chinn Y., Chalouni C., Komuves L. G., Hass P. E., Sandoval W., Peterson A. S. (2012) A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nat. Cell Biol. 14, 61–72 [DOI] [PubMed] [Google Scholar]
- 13. Kee H. L., Dishinger J. F., Blasius T. L., Liu C. J., Margolis B., Verhey K. J. (2012) A size-exclusion permeability barrier and nucleoporins characterize a ciliary pore complex that regulates transport into cilia. Nat. Cell Biol. 14, 431–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hu Q., Milenkovic L., Jin H., Scott M. P., Nachury M. V., Spiliotis E. T., Nelson W. J. (2010) A septin diffusion barrier at the base of the primary cilium maintains ciliary membrane protein distribution. Science 329, 436–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vincensini L., Blisnick T., Bastin P. (2011) 1001 model organisms to study cilia and flagella. Biol. Cell 103, 109–130 [DOI] [PubMed] [Google Scholar]
- 16. Moser J. J., Fritzler M. J., Rattner J. B. (2009) Primary ciliogenesis defects are associated with human astrocytoma/glioblastoma cells. BMC Cancer 9, 448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Molla-Herman A., Ghossoub R., Blisnick T., Meunier A., Serres C., Silbermann F., Emmerson C., Romeo K., Bourdoncle P., Schmitt A., Saunier S., Spassky N., Bastin P., Benmerah A. (2010) The ciliary pocket: an endocytic membrane domain at the base of primary and motile cilia. J. Cell Sci. 123, 1785–1795 [DOI] [PubMed] [Google Scholar]
- 18. Portman N., Gull K. (2010) The paraflagellar rod of kinetoplastid parasites: from structure to components and function. Int. J. Parasitol. 40, 135–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Portman N., Lacomble S., Thomas B., McKean P. G., Gull K. (2009) Combining RNA interference mutants and comparative proteomics to identify protein components and dependences in a eukaryotic flagellum. J. Biol. Chem. 284, 5610–5619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bastin P., Sherwin T., Gull K. (1998) Paraflagellar rod is vital for trypanosome motility. Nature 391, 548. [DOI] [PubMed] [Google Scholar]
- 21. Santrich C., Moore L., Sherwin T., Bastin P., Brokaw C., Gull K., LeBowitz J. H. (1997) A motility function for the paraflagellar rod of Leishmania parasites revealed by PFR-2 gene knockouts. Mol. Biochem. Parasitol. 90, 95–109 [DOI] [PubMed] [Google Scholar]
- 22. Kohl L., Gull K. (1998) Molecular architecture of the trypanosome cytoskeleton. Mol. Biochem. Parasitol. 93, 1–9 [DOI] [PubMed] [Google Scholar]
- 23. Vaughan S., Kohl L., Ngai I., Wheeler R. J., Gull K. (2008) A repetitive protein essential for the flagellum attachment zone filament structure and function in Trypanosoma brucei. Protist 159, 127–136 [DOI] [PubMed] [Google Scholar]
- 24. Zhou Q., Liu B., Sun Y., He C. Y. (2011) A coiled-coil- and C2-domain-containing protein is required for FAZ assembly and cell morphology in Trypanosoma brucei. J. Cell Sci. 124, 3848–3858 [DOI] [PubMed] [Google Scholar]
- 25. Sherwin T., Gull K. (1989) The cell division cycle of Trypanosoma brucei brucei: timing of event markers and cytoskeletal modulations. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 323, 573–588 [DOI] [PubMed] [Google Scholar]
- 26. Branche C., Kohl L., Toutirais G., Buisson J., Cosson J., Bastin P. (2006) Conserved and specific functions of axoneme components in trypanosome motility. J. Cell Sci. 119, 3443–3455 [DOI] [PubMed] [Google Scholar]
- 27. Ralston K. S., Lerner A. G., Diener D. R., Hill K. L. (2006) Flagellar motility contributes to cytokinesis in Trypanosoma brucei and is modulated by an evolutionarily conserved dynein regulatory system. Eukaryot. Cell 5, 696–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kohl L., Robinson D., Bastin P. (2003) Novel roles for the flagellum in cell morphogenesis and cytokinesis of trypanosomes. EMBO J. 22, 5336–5346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vickerman K. (1985) Developmental cycles and biology of pathogenic trypanosomes. Br Med. Bull. 41, 105–114 [DOI] [PubMed] [Google Scholar]
- 30. Rotureau B., Morales M. A., Bastin P., Spath G. F. (2009) The flagellum-mitogen-activated protein kinase connection in Trypanosomatids: a key sensory role in parasite signalling and development? Cell Microbiol. 11, 710–718 [DOI] [PubMed] [Google Scholar]
- 31. Rotureau B., Ooi C. P., Huet D., Perrot S., Bastin P. (2013) Forward motility is essential for trypanosome infection in the tsetse fly. Cell Microbiol. [DOI] [PubMed] [Google Scholar]
- 32. Kisalu N. K., Langousis G., Bentolila L. A., Ralston K. S., Hill K. L. (2013) Mouse infection and pathogenesis by Trypanosoma brucei motility mutants. Cell Microbiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Robinson D., Beattie P., Sherwin T., Gull K. (1991) Microtubules, tubulin, and microtubule-associated proteins of trypanosomes. Method Enzymology 196, 285–299 [DOI] [PubMed] [Google Scholar]
- 34. Bonhivers M., Nowacki S., Landrein N., Robinson D. R. (2008) Biogenesis of the trypanosome endo-exocytotic organelle is cytoskeleton mediated. PLoS Biology 6, e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Absalon S., Blisnick T., Kohl L., Toutirais G., Dore G., Julkowska D., Tavenet A., Bastin P. (2008) Intraflagellar transport and functional analysis of genes required for flagellum formation in trypanosomes. Mol. Biol. Cell 19, 929–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cooper R., de Jesus A. R., Cross G. A. (1993) Deletion of an immunodominant Trypanosoma cruzi surface glycoprotein disrupts flagellum-cell adhesion. J. Cell Biol. 122, 149–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. LaCount D. J., Barrett B., Donelson J. E. (2002) Trypanosoma brucei FLA1 is required for flagellum attachment and cytokinesis. J. Biol. Chem. 277, 17580–17588 [DOI] [PubMed] [Google Scholar]
- 38. Wirtz E., Leal S., Ochatt C., Cross G. A. (1999) A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol. Biochem. Parasitol. 99, 89–101 [DOI] [PubMed] [Google Scholar]
- 39. Tjalsma H., Lambooy L., Hermans P. W., Swinkels D. W. (2008) Shedding & shaving: disclosure of proteomic expressions on a bacterial face. Proteomics 8, 1415–1428 [DOI] [PubMed] [Google Scholar]
- 40. Blonder J., Chan K. C., Issaq H. J., Veenstra T. D. (2006) Identification of membrane proteins from mammalian cell/tissue using methanol-facilitated solubilization and tryptic digestion coupled with 2D-LC-MS/MS. Nat. Protoc. 1, 2784–2790 [DOI] [PubMed] [Google Scholar]
- 41. Blonder J., Conrads T. P., Yu L. R., Terunuma A., Janini G. M., Issaq H. J., Vogel J. C., Veenstra T. D. (2004) A detergent- and cyanogen bromide-free method for integral membrane proteomics: application to Halobacterium purple membranes and the human epidermal membrane proteome. Proteomics 4, 31–45 [DOI] [PubMed] [Google Scholar]
- 42. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 43. Absalon S., Kohl L., Branche C., Blisnick T., Toutirais G., Rusconi F., Cosson J., Bonhivers M., Robinson D., Bastin P. (2007) Basal body positioning is controlled by flagellum formation in Trypanosoma brucei. PLoS ONE 2, e437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kelly S., Reed J., Kramer S., Ellis L., Webb H., Sunter J., Salje J., Marinsek N., Gull K., Wickstead B., Carrington M. (2007) Functional genomics in Trypanosoma brucei: a collection of vectors for the expression of tagged proteins from endogenous and ectopic gene loci. Mol. Biochem. Parasitol. 154, 103–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang Z., Morris J. C., Drew M. E., Englund P. T. (2000) Inhibition of trypanosoma brucei gene expression by RNA interference using an integratable vector with opposing T7 promoters. J. Biol. Chem. 275, 40174–40179 [DOI] [PubMed] [Google Scholar]
- 46. Redmond S., Vadivelu J., Field M. C. (2003) RNAit: an automated web-based tool for the selection of RNAi targets in Trypanosoma brucei. Mol. Biochem. Parasitol. 128, 115–118 [DOI] [PubMed] [Google Scholar]
- 47. Burkard G., Fragoso C. M., Roditi I. (2007) Highly efficient stable transformation of bloodstream forms of Trypanosoma brucei. Mol. Biochem. Parasitol. 153, 220–223 [DOI] [PubMed] [Google Scholar]
- 48. Absalon S., Blisnick T., Bonhivers M., Kohl L., Cayet N., Toutirais G., Buisson J., Robinson D., Bastin P. (2008) Flagellum elongation is required for correct structure, orientation and function of the flagellar pocket in Trypanosoma brucei. J. Cell Sci. 121, 3704–3716 [DOI] [PubMed] [Google Scholar]
- 49. Dacheux D., Landrein N., Thonnus M., Gilbert G., Sahin A., Wodrich H., Robinson D. R., Bonhivers M. (2012) A MAP6-related protein is present in protozoa and is involved in flagellum motility. PLoS One 7, e31344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pradel L. C., Bonhivers M., Landrein N., Robinson D. R. (2006) NIMA-related kinase TbNRKC is involved in basal body separation in Trypanosoma brucei. J. Cell Sci. 119, 1852–1863 [DOI] [PubMed] [Google Scholar]
- 51. Dubois K. N., Alsford S., Holden J. M., Buisson J., Swiderski M., Bart J. M., Ratushny A. V., Wan Y., Bastin P., Barry J. D., Navarro M., Horn D., Aitchison J. D., Rout M. P., Field M. C. (2012) NUP-1 Is a Large Coiled-Coil Nucleoskeletal Protein in Trypanosomes with Lamin-Like Functions. PLoS Biology 10, e1001287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kohl L., Sherwin T., Gull K. (1999) Assembly of the paraflagellar rod and the flagellum attachment zone complex during the Trypanosoma brucei cell cycle. J. Eukaryot. Microbiol. 46, 105–109 [DOI] [PubMed] [Google Scholar]
- 53. Adhiambo C., Blisnick T., Toutirais G., Delannoy E., Bastin P. (2009) A novel function for the atypical small G protein Rab-like 5 in the assembly of the trypanosome flagellum. J. Cell Sci. 122, 834–841 [DOI] [PubMed] [Google Scholar]
- 54. Robinson D. R., Gull K. (1991) Basal body movements as a mechanism for mitochondrial genome segregation in the trypanosome cell cycle. Nature 352, 731–733 [DOI] [PubMed] [Google Scholar]
- 55. Emmer B. T., Daniels M. D., Taylor J. M., Epting C. L., Engman D. M. (2010) Calflagin inhibition prolongs host survival and suppresses parasitemia in Trypanosoma brucei infection. Eukaryot. Cell 9, 934–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ralston K. S., Kabututu Z. P., Melehani J. H., Oberholzer M., Hill K. L. (2009) The Trypanosoma brucei flagellum: moving parasites in new directions. Annu. Rev. Microbiol. 63, 335–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Buisson J., Chenouard N., Lagache T., Blisnick T., Olivo-Marin J. C., Bastin P. (2013) Intraflagellar transport proteins cycle between the flagellum and its base. J. Cell Sci. 126, 327–338 [DOI] [PubMed] [Google Scholar]
- 58. Fenn K., Matthews K. R. (2007) The cell biology of Trypanosoma brucei differentiation. Curr. Opin. Microbiol. 10, 539–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhou Q., Gheiratmand L., Chen Y., Lim T. K., Zhang J., Li S., Xia N., Liu B., Lin Q., He C. Y. (2010) A comparative proteomic analysis reveals a new bi-lobe protein required for bi-lobe duplication and cell division in Trypanosoma brucei. PLoS One 5, e9660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Inoue M., Nakamura Y., Yasuda K., Yasaka N., Hara T., Schnaufer A., Stuart K., Fukuma T. (2005) The 14-3-3 proteins of Trypanosoma brucei function in motility, cytokinesis, and cell cycle. J. Biol. Chem. 280, 14085–14096 [DOI] [PubMed] [Google Scholar]
- 61. Voncken F., Gao F., Wadforth C., Harley M., Colasante C. (2013) The phosphoarginine energy-buffering system of Trypanosoma brucei involves multiple arginine kinase isoforms with different subcellular locations. PLoS One 8, e65908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Miranda M. R., Bouvier L. A., Canepa G. E., Pereira C. A. (2009) Subcellular localization of Trypanosoma cruzi arginine kinase. Parasitology 136, 1201–1207 [DOI] [PubMed] [Google Scholar]
- 63. Pereira C. A., Alonso G. D., Torres H. N., Flawia M. M. (2002) Arginine kinase: a common feature for management of energy reserves in African and American flagellated trypanosomatids. J. Eukaryot. Microbiol. 49, 82–85 [DOI] [PubMed] [Google Scholar]
- 64. Ralston K. S., Kisalu N. K., Hill K. L. (2011) Structure-function analysis of dynein light chain 1 identifies viable motility mutants in bloodstream-form Trypanosoma brucei. Eukaryot. Cell 10, 884–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bastin P., MacRae T. H., Francis S. B., Matthews K. R., Gull K. (1999) Flagellar morphogenesis: protein targeting and assembly in the paraflagellar rod of trypanosomes. Mol. Cell. Biol. 19, 8191–8200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bancaud A., Huet S., Rabut G., Ellenberg J. (2010) Fluorescence perturbation techniques to study mobility and molecular dynamics of proteins in live cells: FRAP, photoactivation, photoconversion, and FLIP. In Goldman R. D., Swedlow J. R., Spector D. L. (Ed.), Live cell imaging: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor NY., pp. 67–93 [DOI] [PubMed] [Google Scholar]
- 67. Bastin P., Ellis K., Kohl L., Gull K. (2000) Flagellum ontogeny studied via an inherited and regulated RNA interference system. J. Cell Sci. 113, 3321–3328 [DOI] [PubMed] [Google Scholar]
- 68. Farr H., Gull K. (2009) Functional studies of an evolutionarily conserved, cytochrome b5 domain protein reveal a specific role in axonemal organisation and the general phenomenon of post-division axonemal growth in trypanosomes. Cell Motil. Cytoskeleton 66, 24–35 [DOI] [PubMed] [Google Scholar]
- 69. Esson H. J., Morriswood B., Yavuz S., Vidilaseris K., Dong G., Warren G. (2012) Morphology of the trypanosome bilobe, a novel cytoskeletal structure. Eukaryot. Cell 11, 761–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lewin R. A., Lee K. W. (1985) Autotomy of algal flagella: electron microscope studies of Chlamydomonas (Chlorophyceae) and Tetraselmis (Prasinophyceae). Phycologia 24, 311–316 [Google Scholar]
- 71. Arnaiz O., Malinowska A., Klotz C., Sperling L., Dadlez M., Koll F., Cohen J. (2009) Cildb: a knowledgebase for centrosomes and cilia. Databas: the journal of biological databases and curation 2009, bap022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Roditi I., Schwarz H., Pearson T. W., Beecroft R. P., Liu M. K., Richardson J. P., Buhring H. J., Pleiss J., Bulow R., Williams R. O., et al. (1989) Procyclin gene expression and loss of the variant surface glycoprotein during differentiation of Trypanosoma brucei. J. Cell Biol. 108, 737–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Demonchy R., Blisnick T., Deprez C., Toutirais G., Loussert C., Marande W., Grellier P., Bastin P., Kohl L. (2009) Kinesin 9 family members perform separate functions in the trypanosome flagellum. J. Cell Biol. 187, 615–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Rotureau B., Blisnick T., Subota I., Julkowska D., Cayet N., Perrot S., Bastin P. (2014) Flagellar adhesion in Trypanosoma brucei relies on interactions between different skeletal structures in the flagellum and cell body. J. Cell Sci. 127, 204–215 [DOI] [PubMed] [Google Scholar]
- 75. Sun S. Y., Wang C., Yuan Y. A., He C. Y. (2013) An intracellular membrane junction consisting of flagellum adhesion glycoproteins links flagellum biogenesis to cell morphogenesis in Trypanosoma brucei. J. Cell Sci. 126, 520–531 [DOI] [PubMed] [Google Scholar]
- 76. Sherwin T., Schneider A., Sasse R., Seebeck T., Gull K. (1987) Distinct localization and cell cycle dependence of COOH terminally tyrosinolated alpha-tubulin in the microtubules of Trypanosoma brucei brucei. J. Cell Biol. 104, 439–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Fliegauf M., Olbrich H., Horvath J., Wildhaber J. H., Zariwala M. A., Kennedy M., Knowles M. R., Omran H. (2005) Mislocalization of DNAH5 and DNAH9 in respiratory cells from patients with primary ciliary dyskinesia. Am. J. Resp. Crit. Care 171, 1343–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Yagi T., Uematsu K., Liu Z., Kamiya R. (2009) Identification of dyneins that localize exclusively to the proximal portion of Chlamydomonas flagella. J. Cell Sci. 122, 1306–1314 [DOI] [PubMed] [Google Scholar]
- 79. Bui K. H., Yagi T., Yamamoto R., Kamiya R., Ishikawa T. (2012) Polarity and asymmetry in the arrangement of dynein and related structures in the Chlamydomonas axoneme. J. Cell Biol. 198, 913–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Johnson K. A., Rosenbaum J. L. (1992) Polarity of flagellar assembly in Chlamydomonas. J. Cell Biol. 119, 1605–1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Archer S. K., Inchaustegui D., Queiroz R., Clayton C. (2011) The cell cycle regulated transcriptome of Trypanosoma brucei. PLoS One 6, e18425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Morga B., Bastin P. (2013) Getting to the heart of intraflagellar transport using Trypanosoma and Chlamydomonas models: the strength is in their differences. Cilia 2, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Liu W., Apagyi K., McLeavy L., Ersfeld K. (2010) Expression and cellular localisation of calpain-like proteins in Trypanosoma brucei. Mol. Biochem. Parasitol. 169, 20–26 [DOI] [PubMed] [Google Scholar]
- 84. Chan K. Y., Ersfeld K. (2010) The role of the Kinesin-13 family protein TbKif13–2 in flagellar length control of Trypanosoma brucei. Mol. Biochem. Parasitol. 174, 137–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Woodward R., Carden M. J., Gull K. (1995) Immunological characterization of cytoskeletal proteins associated with the basal body, axoneme and flagellum attachment zone of Trypanosoma brucei. Parasitology 111 (Pt 1), 77–85 [DOI] [PubMed] [Google Scholar]
- 86. Pedersen L. B., Geimer S., Sloboda R. D., Rosenbaum J. L. (2003) The Microtubule plus end-tracking protein EB1 is localized to the flagellar tip and basal bodies in Chlamydomonas reinhardtii. Curr. Biol. 13, 1969–1974 [DOI] [PubMed] [Google Scholar]
- 87. Bloch M. A., Johnson K. A. (1995) Identification of a molecular chaperone in the eukaryotic flagellum and its localization to the site of microtubule assembly. J. Cell Sci. 108 (Pt 11), 3541–3545 [DOI] [PubMed] [Google Scholar]
- 88. Woolley D., Gadelha C., Gull K. (2006) Evidence for a sliding-resistance at the tip of the trypanosome flagellum. Cell Motil. Cytoskel. 63, 741–746 [DOI] [PubMed] [Google Scholar]
- 89. Haycraft C. J., Banizs B., Aydin-Son Y., Zhang Q., Michaud E. J., Yoder B. K. (2005) Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 1, e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Rotureau B., Subota I., Buisson J., Bastin P. (2012) A new asymmetric division contributes to the continuous production of infective trypanosomes in the tsetse fly. Development 139, 1842–1850 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.