

Abstract
Described here is a quantitative mass spectrometry-based proteomics method for the large-scale thermodynamic analysis of protein-ligand binding interactions. The methodology utilizes a chemical modification strategy termed, Stability of Proteins from Rates of Oxidation (SPROX), in combination with a Stable Isotope Labeling with Amino Acids in Cell Culture (SILAC) approach to compare the equilibrium folding/unfolding properties of proteins in the absence and presence of target ligands. The method, which is general with respect to ligand, measures the ligand-induced changes in protein stability associated with protein-ligand binding. The methodology is demonstrated in a proof-of-principle study in which the well-characterized protein-drug interaction between cyclosporine A (CsA) and cyclophilin A was successfully analyzed in the context of a yeast cell lysate. A control experiment was also performed to assess the method's false positive rate of ligand discovery, which was found to be on the order of 0.4 - 3.5%. The new method was utilized to characterize the adenosine triphosphate (ATP)-interactome in Saccharomyces cerevisiae using the nonhydrolyzable ATP analog, adenylyl imidodiphosphate (AMP-PNP), and the proteins in a yeast cell lysate. The new methodology enabled the interrogation of 526 yeast proteins for interactions with ATP using 2035 peptide probes. Ultimately, 325 peptide hits from 139 different proteins were identified. Approximately 70% of the hit proteins identified in this work were not previously annotated as ATP binding proteins. However, nearly two-thirds of the newly discovered ATP interacting proteins have known interactions with other nucleotides and co-factors (e.g. NAD and GTP), DNA, and RNA based on GO-term analyses. The current work is the first proteome-wide profile of the yeast ATP-interactome, and it is the largest proteome-wide profile of any ATP-interactome generated, to date, using an energetics-based method. The data is available via ProteomeXchange with identifiers PXD000858, DOI 10.6019/PXD000858, and PXD000860.
The characterization of protein-ligand interactions is important in many areas of biochemical research from fundamental studies of biological processes to understanding drug action. Currently, the most widely used methods for proteome-wide analyses of protein-ligand binding interactions are those that combine an affinity purification step with a mass spectrometry-based proteomics analysis. Such methods have provided a wealth of information about protein-protein interaction networks in different proteomes (1–4), and they have helped identify the protein targets of small molecules (5–7). However, a significant drawback to their use is the need for specially designed ligands to facilitate the affinity purification. This has prompted the development of more general methods for protein-ligand binding analyses that can be performed directly in solution and do not require derivatization and/or immobilization of the ligand. Several such methods involving the use of chromatography co-elution (8), protease susceptibility (9), and energetics-based approaches (10–15) have recently been reported.
Energetics-based approaches are especially attractive for protein-ligand binding analyses because they can be both quantitative and general with respect to ligand class. Two energetics-based approaches, the stability of proteins from rates of oxidation (SPROX)1 (10, 16, 17) and pulse proteolysis techniques (13, 18), have shown promise for protein-ligand binding analyses on the proteomic scale, but so far have been limited in their proteomic coverage. Although the pulse proteolysis technique does utilize targeted mass spectrometry-based proteomics analyses for the identification of hit proteins, the technique relies on gel-based strategies for the resolution, detection, and quantitation of potential protein targets (13, 18). This reliance on gel-based strategies for protein resolution, detection, and quantitation, ultimately limits the complexity of protein samples that can be interrogated for ligand binding. In contrast, the SPROX technique has been interfaced with conventional bottom-up shotgun proteomics platforms that exploit the capabilities of modern LC-MS/MS systems to resolve, detect, and quantify the protein components of complex biological mixtures (10, 16, 17).
A key limitation to the bottom-up shotgun proteomics protocols developed for SPROX analyses, to date, is that they require the detection and quantitation of methionine-containing peptides to report on the thermodynamic stability of the proteins to which they map. Although the frequency of methionine residues in proteins is relatively low (∼2.5%) (19), the large majority of proteins have at least one methionine. Because one methionine residue can report on the global equilibrium folding/unfolding properties of the protein or protein domain to which it maps, the scope of SPROX is not fundamentally limited by the relatively low frequency of methionine residues in proteins. Rather, the protein coverage in proteome-wide SPROX experiments is limited by the practicalities associated with the comprehensive detection and quantitation of methionine-containing peptides in the bottom-up shotgun proteomics experiment.
The SPROX protocol described here utilizes a stable isotope labeling with amino acids in cell culture (SILAC)-based strategy to expand the protein coverage in proteome-wide SPROX experiments by enabling any peptide (i.e. methionine-containing or not) that is identified and quantified in a bottom-up shotgun proteomics experiment to report on the stability of the protein to which it maps. As part of the work described here the capabilities of this new method for protein-ligand binding analysis (referred to hereafter as SILAC-SPROX) are demonstrated and benchmarked in two protein-ligand binding studies. In the first part of this work, the endogenous proteins in a yeast cell lysate are analyzed for binding to cyclosporine A (CsA), an immunosuppressant with well-characterized protein targets (5, 20). In the second part of this work, the endogenous proteins in a yeast cell lysate are analyzed for binding to adenylyl imidodiphosphate (AMP-PNP), a nonhydrolyzable analog of the ubiquitous enzyme co-factor, adenosine triphosphate (ATP), which has less well-characterized protein targets. In the CsA binding study, the already well-characterized tight-binding interaction between CsA and cyclophilin A (21–23) was successfully detected and quantified using the methodology. A number of known and unknown protein binding interactions of ATP were identified and quantified in the ATP-binding experiments described here. The SILAC-SPROX approach shows promise for future studies of protein-ligand interactions at the systems level (e.g. in cellular processes and disease states).
EXPERIMENTAL PROCEDURES
Yeast Cell Lysate Preparation
A glycerol stock of the Saccharomyces cerevisiae deletion strain BY4739 (Open Biosystems, Lafayette, CO), an auxotroph for lysine, was streaked on a Petri-dish containing synthetic complete (SC) media and 30 mg/L of light l-Lysine. The SC-media was comprised of 1.7 g of yeast nitrogen base, 5 g of ammonium sulfate, 20 g of bacto-agar, 2 g glucose, and 1.92 g of synthetic drop out mix without lysine (Sunrise Science Product, San Diego, CA). After a 3-day incubation at 30 °C, one colony was selected and inoculated into 10 ml of SC-media and 30 mg/L of light l-lysine (Sigma Aldrich, St Louis, MO). After an overnight incubation at 30 °C, 5 μl of the cell culture was transferred into 50 ml (or 100 ml) of SC-media containing light l-lysine (Sigma Aldrich, Sigman, St. Louis, MO) at final concentration of 30 mg/ml; while another 5 μl portion of the culture was transferred into 50 ml (or 100 ml) of SC-media containing same concentration of heavy lysine (13C6 15N2.Cl). The two cell cultures were incubated at 30 °C, and 5 μl portions of each culture were transferred to 50 ml (or 100 ml) volumes of corresponding light and heavy media twice before they were ultimately each transferred to 1 L of the corresponding light and heavy media and grown overnight at 30 °C. Approximately a dozen, 60–80 ml portions of each cell culture were centrifuged at 1000 × g at 4 °C for 10 min to generate about a dozen cell pellets of the light lysine labeled yeast cells and about a dozen cell pellets of the heavy lysine labeled yeast cells.
The light and heavy lysine labeled cell lysates used in the SILAC-SPROX analyses were prepared by adding ∼500 μl of lysis buffer to a cell pellet. The lysis buffer in the CsA-binding experiments was composed of 20 mm phosphate buffer (pH 7.4) and a protease inhibitor mixture containing 1 mm AEBSF, 500 μm Bestatin, 15 mm E-64, 20 μm Leupeptin, and 10 μm Pepstatin A. The lysis buffer in the ATP-binding experiments included the same protease inhibitor mixture but was composed of 0.1 m Tris buffer (pH 7.4). In all cases, the cells were lysed by mechanical disruption using a disruptor genie (Scientific Industries, Bohemica, NY) and 400–600 μm diameter glass beads. A total of 15 cycles consisting of mechanical disruption for 30 s and cooling on ice for 1 min were used to lyse the cells in each cell pellet. Ultimately, the lysed cells were centrifuged at 15,000 × g for 10 min at 4 °C, and the supernatant was collected for use in SILAC-SPROX analyses. The concentration of protein in each light and heavy lysate was typically 10–20 mg/ml total protein as determined in a Bradford assay (24). If the protein concentration in the light and heavy lysates were unequal, the protein concentrations in the two samples were normalized to the lower concentration prior to the SILAC-SPROX analysis.
SILAC-SPROX Analysis
For each of the seven SILAC-SPROX analyses performed here, a pair of heavy and light labeled yeast cell lysates with matched protein concentrations (∼15 mg/ml) was prepared and used to generate (−) and (+) ligand stock solutions for each experiment. In the two CsA-binding experiments, nine volumes of the heavy and light yeast cell lysates were mixed with 1 volume of DMSO and with one volume 10 mm CsA in DMSO to generate the (−) and (+) ligand stock solutions, respectively. In the Control Experiment, nine volumes of the heavy and light yeast cell lysates were mixed with one volume of water to generate the two (−) ligand samples analyzed in the Control experiment. For the ATP-binding studies in Solution Experiment 1A/B and Gel Experiment 1, one volume of 100 mm MgCl2 was added to eight volumes of both the light and heavy lysate, one volume of aqueous 828 mm AMP-PNP was added to the heavy lysate to create the (+) ligand stock solution, and one volume of lysis buffer was added to the light lysate to generate the (−) ligand stock solution. The (−) and (+) ligand stock solutions for the ATP-binding studies in Solution Experiment 2 and Gel Experiment 2 were prepared as in Solution Experiment 1 and Gel Experiment 1, with the exception that the concentration of the MgCl2 solution used to prepare the samples was 1 m instead of 100 mm.
In each SILAC-SPROX experiment the (−) and (+) ligand stock solutions were equilibrated for 30–60 min, before 20 μl aliquots of the samples (containing ∼200–400 μg of total protein each) were diluted into a series of denaturant containing buffer stock solutions. In the CsA-binding experiments, the denaturant-containing buffer stock solutions contained 20 mm phosphate buffer (pH 7.4) with GdmCl concentrations ranging from 0 to 7 m, and 25 μl volumes were combined with the 20 μl sample aliquots of the (−) and (+) ligand stock solutions. In the ATP-binding experiments the denaturant-containing buffer stock solutions contained 0.1 m Tris●HCl buffer (pH 7.4) with urea concentrations ranging from 0 to 8 m, and 75 μl volumes were combined with the 20 μl sample aliquots. In both cases, the exact concentration of GdmCl or urea in each denaturant-containing buffer stock solution was determined from a refractive index measurement of the buffer as described previously (25, 26). The final GdmCl concentrations in the 12 denaturant-containing buffers used in the CsA-binding and control experiments were: 0, 0.6, 1.1, 1.4, 1.6, 1.9, 2.1, 2.4, 2.6, 2.9, 3.1, and 3.4 m. The final urea concentrations in the eight denaturant-containing buffers used for the ATP binding studies in Solution Experiment 1A/B and Gel Experiment 1 were 0, 1, 2, 2.7, 3.3, 4, 4.8, and 6 m. The final urea concentrations in the 10 denaturant-containing buffers used for the ATP binding studies in Solution Experiment 2 and Gel Experiment 2 were 0.0, 1.0, 1.5, 2.0, 2.5, 2.8, 3.3, 4.0, 4.9, and 6.0 m.
The (−) and (+) ligand samples in each SILAC-SPROX analysis were equilibrated for 30–60 min in the denaturant containing buffers before the methionine oxidation reaction in SILAC-SPROX was initiated by addition of hydrogen peroxide to the protein and protein-ligand samples in each denaturant-containing buffer. The final hydrogen peroxide concentration in each reaction was 0.98 m or 0.54 m, depending on the experiment (see Table S1). The oxidation reactions were allowed to proceed for 3, 6, or 24 min, depending on the experiment (see Table S1). The oxidation reactions were each quenched with the addition of 760 μl of a 375 mm solution of l-methionine. In each SILAC-SPROX analysis, the (−) and (+) ligand samples (i.e. the heavy and light yeast cell lysate sample) in buffers containing the same denaturant concentration were combined (see Step 4 in Fig. 1), and the light and heavy-labeled proteins in each sample were precipitated with TCA. The samples were centrifuged, and the resulting protein pellets were washed with ice-cold ethanol and acetone before they were dried.
Fig. 1.
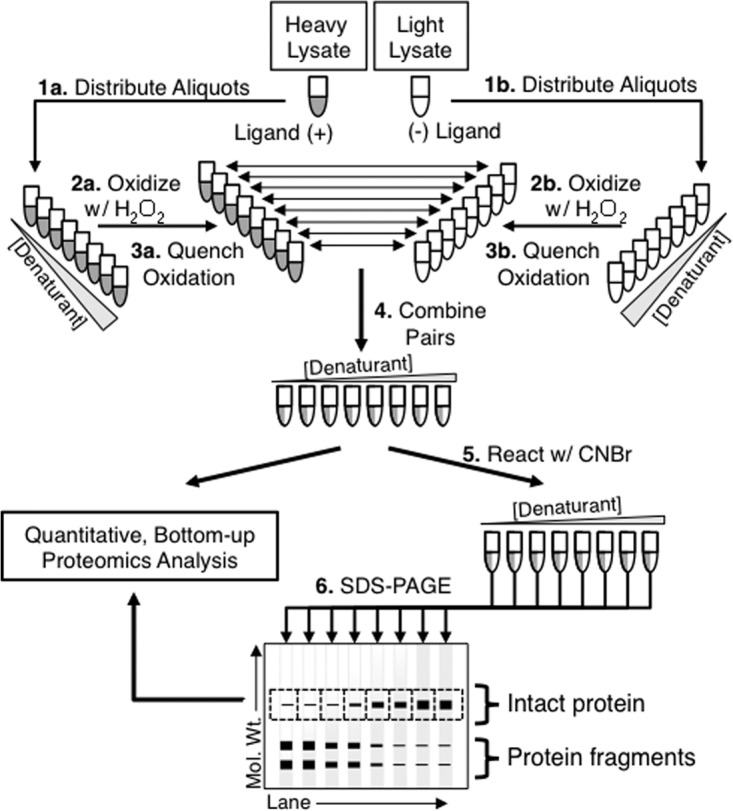
Schematic representation of the SILAC-SPROX protocol developed in this work. The protocol requires the preparation of light and heavy cell lysates. The test ligand is spiked into one of the cell lysates. In Steps 1–3, the two cell lysate samples are subjected to a SPROX analysis in which aliquots of each cell lysate are distributed into a series of denaturant-containing buffers and reacted with hydrogen peroxide under conditions that enable the selective oxidation of exposed methionine residues in proteins. The oxidation reactions are quenched with the addition of excess methionine, and the appropriate light and heavy samples with matching denaturant concentrations are combined (see Step 4). The protein samples are then directly submitted to a conventional bottom-up proteomics analysis in solution, (referred to in the text as the solution-based approach) and/or reacted with CnBr (Step 5) before they are fractionated by SDS-PAGE and submitted to a conventional gel-based proteomics analysis (referred to in the text as the gel-based approach) (Step 6).
Cyanogen Bromide Treatment
For the gel-based experiments performed in the ATP-binding study, the combined (−) and (+) ligand protein pellets from the SPROX analyses were re-dissolved in 100 μl of a 70% (v/v) solution of formic acid (TCI America, Portland, OR). In Gel Experiment 1, 1 mg of crystalline cyanogen bromide (CnBr) (Sigma Aldrich, St. Louis, MO) was added to each solution. This corresponded to an estimated CnBr:methionine mole ratio of ∼70:1 based on the measured protein concentration of the initial lysate and a frequency of methionine residues in proteins of 2.5%. In Gel Experiment 2, aliquots (∼4 μl) of a 5 m CnBr solution of CnBr in ACN were added to each protein solution. This corresponded to an estimated CnBr:methionine mole ratio of ∼140:1 in Gel Experiment 2. In both Gel Experiments 1 and 2, the CnBr treated samples were incubated with frequent shaking at room temperature for 4 h before any unreacted CnBr was evaporated by heating samples with open caps at 50 °C for 5 min in a chemical fume hood. The protein samples were neutralized by adding 200 μl of 1.7 m of 4-ethylmorpholine (NEM) (Sigma Aldrich, St. Louis, MO). The samples were diluted with 1.5 ml of distilled water and the proteins in each sample were precipitated with TCA (10% w/v). The resulting protein pellets were washed with ice-cold ethanol and acetone, and dried.
Gel-Based Proteomics Sample Preparation
In the gel-based experiments performed in the ATP-binding study, the TCA precipitated protein pellets obtained after the CnBr treatment were re-dissolved in 40 μl of freshly made 8–10 M urea and 20 μl of 6× Laemmli sample buffer comprised of 375 mm Tris●HCl pH 6.8, 6% SDS, 50% Glycerol and 0.045% Bromphenol (Boston Bioproducts, Boston, MA). A 3 μl aliquot of β-mercaptoethanol (BME) was added to each sample and the samples were heated at 95 °C for 5 min to reduce disulfide bonds. In Gel Experiment 1, a 20 μl volume of each sample was loaded into one lane of a mini polyacrylamide gel 10 × 8.5 cm (NUsep, Bogart, GA), and in Gel Experiment 2 a 15 μl volume of each sample was loaded onto a midsize polyacrylamide gel (BioRad Criterion, Hercules, CA). After electrophoretic separation, the gels were fixed for 20 min by treatment with a solution containing 25% isopropanol, 10% acetic acid, and 65% distilled water; was stained overnight by treatment with a solution containing 0.01% R-250 (Bio Rad, Hercules, CA) in 10% acetic acid; and was de-stained upon repeated treatments with 10% acetic acid until the gel image was clear. In Gel Experiment 1, a portion of the gel corresponding to a molecular weight range of 20–30 kDa was excised resulting in eight different gel pieces (see Fig. 3A below). In Gel Experiment 2, the gel was extensively cut into 14 different molecular weight fractions corresponding to: 0–6, 6–12, 12–15, 15–18, 18–19, 19–20, 20–24, 24–30, 30–37, 37–50, 50–75, 75–100, 100–250, and larger than 250 kDa (see Fig. 3B below). The eight gel bands generated in Gel Experiment 1 and the 140 gel bands generated in Gel Experiment 2 were each subjected to a standard in-gel digestion protocol (27) using Lys-C as the protease. Prior to LC-MS/MS analysis, the protease digested samples were each concentrated and desalted using C18 resin (The Nest Group, Southborough, MA) according to the manufacturer's protocol.
Fig. 3.
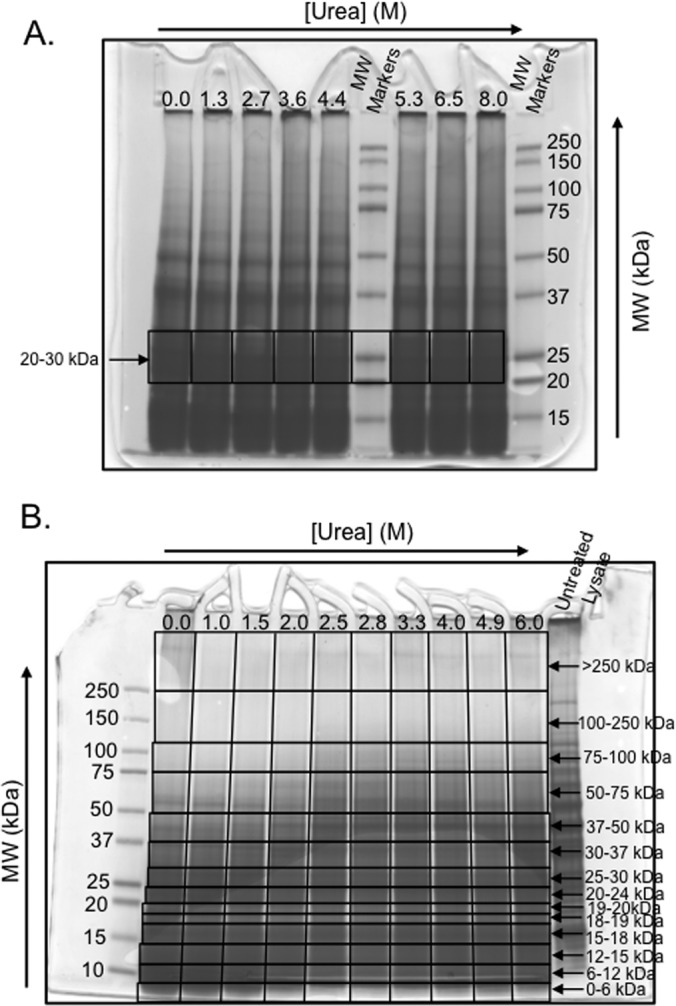
SDS-PAGE results obtain in gel-based SILAC-SPROX experiments. The results obtained in Gel Experiments 1 and 2 are shown in A, and B, respectively. In each case the black boxes indicate the gel-bands that were excised and submitted to bottom-up proteomics analyses. The arrows indicate the rows of gel-bands that were used to generate the SILAC-SPROX data in each experiment.
Gel-Free Proteomics Sample Preparation: CsA-Binding Experiments
The dried protein pellets from the SILAC-SPROX analyses were dissolved in 60 μl (CsA-Binding Experiment 1) or 30 μl (Control Experiment and CsA-Binding Experiment 2) of 0.5 m triethyl ammonium bicarbonate (TEAB) containing 3 μl (Experiment 1) or 1.5 μl (CsA-Binding Experiment 2 and Experiment 2) of a 2% stock solution of SDS. The disulfide bonds in each protein sample were reduced upon addition of 5 μl of 50 mm TCEP and treatment for 1 h at 60 °C. The protein samples were each reacted with 2.5 μl of 200 mm methyl methane thiosulfonate (MMTS) for 10 min at room temperature to block cysteine side chains. Ultimately, 3 μl of a 1 μg/μl trypsin solution was added to the protein sample in each tube and the samples were incubated overnight (∼15 h) at 37 °C before 5 μl of 10% trifluoroacetic acid (TFA) was added to quench the trypsin digestion. The samples were desalted using C18 resin (The Nest Group, Southborough, MA) according to the manufacturer's protocol, and an estimated 6 μg of total peptide from each sample, was subjected to an LC-MS/MS analysis.
Gel-Free Proteomics Sample Preparation: ATP-Binding Study
The dried pellets from the SILAC-SPROX analyses were dissolved in 60 μl of 0.1 m Tris buffer (pH 8.0) containing 8 m urea, heated at 37 °C for 5 min, vortexed for 10 min, and heated again at 37 °C for 5 min. A 6 μl aliquot of 50 mm TCEP.HCl and 3 μl aliquot of 2% SDS solution were added to each re-dissolved pellet and the samples were incubated for 1 h at 37 °C before 6 μl of a 100 mm solution of MMTS was added to each protein sample. After a 10 min incubation at room temperature, ∼99 μl of Tris buffer (pH 8.0) containing 4 μg of Lys-C was added to each sample and the proteins were digested overnight at 37 °C. Each sample was centrifuged at 15,000 × g for 5 min to precipitate undigested protein. The supernatant was desalted with C18 resin (The Nest Group, Southborough, MA), and an estimated 2 μg of total peptide from each sample was subjected to an LC-MS/MS analysis.
LC-MS/MS Analyses
The proteomics samples generated in the Control Experiment, CsA-Binding Experiments 1 and 2, Solution Experiment 1A, and Gel Experiment 2 were analyzed using an Agilent 6520 Q-TOF mass spectrometer (Agilent Technologies, Inc, San Jose, CA) equipped with a Chip Cube interface. The HPLC chip used for all the analyses, except those in Gel Experiment 2, included a 160 nL enrichment column and a 75 μm X 150 mm analytical column with Zorbax 300SB-C18 5 μm packing material. Sample were loaded onto the column and peptides were eluted using linear gradients of from 0% to 3% Solvent B (ACN in 0.1% formic acid (FA)) in Solvent A for the 5 min, 3% to 15% Solvent B in A for 2.5 min, 15% to 45% Solvent B in A for 78 min, and 45% to 100% Solvent B in A for 10 min. The column was flushed with 100% Solvent B for the 10 min and equilibrated with 3% Solvent B for 10 min before the next sample was injected. The HPLC chip used in Gel Experiment 2 was a short chip with a 40 nL enrichment column and a 73 μm × 43 mm analytical column packed with Zorbax 80SB-C18 5 μm material. The gradient was: 3%-5% Solvent B over 2 min, 5%-15% over 2 min, 15%-50% over 18 min 60%-90% over 3 min, 100% over 2 min and 5% over 3 min.
In all analyses on the Agilent Q-TOF system, the LC flow rate was 0.4 μl/min. The capillary voltage ranged from 1800 to 1850 V. The flow rate of the drying gas was set at 6 L/min at 350 °C. The skimmer and fragmentor were set at 65 and 175 V, respectively. The collision energy was as determined by the equation 3.5 V/100 Da with a −4.8 V offset. The inclusion window width for precursor ions was 4 m/z. The precursor purity stringency and purity cutoff were set to 100 and 30% respectively. The scan rate was three scans per second in the mass spectra and two scans per second in the product ion spectra. In every cycle, four precursors were selected for fragmentation.
Agilent's Spectrum Mill MS Proteomics Workbench software, Rev B.04.00.122 PreRelease was used to extract peak lists from the LC-MS/MS data generated on the Agilent Q-TOF system and to search the data against the 6619 Saccharomyces cerevisiae proteins in the UniProttKB/Swiss-Prot database (uniprot_sprot_fasta.gz Release 2012_01/modified on 2/22/12 downloaded on 2/28/2012). The search parameters used in the Control Experiment and the CsA-Binding Experiments 1 and 2 were: Saccharomyces cerevisiae as the organism, SILAC heavy labeled lysine-8 and cysteine modification with MMTS as a fixed modification, oxidation of methionine and deamidation of asparagine (N) as variable (0–1) modifications, the precursor and product mass ion tolerances as 20 and 50 ppm (respectively), the protease as trypsin with a maximum of three missed cleavages, and the maximum product ion charge as 7. The same search parameters were used in the Solution Experiment 1A and Gel Experiment 2 with the exception that the protease was Lys-C. The L/H ratios of all peptides output from the Spectrum Mill searches, regardless of score, were used in the subsequent data analysis steps. Our justification for using all peptide identifications in subsequent data analysis steps was that true false positives would be best filtered out at later steps (e.g. would not be identified in samples from multiple denaturant concentrations, and/or not yield meaning L/H ratios).
The LC-MS/MS analyses in Solution Experiment 1B, Solution Experiment 2, and Gel Experiment 1 were performed using an LTQ Orbitrap XL mass spectrometer (Thermo-Scientific, Inc, San Jose, CA). The LC system was configured in a vented format and consisted of a fused-silica nanospray needle packed in-house with Magic C18 AQ 100A reverse-phase media (Michrom Bioresources Inc., Auburn, CA) in the column (25 cm) and Magic C18 AQ 200A reverse-phase media in the trap (2 cm). The peptide samples were loaded onto the column and chromatographic separation was performed using a two-mobile-phase solvent system consisting of 0.1% formic acid in water (A) and 0.1% acetic acid in acetonitrile (B). The mass spectrometer operated in a data-dependent MS/MS mode over the m/z range of 400–1800. For each cycle, the five most abundant ions from each MS scan were selected for MS/MS analysis using 35% normalized collision energy. Selected ions were dynamically excluded for 45 s.
Peak lists were extracted from the raw LC-MS/MS data files generated on the Orbitrap system and the data were searched against the 6651 Saccharomyces cerevisiae proteins in the 2013_09 release of the UniProt Knowledgebase downloaded at ftp://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/proteomes/. using Maxquant 1.3.0.5 (28). The following modifications were considered: methyl methanethiosulfonate at cysteine as a fixed modification for Solution Experiments 1B and 2 and carbamidomethylation at cysteines in Gel Experiment 1; SILAC labeling of lysine, and variable (0–1) oxidation of methionine and deamidation of Asparagine and Glutamine (N and Q). The enzyme was set as Lys-C, and up to two missed cleavages were permitted for Solution Experiment 1B and Gel Experiment 1 (up to three missed cleavages were allowed for Solution Experiment 2). The false discovery rate for peptide and protein identifications was set to 1%, and all other settings were set at the default parameters. As part of the default settings, the mass tolerance for precursor ions was 10 ppm and the mass tolerance for fragment ions was 0.5 Da. The search results were exported to Excel for further data analysis as described below.
In cases where identified peptides could be matched to multiple protein isoforms or multiple members of a protein family, all potential proteins are listed with the initial identification. Such redundancy is not expected to be a major problem in SILAC-SPROX experiments as the ligand-binding properties of highly homologous proteins are likely to be similar.
Quantitative Proteomic Data Analysis
The protein and peptide identifications and L/H (or H/L) ratios obtained from the LC-MS/MS data analyses (see Tables S3–S23) were filtered to show only those that contained lysine residues. In the CsA binding study only the identified peptides (and corresponding protein identifications) with L/H ratios >0 were used in subsequent data analysis steps. In the ATP-binding study, identified peptides (and corresponding protein identifications) with L/H (or H/L) ratios ≥0 were used in subsequent data analysis steps. If multiple L/H ratios were calculated for a given peptide sequence and charge state at a specific denaturant concentration (e.g. both the light and heavy version of a peptide and charge state was identified in the LC-MS/MS analysis and/or a peptide and charge state was identified in multiple product ion scans) they were averaged to give a single L/H ratio for the peptide and charge state at that denaturant concentration. Ultimately, these averaged L/H ratios were used to generate SILAC-SPROX data sets.
In the data analysis, only peptides that were identified in multiple runs (i.e. in samples from multiple denaturant concentrations) were considered (see Tables S3–S23). In the CsA binding experiments, methionine-containing peptides that were identified in the protein samples derived from four or more different denaturant-containing buffers were assayed for potential change in thermodynamic stability because of ligand binding. In ATP binding experiments, peptides that were present in six or more denaturant containing buffers were included in the assay except in the cases of Solution Experiment 1A and Solution Experiment 2, where peptides that were present in four and eight (respectively) or more denaturant containing buffers were included in the assay.
Using Microsoft Excel, peptides were filtered to identify those with significantly altered L/H ratios at two or more consecutive denaturant concentrations. Significantly altered L/H (or H/L) ratios in this work were taken to be those that changed by >1.7-fold from the baseline value observed for each peptide. The selection of this 1.7-fold cut-off value was based on a global analysis of the L/H (or in some experiments H/L) ratios recorded in the CsA and ATP binding experiments described here. The distributions of the L/H (or in some experiments H/L) ratios measured for the lysine- and nonmethionine-containing peptides identified in the CsA and ATP binding experiments performed here are shown in Figs. S3 and S4. In all the binding experiments performed here ≥90% of the measured L/H (or in some experiments L/H) ratios were within 1.7-fold of the median value, with the exception of Gel Experiment 2 in which ≥83% of the measured L/H ratios were within 1.7-fold of the median value (see Figs. S3 and S4). Based on this global analysis and our requirement that hit peptides must have significantly altered L/H ratios at two or more consecutive denaturant concentrations, the estimated p value associated with each peptide hit in this study was <0.01, except in the case of peptide hits identified in Gel Experiment 2, where the estimated p value associated with each peptide hit was <0.03.
RESULTS
General Strategy
The SILAC-SPROX protocol developed in this work is outlined in Fig. 1. A unique aspect of the SILAC-SPROX protocol is the use of cyanogen bromide (CnBr) in the gel-based approach. The CnBr reaction (Step 5 in Fig. 1) is used to cleave the polypeptide chain of proteins after methionine residues that were not oxidized in the SPROX analysis (Steps 1–3 in Fig. 1). Methionine residues in proteins that were oxidized in the SPROX analysis are protected from CnBr cleavage. As proteins are unfolded in the presence of increasing concentrations of the chemical denaturant used in the SPROX analysis, the “buried” methionine residues in a protein's three-dimensional structure are exposed to solvent, get oxidized in the SPROX analysis, and become protected from cyanogen bromide cleavage. Thus, as depicted in the gel-based SDS-PAGE readout in Fig. 1, intact proteins will predominantly appear in SPROX samples from higher denaturant concentrations, and the CnBr fragments of proteins will predominantly appear in SPROX samples from lower concentrations of denaturant. In the gel-based readout, rows of gel-bands corresponding to specific molecular weight ranges are excised, and subjected to proteomics protocols to quantify, in each gel-band, the relative amount of protein (or cyanogen bromide cleaved fragments) from the (−) and (+) ligand samples.
The CnBr digestion and subsequent gel-based separation of the intact proteins from their respective CnBr fragments is important in the gel-based approach because it enables every peptide that is successfully identified and quantified in the gel-based proteomics readout (not just the methionine-containing peptides) to report on the ligand binding properties of the protein from which it was derived. However, the SILAC-SPROX behavior of peptides extracted from intact protein bands will be opposite that of peptides extracted from gel bands containing CnBr fragments. That is, the amount of an intact protein will increase as a function of denaturant concentration; whereas, the amount of CnBr digested fragments of a protein will decrease as a function of denaturant concentration (see Fig. 1). Therefore, to minimize false negatives it is important that the gel be sectioned in molecular weight ranges that effectively separate intact proteins from their CnBr fragments. Otherwise, false negatives will result.
In both the solution- and gel-based experiments, the denaturant dependence of the L/H ratios obtained for the methionine-containing peptides is evaluated to identify hit peptides and the hit proteins to which they map (see Fig. 2). Unique to the gel-based experiment is that the denaturant dependence of the L/H ratios obtained from the nonmethionine-containing peptides generated in the bottom-up proteomics experiments can be used to identify protein hits. As with the methionine-containing peptides from protein hits in the solution-based experiment, the nonmethionine-containing peptides from protein hits in the gel-based experiment will have either high or low L/H ratios at intermediate denaturant concentrations, depending on whether they are identified in gel-bands containing the intact protein or CnBr cleaved fragments and on whether they are the result of a ligand induced stabilization or destabilization. The altered L/H ratios result from the differential denaturant dependence of the oxidation reaction and CnBr digestion in SILAC-SPROX because of stabilizing (or destabilizing) effects of the ligand on target proteins (see Fig. 2).
Fig. 2.
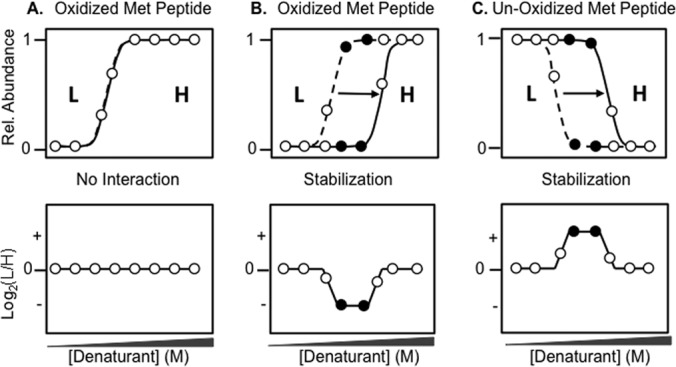
Schematic representation of SILAC-SPROX data expected for methionine-containing peptides in a SILAC-SPROX experiment in which the test ligand is spiked into the heavy lysate. Shown is the expected SPROX data (top panels) and the corresponding SILAC-SPROX data (bottom panels) for an oxidized methionine-containing peptide derived from a protein that does not interact with the ligand, A, for an oxidized interact-containing peptide derived from a protein that is stabilized in the presence of the ligand, B, and for a nonoxidized methionine-containing peptide derived from a protein that is stabilized in the presence of ligand, C, The SPROX curves represented with the dotted and solid lines in the top panels indicate those expected for a protein in the light and heavy lysate (respectively). Open circles represent data points at denaturant concentrations where the L/H ratio is unchanged in SILAC-SPROX. Closed circles represent data points at denaturant concentrations where the L/H ratio is indicative of a peptide (protein) hit. SILAC-SPROX data generated from ligand-induced destabilizations would be similar to that shown, except the sign of the altered Log2(L/H) ratios would be opposite from that shown in each case.
CsA Binding Experiment
The experimental protocol outlined in Fig. 1 was initially used in a proof-of-principle study to detect and quantify the direct-binding interaction between CsA and cyclophilin A in an endogenous yeast cell lysate. In this CsA binding study the combined sample pairs generated in Step 4 of the protocol (see Fig. 1) were only submitted to a bottom-up shotgun proteomics analysis using the LC-MS/MS readout. Summarized in Table I are the proteomics results obtained from the LC-MS/MS analyses of the combined sample pairs generated in two replicate CsA binding experiments, and in a control experiment in which no ligand was used.
Table I. Summary of the proteomic results obtained in the SILAC-SPROX experiments to characterize the CsA binding properties of the proteins in a yeast cell lysate.
| SILAC-SPROX experiment | Total Lys-containing peptides (proteins) identified | Total Lys- and Met-containing peptides (proteins) identified | Total peptides (proteins) assayed for bindinga | Hit peptides (Proteins) |
|---|---|---|---|---|
| Control (No Ligand) | 1471 (546) | 415 (203) | 119 (58) | 4 (4) |
| CsA Binding Exp. 1 | 982 (429) | 298 (144) | 69 (25) | 3 (2) |
| CsA Binding Exp. 2 | 1149 (493) | 328 (165) | 104 (48) | 2 (1) |
a Includes peptides (and the proteins to which they map) that were successfully identified and quantified in samples from at least four denaturant concentrations.
The only hit protein identified in both of the CsA binding experiments was cyclophilin A. The one lysine- and methionine-containing peptide from cyclophilin A, VIPDFMLQGGDFTAGNGTGGK, that was assayed in the analyses yielded SILAC-SPROX data consistent with the known CsA-induced stabilization of this protein (Fig. 4A). Both the oxidized and nonoxidized version of the VIPDFMLQGGDFTAGNGTGGK peptide were found to have significantly altered (i.e. >1.7-fold change as described in the Experimental Procedures) L/H ratios at 4 different denaturant concentrations (Fig. 4A). Two peptides, FVPSKPMCVEAFSEYPPLGR and SVEM(Ox)HHEQLEQGVPGDNVGFNVK, from elongation factor 1α were also identified as hits with two significantly altered L/H ratios, but only in Experiment 1 (see Fig. S1). Four of the 119 lysine- and methionine-containing peptides assayed in the control experiment were identified as hits (i.e. had significantly altered L/H ratios at two or more denaturant concentrations) (see Fig. S2).
Fig. 4.
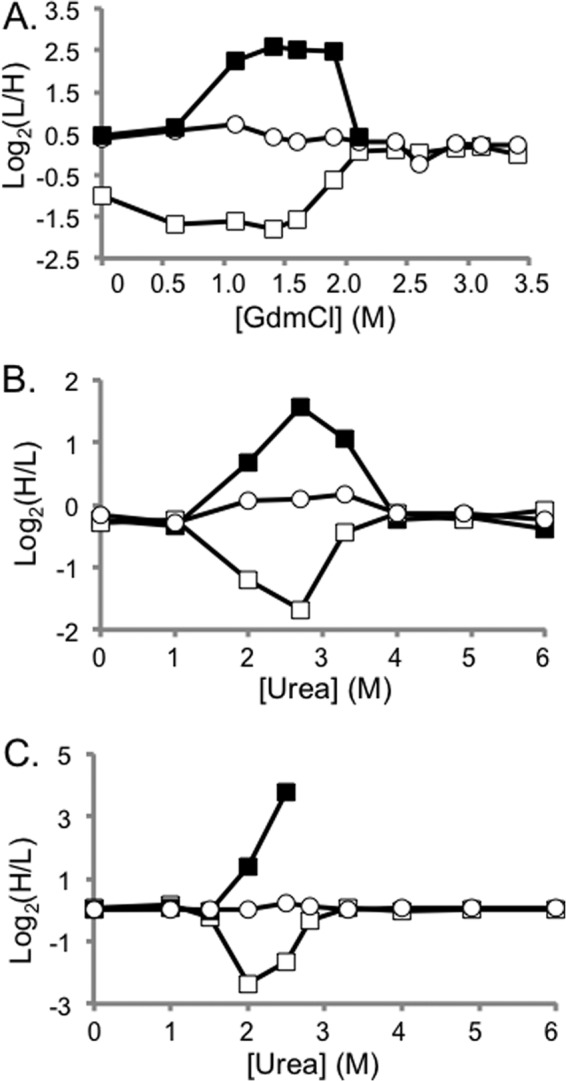
Representative SILAC-SPROX data obtained on protein hits identified in the CsA- and ATP-binding experiments using the solution-based approach. Shown in A, are CsA-binding results from Replicate 2 that were obtained on CypA peptides including the +3 charge state of the nonmethionine containing peptide HVVFGEVVDGYDIVK (open circles) and the oxidized and nonoxidized versions (albeit the +2 and +3 charge states, respectively) of a methionine-containing peptide VIPDFMLQGGDFTAGNGTGGK (open and closed squares, respectively). Shown in B, are ATP-binding results from Solution Experiment 1B that were obtained on PGM-1 peptides including a nonmethionine containing peptide, LSRAIQTANIALEK (open circles) and the oxidized and nonoxidized version of a methionine-containing peptide TVMIAAHGNSLRGLVK (open and closed squares, respectively). Shown in C, are the ATP-binding results from Solution Experiment 2 that were obtained on the same PGM-1 peptides as in B. In all cases the methionine-containing peptide data are consistent with that expected for ligand induced stabilizations. A comparison of the data in B, and C, shows that the more aggressive reaction conditions and increased ligand concentration produced more dramatically altered H/L ratios.
ATP Binding Study
The SILAC-SPROX protocol in Fig. 1 was used to characterize the ATP binding properties of the proteins in a yeast cell lysate. The ligand in this ATP binding study was AMP-PNP (a nonhydrolysable ATP mimic), and it was spiked into the heavy lysate. Important experimental parameters (e.g. the ATP concentration, oxidation reaction time, and mass spectrometry instrument platform) used in the four experiments are described in the Experimental Procedures and are summarized in Table S1. The solution-based readout was used in Solution Experiments 1A/B and 2, and the gel-based readout was used in Gel Experiments 1 and 2. The gel cutting strategies used in Gel Experiments 1 and 2 are shown in Fig. 3. The proteomic results obtained in the different ATP-binding experiments are summarized in Table II.
Table II. Summary of the proteomic results obtained in the SILAC-SPROX experiments to characterize the ATP binding properties of the proteins in a yeast cell lysate.
| Experiment | Total peptides (proteins) assayed for binding | Hit peptides (proteins) | Known ATP binding proteins assayed (hits)a |
|---|---|---|---|
| Solution Exp. 1A | 93 (38) | 5 (3) | 6 (0) |
| Solution Exp. 1B | 526 (209) | 55 (27) | 56 (4) |
| Gel Exp. 1 | 1346 (354) | 23 (12) | 73 (1) |
| Solution Exp. 2 | 353 (216) | 137 (99) | 61 (29) |
| Gel Exp. 2 | 431 (171) | 205 (47) | 27 (8) |
| Total | 2035 (526) | 325 (139) | 109 (37) |
a Known ATP-binding proteins were identified using the yeast genome database and the Gene Ontology term, “ATP Binding” (code 0005524).
In total, 526 proteins in the yeast cell lysate were assayed for binding to the ATP analog used in this study, and 325 peptide hits from 139 different proteins were identified (see Table S2). Shown in Figs. 4B, 4C, and 5 are representative SILAC-SPROX data obtained on one of the hit proteins, phosphoglycerate mutase (PGM-1), which was identified as a hit in both the solution- and gel-based experiments. As expected, the only hit peptides detected for PGM-1 in Solution Experiments 1 and 2 were methionine-containing peptides (Figs. 4B and 4C). Interestingly, PGM-1 was only identified as a hit protein in Gel Experiment 1 using methionine-containing peptide data (data not shown). Presumably, PGM-1 was not identified as a hit using nonmethionine-containing peptide data in Gel Experiment 1 because the excised gel-bands in this experiment, which covered the 20–30 kDa molecular weight range of the gel (see Fig. 3A), included both the intact 27 kDa protein and the 21 kDa CnBr fragment of this protein. However, in Gel Experiment 2, where the gel cutting strategy effectively isolated the intact protein from its respective CnBr fragments (see Fig. 3B), all 10 of the nonmethionine containing peptides identified from PGM-1 had significantly altered H/L values in the 2–3 M urea range (Fig. 5). As expected for a ligand-induced stabilization, the altered Log2(H/L) values from peptides originating from the intact protein bands in the 25–30 kDa range were negative (Fig. 5B); whereas the altered Log2(H/L) values from peptides originating from the fragment band in the 20–24 kDa range were positive (Fig. 5C), with one exception that is described in the figure legend.
Fig. 5.
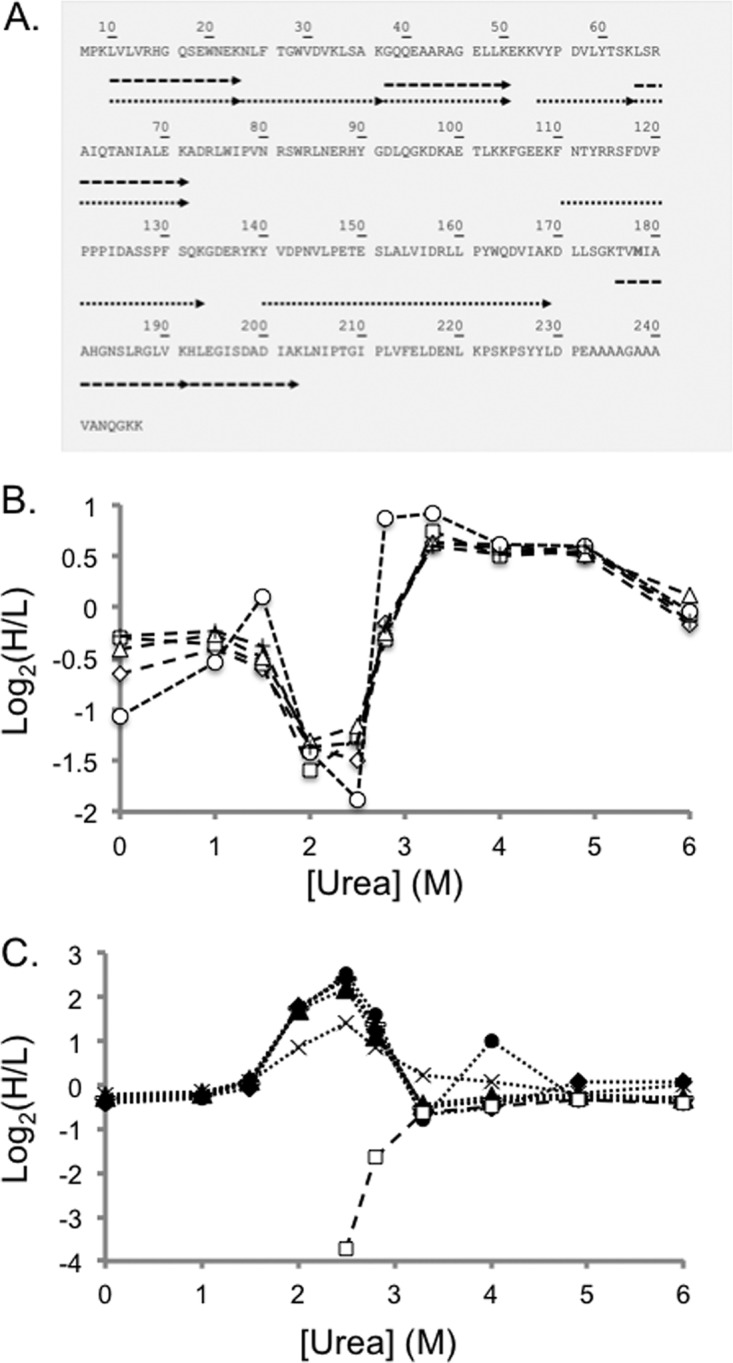
Representative SILAC-SPROX data obtained on a protein hit, PGM-1, identified in the ATP-binding experiments using the gel-based approach. Shown in A, is a schematic representation of the sequence coverage obtained for PGM-1 in Gel Experiment 2. Each arrow represents a peptide identified in the bottom-up proteomics analysis. The dashed and dotted arrows represent peptides detected from the full-length protein and CnBr fragments, respectively. Shown in B, are the SILAC-SPROX data obtained in Gel Experiment 2 on the five nonmethionine containing peptides identified from gel bands in the 25–30 kDa size range that includes intact PGM-1. These peptides included: GQQEAARAGELLK (open diamonds), LSRAIQTANIALEK (open triangles), VYPDVLYTSK (crosses), HLEGISDADIAK (open squares), and LVLVRHGQSEWNEK (open circles). Shown in C, are the SILAC-SPROX data obtained in Gel Experiment 2 on the seven the nonmethionine containing peptides identified from gel bands in the 20–25 kDa size range that included the 21 kDa CnBr fragment of PGM-1. These peptides included all the peptides in B, (same symbols but filled) and two additional peptides, NLFTGWVDVK (bars) and YVDPNVLPETESLALVIDRLLPYWQDVIAK (x's). The HLEGISDADIAK peptide, which is unique to the full-length 27 kDa protein was identified in both A, and B, but in both cases displayed the behavior expected of being derived from the intact protein. This suggests that the gel electrophoresis and cutting strategy may not have completely separated all the intact protein from the CnBr-fragment. Nevertheless, the ability of the nonmethionine containing peptide to report on the ATP binding properties of the protein was not compromised.
False Positive/Negative Rate
Based on the results of the Control Experiment in which four peptide hits were identified from the 119 lysine- and methionine-containing peptides assayed, a false positive rate of 3.4% can be estimated for ligand discovery using SILAC-SPROX and the solution-based readout. Another way to assess the false positive rate in SILAC-SPROX experiments using the solution-based readout is to ask what fraction of hit peptides would be selected from all the nonmethionine-containing peptides assayed in the solution-based experiments. Such an analysis of the data in the control experiment, the two CsA binding experiments and the two solution-based ATP binding experiments yielded false positive rates ranging from 0.4 to 2.0%. Additionally, we note that false positives appear to result from random errors, as the large majority of the false positives detected in these experiments were unique. Consistent with such random error is that about half the false positives had significantly altered Log2(H/L) values that were negative and the other half had significantly altered Log2(H/L) values that were positive.
The false negative rate associated with SILAC-SPROX experiments is more difficult to determine and it is likely to depend on the system under study. There are several caveats to the use of SILAC-SPROX in protein-ligand binding experiments. One caveat is that the ligand binding event must shift the SPROX transition midpoint by a measurable amount (i.e. at least 0.5 m GdmCl or at least 1 m urea based on the denaturant concentration spacing used in these experiments). The magnitude of a SPROX transition midpoint shift for a given protein-ligand interaction depends on the affinity of the interaction and the concentration of free ligand used in the SPROX analysis (see “Quantitation of Ligand Binding” section below). Based on free ligand concentrations used in these experiments the selected hits in our experiments should have been CsA binding proteins with Kd values < 50 μm, and ATP-binding proteins with Kd values <0.25 mm in Experiment 1 and < 2 mm in Experiment 2.
It is also important that potential protein target have buried methionine residue(s) in the domain(s) involved in ligand binding. However, it is not necessary that the methionine residue be located at the binding site of the ligand. In the SPROX experiment the chemical-denaturant dependence of the hydrogen peroxide-mediated oxidation reaction with methionine is evaluated. This means that the unfolding/refolding transitions probed with SPROX are those that result from the global and subglobal unfolding/refolding of proteins and protein domains, respectively. Therefore, any buried methionine in the target protein can report on the global or subglobal unfolding/refolding properties of the protein or protein domain in which it resides. However, it is important that oxidation of a protein's methionine residues does not preclude the ligand binding interaction.
Proteome Coverage
The proteome coverage obtained in the large-scale gel-based experiment using the Q-TOF (i.e. 431 peptides and 171 proteins in Gel Experiment 2) was increased ∼fivefold over that in the solution-based experiment using the same instrument platform (i.e. 93 peptides and 38 proteins in Solution Experiment 1A) (see Table II). The analytical capabilities (e.g. speed and sensitivity) of the mass spectrometry instrument used in the SILAC-SPROX experiment clearly impact the proteome coverage. For example, the total number of peptides (and proteins) assayed using the orbitrap instrument in Solution Experiment 1B, 526 peptides (and 209 proteins), was about fivefold greater than that obtained using the Q-TOF instrument to analyze the samples in Solution Experiment 1A, 93 peptides (and 38 proteins) (see Table II). If a similar fivefold improvements were realized on the orbitrap instrument, our data suggest that a large-scale gel-based experiment using the orbitrap could potentially enable the analysis of ∼1000 proteins using ∼1800 to 2500 peptide probes (i.e. five times the number of peptide and proteins assayed in Gel Experiment 2, Solution Experiment 1B, or in Solution Experiment 2). This is ∼three- fivefold more peptide and protein coverage than that previously reported in SPROX experiments using an isobaric mass tagging strategy (16). It is also noteworthy that the solution and gel-based protocols were to some degree complementary in their peptide and protein coverage. Only about 35% of the proteins were assayed in both the gel- and solution-based experiments (Fig. 6A). Also, only about 17% of the hits were identified in both the gel and solution phase experiments (Fig. 6B).
Fig. 6.
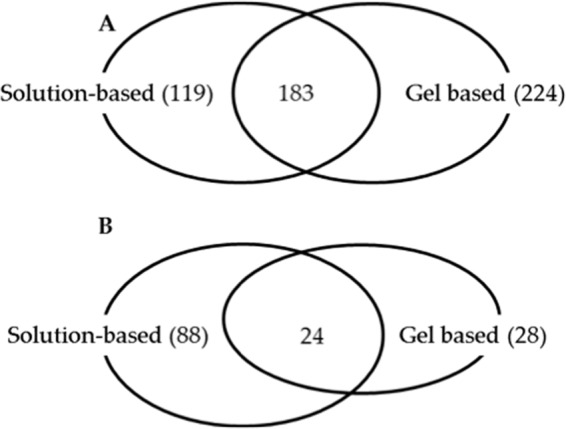
Venn diagram showing overlap of results obtained in the gel- and solution-based experiments. Shown in A, are the overlapping proteins that were assayed in the gel- and solution-based experiments. Shown in B, are the overlapping protein hits that were identified in the gel- and solution-based experiments.
One important feature of the gel-based SILAC-SPROX protocol is that it increases the number of peptide probes per protein. In the two gel-based data sets in this work there were an average of 2.5 and 3.8 unique peptide probes per protein, whereas in the solution-based data sets there were on average between 1.6 and 2.5 peptide probes per protein (see Table II). The increased number of peptide probes in the gel-based experiment is advantageous because it raises the confidence level of protein hits. However, it is important to note that in the SILAC-SPROX experiment, even protein hits identified with one hit peptide are based on the identification and quantitation of that peptide in multiple LC-MS/MS analyses. The requirement for hit peptides to have altered L/H ratios at two or more denaturant concentrations also builds technical replicates into the SILAC-SPROX experiment.
Overall, 59 of the 139 protein hits in Table II, were identified as hits based on data from multiple peptide probes (see Table S2). A total of 75 protein hits, including 62 from solution phase experiments and 15 from gel-based experiments, were identified as hits based on quantitative data from a single peptide probe. However, in all but nine of these 75 cases the protein hits were in fact identified in the mass spectral readout based on MS/MS data obtained on two or more unique peptides. For example, the large majority of protein hits identified in the solution-based experiment were successfully identified based on MS/MS data from nonmethionine-containing peptides. It is also important to note all the proteins included in the SILAC-SPROX assay were identified on the basis of multiple peptide mass spectra that were collected in the LC-MS/MS analyses. This is because the successful identification and quantitation in multiple LC-MS/MS analyses was a requirement for every peptide probe used in the assay.
Quantitation of Ligand Binding
The L/H versus [Denaturant] plots obtained for peptide hits in the solution-based experiments were visually inspected to determine the range of denaturant concentrations at which the L/H ratios deviated from the baseline value established for each peptide. This range was taken to be the ΔCSPROX1/2 value, which was given a positive sign for stabilizations and a negative sign for destabilizations (see Table S2). In the case of each ligand-induced stabilization the ΔCSPROX1/2 value was used to calculate a dissociation constant (Kd value) (see Table S2). This was accomplished by first calculating a binding free energy using equation 1, which has been previously described (11).
In equation 1, ΔΔGf is the binding free energy, m is the δΔGf/δ[Denaturant] (where ΔGf is the protein folding free energy), and ΔCSPROX1/2 is the SPROX transition midpoint shift observed upon ligand binding. The m-value used in this work to calculate the binding affinity of CsA to Cyclophilin A was the experimentally reported value of 3.7 kcal mol−1 M−1. All other m-values were estimated to be 2.6 and 1.3 kcal mol−1 M−1 for GdmCl and urea, respectively. These estimates were based on an average protein domain size of 100 amino acids and an average contribution to the m-value per residue of 0.013 and 0.026 kcal mol−1 M−1 for urea and for GdmCl (respectively). These per residue values were empirically derived by Myers et al. (29).
Ultimately, equation 2 was used to calculate the Kd values in this work.
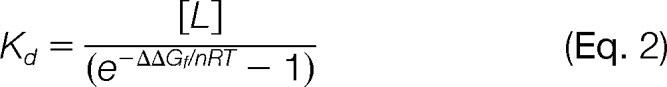 |
In Eq. 2, [L] is the molar concentration of the free ligand, n is the number of independent binding sites, R is the universal gas constant, and T is the temperature in Kelvin (K), which was 298 K in this work. In both the CsA and ATP binding studies the total ligand concentration (see Table S1) was much greater than any individual target protein concentration, therefore the total ligand concentration was used for the free ligand concentration. The effective free ligand concentration in the two CsA binding experiments was 400 μm. The free ligand concentration in ATP binding Experiments 1 and 2 was 2 and 16 mm (respectively), which corresponded to the effective concentration of the ATP-Mg complex in each experiment. In all cases, it was also assumed that n = 1.
The range of ΔΔGf values that can be detected in SILAC-SPROX is generally unbounded on the low (i.e. more negative and tight binding) end. However, the high (i.e. less negative and weak binding) end is bound by the minimum ΔCSPROX1/2 value that can be detected. The number and spacing of the denaturant concentrations used in the SPROX analysis will determine the minimum ΔCSPROX1/2 value that can be detected in SILAC-SPROX experiments. The minimum ΔCSPROX1/2 value that could be detected in these SILAC-SPROX experiments was 0.5 m GdmCl and 1.0 m urea. Thus, considering the free ligand concentrations and estimated m-values used in our analyses (see above) the selected hits in our experiment should have been CsA binding proteins with Kd values < 50 μm, and ATP-binding proteins with Kd values <0.250 mm in Experiment 1 and < 2 mm in Experiment 2.
The above calculations of the binding affinities expected for the protein hits in our SILAC-SPROX experiments assume a single, site-specific interaction of the ligand with the protein domain from which the peptide probe is derived. The ligand concentrations used in the SILAC-SPROX experiment are necessarily in large excess over the protein concentration. This creates the possibility of selecting protein hits that have nonspecific interactions with the ligand (i.e. multiple, low affinity binding sites for ligand). For example, using a 16 mm ligand concentration (as was done in the second ATP-binding experiment) protein hits can not only result from a single, site-specific binding interaction with a Kd value < 2 mm (as described above), they can also result from ATP binding at two or more sites in a given protein folding domain, each with a Kd value of 10 mm. The likelihood of selecting protein hits with such nonspecific ligand binding interactions is decreased when lower ligand concentrations are used in the SILAC-SPROX experiment. For example, using a 400 μm ligand concentration (as was done in the CsA binding experiment) the detection of protein hits with nonspecific ligand interactions (e.g. multiple binding sites with Kd values of 10 mm) would require the existence of 60 such sites in one protein domain to produce a measurable ΔCSPROX1/2 value in the SILAC-SPROX experiment. The presence of such a large number of binding sites in a given protein folding domain is unlikely. Thus, using ligand concentrations in the hundreds of micromolar range in SILAC-SPROX experiments is unlikely to result in the selection of protein hit with nonspecific binding interactions with the ligand. One important aspect of the SILAC-SPROX approach (as well as other energetics-based approaches) is that the ligand concentration can be tuned to select for proteins with different binding affinities to the target ligand.
DISCUSSION
A search of the yeast genome database using the Gene Ontology term, “ATP Binding” (code 0005524) identifies ∼660 ATP binding proteins. A total of 109 of these proteins were effectively assayed in the experiments described here, and 37 of these known ATP binding proteins were identified as hits. Based on the experimental conditions used in our assays we estimate these 37 proteins bind ATP with Kd values < 2 mm (see Quantitation of Ligand Binding). We hypothesize that the other 72 known ATP binding proteins assayed here did not yield a measurable transition midpoint shift in the SPROX experiment because they have a relatively low affinity for ATP (e.g. Kd > 2 mm). It is also possible that these 72 proteins were missed in our assay because they were not correctly annotated, lacked appropriate methionine residues in their sequence, or the oxidation reaction products perturbed the binding interaction. Interestingly, the results of a recent ATP-binding study in Mycobacterium tuberculosis (Mtb) using a chemical proteomics approach also revealed ∼30% of the ATP binding proteins in the Mtb proteome were tight binding, as judged by a qualitative competitive binding assay (30).
The 37 known ATP binding proteins detected as hits in the SILAC-SPROX experiments described here constitute 28% of the identified protein hits in this work. This result is similar to that reported in a recent study of ATP-binding protein in Arabidopsis thaliana using a chemical proteomics approach, in which a large fraction (∼60%) of the identified protein hits were not previously known to bind ATP, ADP, or AMP (31). Many of the previously unknown ATP binding yeast proteins identified here (see Table S2) are known to bind other nucleotides and co-factors (e.g. NAD and GTP), DNA, and RNA based on GO-term analyses (see Fig. 7). The mode of protein-adenine recognition for ATP is similar to that of other adenine containing cofactors including coenzyme A (CoA) and NAD+/NADP. Therefore, it is not surprising that some of the protein hits in this work were known to bind co-factors with the adenosine moiety. It is also not surprising that GTP-binding proteins were identified as hits in the ATP-binding experiments described here. GTP-binding proteins such as Elongation factors (i.e. EF-Tu, EF-1α, EF-2, and EF-G) contain the so-called P-loop that is a known phosphate binding motif (32). Two GTP-binding proteins, EF-Tu and EF-G, were also shown to bind ATP in a previous energetics-based study using the pulse proteolysis method to characterize ATP binding to the proteins in an E. coli cell lysate (18). The yeast homologs of these proteins, EF-1α and EF-2, were also among the hits in our SILAC-SPROX study as were other GTPase Elongation factors such as EF-1β, EF-3A (see Table S2). A GTP binding elongation factor 2-like protein was also among the hits in a chemical proteomics-based ATP binding study in Arabidopsis thaliana (31).
Fig. 7.
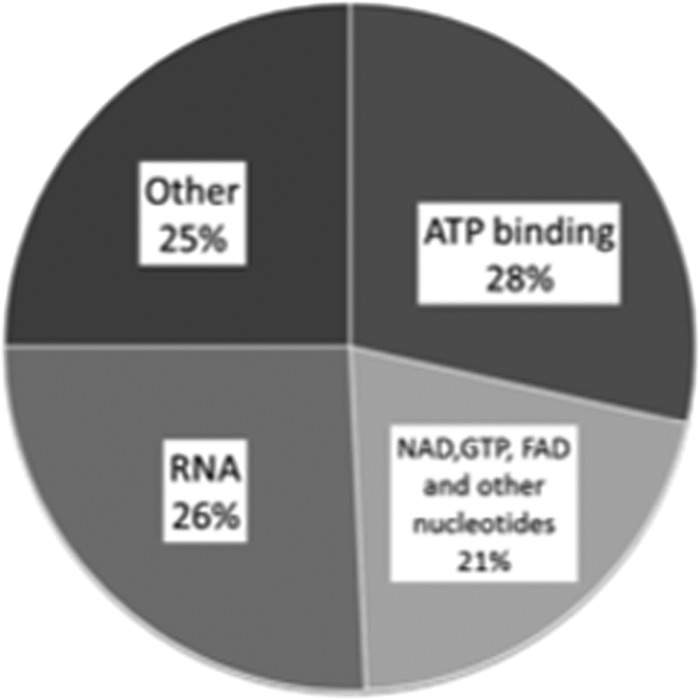
Distribution of known ligands for the hit proteins identified in the ATP-binding study.
Nearly two-thirds of the newly discovered ATP binding yeast proteins in this work have known interactions with other nucleotides and co-factors (e.g. NAD, FAD, and GTP), DNA and RNA (see Table S2). It is possible that the high ligand concentrations used in SILAC-SPROX may have caused some of these proteins to appear as hits as a result of nonspecific binding interactions (see Quantitation of Ligand Binding above). However, it is noteworthy that a significant fraction (∼25%) of the newly discovered ATP binding proteins identified in a recent global analysis of ATP-binding proteins in Arabidopsis thaliana also had previously known interaction with other nucleotides and co-factors (31). In theory, the chemical proteomics approach should be less prone to the detection of nonspecific ATP binding interactions than our energetics-based SILAC-SPROX approach. This is because the acyl-ATP probe concentrations used in the chemical proteomics approach, ∼5–10 μm, are much lower than the free ligand concentrations used in SILAC-SPROX. Thus, the prevalence of other nucleotide (e.g. NAD, FAD, GTP, DNA, and RNA) binding proteins in the group of ATP-binding yeast protein hits identified here is not likely to be entirely because of nonspecific binding. Our results in yeast, together with recent results in E. coli and Arabidopsis thaliana, suggest that there is some promiscuity to nucleotide binding proteins in terms of the specific types of nucleotides they bind (18, 31).
Six of the previously unknown ATP binding yeast proteins in this study are homologous to E. coli proteins that were recently reported to have ATP binding interactions based on results of a pulse proteolysis study (see Table S2) (18). Eight of the previously unknown ATP binding yeast proteins in this study are homologous to Arabidopsis thaliana proteins recently reported to have ATP binding interactions based on a chemical proteomics study (see Table S2) (31). One of the previously unknown ATP binding yeast proteins, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), has protein homologues that were identified as protein hits in both the Arabidopsis thaliana and E. coli ATP binding studies. Moreover, the results of the E. coli study showed that the GAPDH protein was destabilized in the presence of ATP and that the destabilization was caused by ATP stabilizing an unfolding transition state of GAPDH (33). Our results show that yeast GAPDH is also destabilized in the presence of ATP, and suggest that the ATP binding behavior of this protein is conserved in both E. coli and yeast.
Unfortunately, it is difficult to distinguish if hit proteins identified in the gel-based readout are stabilized or destabilized in the SILAC-SPROX experiment because of potential complications associated with the electrophoretic resolution and cutting strategy (see e.g. Fig. 5C). However, ligand-induced stabilizations and destabilizations in SILAC-SPROX can be differentiated in the solution-based readout (see Fig. 2). Interestingly, only seven of the 111 ATP-binding proteins identified in the solution-based experiments were stabilized in the presence of ATP; and four of these seven proteins also had some peptide probes that showed a destabilization (see Table S2). The large majority of the ATP-binding hits found in this study showed only a decrease in thermodynamic stability in the presence of ATP. ATP-induced destabilizations were also observed in a recent study of ATP binding in the E. coli proteome using an energetics-based approach that employed pulse proteolysis, albeit with a much lower frequency (i.e. only three out of the 30 hits detected were destabilized) (18). Although the biophysical reason for the ATP-induced destabilization of yeast GAPDH is likely the same as that previously reported for the ATP-induced destabilization of E. coli GAPDH (18), the biophysical phenomena associated with the other destabilizations observed in this study are currently unclear.
Based on the linkage between protein folding and ligand binding (34), the direct binding of a ligand to the native state of a protein should stabilize the protein domain in which ligand binding occurs. In such cases the ligand-induced stabilization measured in SILAC-SPROX can be used to calculate a Kd value (see Quantitation of Ligand Binding). For example, the ΔCSPROX1/2 value of 1.5 m determined from the nonoxidized methionine-containing peptide data for cyclophilin A in the proof-of-principle experiment (Fig. 4A) can be used to calculate a Kd value of 33 nm that is in reasonable agreement with previously reported values, which have ranged from 30–200 nm (21–23).
Ligand-induced destabilizations can also be observed in the SILAC-SPROX experiment for several different reasons. As noted above, such destabilizations can be observed in cases where the ligand interacts with nonnative state(s) of the protein (see discussion of GAPDH above). Ligand-induced destabilizations can also be observed in cases where ligand binding to one domain of a protein causes a conformational change in another domain of the protein. This phenomenon was observed for one of the previously known ATP-binding proteins, the ATP-dependent molecular chaperone Hsc82, which was identified as a hit protein in this study. In Solution Experiment 2, a methionine-containing peptide from the ATP-binding domain of Hsc82, VLEIRDSGIGMTK, showed a stabilization; whereas three other peptide probes (LLDAPAAIRTGQFGWSANM(ox)ERIM(ox)K, GVVDSEDLPLNLSREMLQQNK, and GVVDSEDLPLNLSREM(ox)LQQNK) from other regions of the protein showed destabilizations (see Table S2). Another phenomenon that can produce ligand-induced destabilizations is that related to indirect effects of ligand binding where the binding of ligand to a protein disrupts protein-protein interactions involving that protein. The disrupted interactions can involve those with itself (e.g. the oligomeric structure could be disrupted) or with other proteins. One strength of the SILAC-SPROX protocol and energetics-based methods in general, is that they can report on such a wide variety of ligand-induced effects. However, this means that the specific biophysical phenomena responsible for producing hits using energetics-based screens must be dissected in additional experiments.
A total of 139 protein hits were identified in the ATP-binding experiments using the SILAC-SPROX protocol described here. With the exception of a recent study that reported the discovery of over 800 ATP-interacting proteins in several different breast cancer cell lines using a specially designed ATP probe in combination with a targeted mass spectrometry-based approach (35), the number of protein hits identified in the current study is comparable to the 100–200 ATP interacting proteins identified in several recent chemical proteomics studies, which also relied on specially designed ATP probes to characterize ATP-interacting proteins (30, 31, 36, 37). The current study is the largest proteome-wide profile of an ATP-interactome using an energetics-based method, to date. A major advantage of energetics-based methods, like the SILAC-SPROX approach described here, is that they do not require the synthesis of specially designed ligands. This makes the methodology easy to adapt for the study of other ligands.
Supplementary Material
Acknowledgments
We thank Dr. Yuko Ogata and the Proteomics Facility at the Fred Hutchinson Cancer Research Center for the collection of orbitrap data. The mass spectrometry data have been deposited to the ProteomeXchange Consortium (http://prorteomecentral.proteomeexchange.org) via the PRIDE partner repository (38) with the data set identifiers PXD000858, DOI 10.6019/PXD000858, and PXD000860. The opinions, findings, and conclusions stated herein are those of the authors and do not necessarily reflect those of VEF.
Footnotes
Author contributions: D.T.T., J.A., and M.C.F. designed research; D.T.T. and J.A. performed research; D.T.T., J.A., and M.C.F. analyzed data; D.T.T., J.A., and M.C.F. wrote the paper.
* This work was supported in part by National Institutes of Health Grants GM084174 and S10RR027746 (to M.C.F.) and in part by a National Science Foundation Grant CHE-1308093 (to M.C.F.). This work was also funded in part by a grant from the Vietnam Education Foundation (VEF) (to D.T.T.).
 This article contains Supplemental Figs. S1 to S4 and Tables S1 to S23.
This article contains Supplemental Figs. S1 to S4 and Tables S1 to S23.
1 The abbreviations used are:
- SPROX
- stability of proteins from rates of oxidation
- AMP-PNP
- adenylyl imidodiphosphate
- BME
- β-mercaptoethanol
- CnBr
- cyanogen bromide
- CsA
- cyclosporine A
- GdmCl
- guanidinium chloride
- GTP
- guanosine triphosphate
- MMTS
- methyl methane thiosulfonate
- NAD
- nicotinamide dinucleotide
- SC
- synthetic complete
- SILAC
- stable isotope labeling with amino acids in cell culture
- NEM
- 4-Ethylmorpholine.
REFERENCES
- 1. Ho Y., Gruhler A., Heilbut A., Bader G. D., Moore L., Adams S. L., Millar A., Taylor P., Bennett K., Boutilier K., Yang L. Y., Wolting C., Donaldson I., Schandorff S., Shewnarane J., Vo M., Taggart J., Goudreault M., Muskat B., Alfarano C., Dewar D., Lin Z., Michalickova K., Willems A. R., Sassi H., Nielsen P. A., Rasmussen K. J., Andersen J. R., Johansen L. E., Hansen L. H., Jespersen H., Podtelejnikov A., Nielsen E., Crawford J., Poulsen V., Sorensen B. D., Matthiesen J., Hendrickson R. C., Gleeson F., Pawson T., Moran M. F., Durocher D., Mann M., Hogue C. W. V., Figeys D., Tyers M. (2002) Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 415, 180–183 [DOI] [PubMed] [Google Scholar]
- 2. Gavin A. C., Bosche M., Krause R., Grandi P., Marzioch M., Bauer A., Schultz J., Rick J. M., Michon A. M., Cruciat C. M., Remor M., Hofert C., Schelder M., Brajenovic M., Ruffner H., Merino A., Klein K., Hudak M., Dickson D., Rudi T., Gnau V., Bauch A., Bastuck S., Huhse B., Leutwein C., Heurtier M. A., Copley R. R., Edelmann A., Querfurth E., Rybin V., Drewes G., Raida M., Bouwmeester T., Bork P., Seraphin B., Kuster B., Neubauer G., Superti-Furga G. (2002) Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 415, 141–147 [DOI] [PubMed] [Google Scholar]
- 3. Glatter T., Wepf A., Aebersold R., Gstaiger M. (2009) An integrated workflow for charting the human interaction proteome: insights into the PP2A system. Mol. Syst. Biol. 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gingras A. C., Gstaiger M., Raught B., Aebersold R. (2007) Analysis of protein complexes using mass spectrometry. Nat. Rev. Mol. Cell Bio. 8, 645–654 [DOI] [PubMed] [Google Scholar]
- 5. Schreiber S. L., Crabtree G. R. (1992) The mechanism of action of cyclosporine-A and FK506. Immunol. Today 13, 136–142 [DOI] [PubMed] [Google Scholar]
- 6. Ong S. E., Schenone M., Margolin A. A., Li X. Y., Do K., Doud M. K., Mani D. R., Kuai L., Wang X., Wood J. L., Tolliday N. J., Koehler A. N., Marcaurelle L. A., Golub T. R., Gould R. J., Schreiber S. L., Carr S. A. (2009) Identifying the proteins to which small-molecule probes and drugs bind in cells. Proc. Natl. Acad, Sci. U.S.A. 106, 4617–4622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McFedries A., Schwaid A., Saghatelian A. (2013) Methods for the elucidation of protein-small molecule interactions. Chem. Biol. 20, 667–673 [DOI] [PubMed] [Google Scholar]
- 8. Chan J. N. Y., Vuckovic D., Sleno L., Olsen J. B., Pogoutse O., Havugimana P., Hewel J. A., Bajaj N., Wang Y., Musteata M. F., Nislow C., Emili A. (2012) Target identification by chromatographic co-elution: monitoring of drug-protein interactions without immobilization or chemical derivatization. Mol. Cell. Proteomics 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lomenick B., Hao R., Jonai N., Chin R. M., Aghajan M., Warburton S., Wang J. N., Wu R. P., Gomez F., Loo J. A., Wohlschlegel J. A., Vondriska T. M., Pelletier J., Herschman H. R., Clardy J., Clarke C. F., Huang J. (2009) Target identification using drug affinity responsive target stability (DARTS). Proc. Natl. Acad. Sci. U.S.A. 106, 21984–21989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. West G. M., Tucker C. L., Xu T., Park S. K., Han X. M., Yates J. R., Fitzgerald M. C. (2010) Quantitative proteomics approach for identifying protein-drug interactions in complex mixtures using protein stability measurements. Proc. Natl. Acad. Sci. U.S.A. 107, 9078–9082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. West G. M., Tang L., Fitzgerald M. C. (2008) Thermodynamic analysis of protein stability and ligand binding using a chemical modification- and mass-spectrometry based strategy. Anal. Chem. 80, 4175–4185 [DOI] [PubMed] [Google Scholar]
- 12. Park C. W., Marqusee S. (2005) Pulse proteolysis: A simple method for quantitative determination of protein stability and ligand binding. Nat. Methods 2, 207–212 [DOI] [PubMed] [Google Scholar]
- 13. Liu P. F., Kihara D., Park C. (2011) Energetics-based discovery of protein-ligand interactions on a proteomic scale. J. Mol. Biol. 408, 147–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xu Y., Falk I. N., Hallen M. A., Fitzgerald M. C. (2011) Mass spectrometry- and lysine amidination-based protocol for thermodynamic analysis of protein folding and ligand binding interactions. Anal. Chem. 83, 3555–3562 [DOI] [PubMed] [Google Scholar]
- 15. Tran D. T., Banerjee S., Alayash A. I., Crumbliss A. L., Fitzgerald M. C. (2012) Slow histidine H/D exchange protocol for thermodynamic analysis of protein folding and stability using mass spectrometry. Anal. Chem. 84, 1653–1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. DeArmond P. D., Xu Y., Strickland E. C., Daniels K. G., Fitzgerald M. C. (2011) Thermodynamic analysis of protein-ligand interactions in complex biological mixtures using a shotgun proteomics approach. J. Proteome Res. 10, 4948–4958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Strickland E. C., Geer M. A., Tran D. T., Adhikari J., West G. M., DeArmond P. D., Xu Y., Fitzgerald M. C. (2013) Thermodynamic analysis of protein-ligand binding interactions in complex biological mixtures using the stability of proteins from rates of oxidation. Nat. Protoc. 8, 148–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chang Y., Schlebach J. P., VerHeul R. A., Park C. (2012) Simplified proteomics approach to discover protein-ligand interactions. Protein Sci. 21, 1280–1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Baud F., Karlin S. (1999) Measures of residue density in protein structures. Proc. Natl. Acad. Sci. U.S.A. 96, 12494–12499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schreiber S. L. (1991) Chemistry and biology of the immunophilins and their immunosuppressive ligands. Science 251, 283–287 [DOI] [PubMed] [Google Scholar]
- 21. Liu J., Albers M. W., Chen C. M., Schreiber S. L., Walsh C. T. (1990) Cloning, expression, and purification of human cyclophilin in Escherichia coli and assessment of the catalytic role of cysteines by site-directed mutagenesis. Proc. Natl. Acad. Sci. U.S.A. 87, 2304–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Harding M. W., Handschumacher R. E. (1988) Cyclophilin, a primary target for cyclosporine - structural and functional implications. Transplantation 46, S29–S35 [DOI] [PubMed] [Google Scholar]
- 23. Handschumacher R. E., Harding M. W., Rice J., Drugge R. J. (1984) Cyclophilin a specific cytosolic binding-protein for cyclosporin-A. Science 226, 544–547 [DOI] [PubMed] [Google Scholar]
- 24. Bradford M. M. (1976) Rapid and sensitive method for quantitation of microgram quantities of protein utilizing principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 25. Nozaki Y. (1972) The preparation of guanidine hydrochloride. Method Enzymol. 26, 43–50 [DOI] [PubMed] [Google Scholar]
- 26. Pace C. N. (1986) Determination and analysis of urea and guanidine hydrochloride denaturation curves. Method Enzymol. 131, 266–280 [DOI] [PubMed] [Google Scholar]
- 27. Shevchenko A., Tomas H., Havlis J., Olsen J. V., Mann M. (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 [DOI] [PubMed] [Google Scholar]
- 28. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 29. Myers J. K., Pace C. N., Scholtz J. M. (1995) Denaturant m values and heat capacity changes: relation to changes in accessible surface areas of protein unfolding. Protein Sci. 4, 2138–2148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wolfe L. M., Veeraraghavan U., Idicula-Thomas S., Schuerer S., Wennerberg K., Reynolds R., Besra G. S., Dobos K. M. (2013) A chemical proteomics approach to profiling the ATP-binding proteome of Mycobacterium tuberculosis. Mol. Cell. Proteomics 12, 1644–1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Villamor J. G., Kaschani F., Colby T., Oeljeklaus J., Zhao D., Kaiser M., Patricelli M. P., van der Hoorn R. A. L. (2013) Profiling protein kinases and other ATP binding proteins in Arabidopsis using acyl-ATP probes. Mol. Cell. Proteomics 12, 2481–2496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Saraste M., Sibbald P. R., Wittinghofer A. (1990) The P-loop - A common motif in ATP binding and GTP-binding proteins. Trends Biochem. Sci. 15, 430–434 [DOI] [PubMed] [Google Scholar]
- 33. Liu P.-F., Park C. (2012) Selective stabilization of a partially unfolded protein by a metabolite. J. Mol. Biol. 422, 403–413 [DOI] [PubMed] [Google Scholar]
- 34. Schellman J. A. (1975) Macromolecular binding. Biopolymers 14, 999–1018 [Google Scholar]
- 35. McAllister F. E., Niepel M., Haas W., Huttlin E., Sorger P. K., Gygi S. P. (2013) Mass spectrometry based method to increase throughput for kinome analyses using ATP probes. Anal. Chem. 85, 4666–4674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Patricelli M. P., Szardenings A. K., Liyanage M., Nomanbhoy T. K., Wu M., Weissig H., Aban A., Chun D., Tanner S., Kozarich J. W. (2007) Functional interrogation of the kinome using nucleotide acyl phosphates. Biochemistry-Us 46, 350–358 [DOI] [PubMed] [Google Scholar]
- 37. Patricelli M. P., Nomanbhoy T. K., Wu J., Brown H., Zhou D., Zhang J., Jagannathan S., Aban A., Okerberg E., Herring C., Nordin B., Weissig H., Yang Q., Lee J.-D., Gray N. S., Kozarich J. W. (2011) In situ kinase profiling reveals functionally relevant properties of native kinases. Chem. Biol. 18, 699–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vizcaino J. A., Cote R. G., Csordas A., Dianes J. A., Fabregat A., Foster J.M., Griss J., Alpi E., Birim M., Contell J., O'Kelly G., Schoenegger A., Ovelleiro D., Perez-Riverol Y., Reisinger F., Rios D., Wang R., Hermjakob H. (2013) The Proteomics Identifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 41, D1063–D1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.