

Abstract
PKCε, a DAG-dependent, Ca2+- independent kinase attenuates extent of fibrosis following tissue injury, suppresses apoptosis and promotes cell quiescence. In crescentic glomerulonephritis (CGN), glomerular epithelial cells (GEC) contribute to fibro-cellular crescent formation while they also transdifferentiate to a mesenchymal phenotype. The aim of this study was to assess PKCε expression in CGN. Using an antibody against PKC-ε phosphorylated at Ser729, we assessed its localization in rat model of immune-mediated rapidly progressive CGN. In glomeruli of control animals, pPKCε was undetectable. In animals with CGN, pPKCε was expressed exclusively in glomerular epithelial cells (GEC) and in GEC comprising fibrocellular crescents that had acquired a myofibroblasttype phenotype. In non-immune GEC injury induced by puromycin aminonucleoside and resulting in proteinuria of similar magnitude as in CGN, pPKCε expression was absent. There was constitutive pPKCε expression in distal convoluted tubules, collecting ducts and thick segments of Henley’s loops in both control and experimental animals. We propose that pPKCε expression occurring in GEC and in fibrocellular crescentic lesions in CGN may facilitate PKCε dependent pathologic processes.
Keywords: glomerulonephritis, crescents, PKCepsilon, podocytes, phosphorylation
Introduction
Crescentic glomerulonephritis is a rapidly progressive form of glomerular inflammation resulting in end-stage renal disease.1,2 Histologically, it is characterised by glomerular cell proliferation and crescent formation, infiltration of glomerular capillaries by inflammatory cells (primarily activated macrophages), and fibrosis (scarring). These processes have been mechanistically linked with specific Th1 and Th2 cytokines and with macrophage-derived mediators of injury,34 a number of which can activate or regulate protein kinase C (PKC).56 PKC includes at least 12 isoforms that can be divided in three subgroups: classical PKCs (PKCα, PKCβ1, PKC(β2, and PKCγ), novel PKCs (PKCδ, PKCε, PKCη, and PKCθ), and atypical PKCs (PKCζ and PKCλ/ι).7 PKC is the cellular receptor for the lipid second messenger diacylglycerol (DAG) and, therefore, a key enzyme in signaling mechanisms triggered by activation of receptors coupled to phospholipase C.7 The novel PKCs (δ, ε, ηand θ) are dependent on DAG but not Ca2+ whereas the atypical PKCs (ζ and λ/ι) are independent of both Ca2+ and DAG.8 PKC isoforms display a distinct cell- and tissue-type expression pattern of localization while PKC activation is associated with membrane translocation of the enzyme.9,10
The role of individual PKC isoforms in mediating cellular responses to injury such as proliferation, apoptosis, cytoskeleton rearrangement, cell motility and scarring is currently being elucidated. In experimental models of tissue injury/disease, recent evidence points to PKCε activation as a protective mechanism against fibrosis as shown in the myocardium,11 the lung12 and the diabetic kidney.13 Activity and intracellular localization of PKCε is controlled by phosphorylation at highly conserved sites present in the catalytic kinase domain. These sites are Thr566 in the activation loop, Thr710 in the turn motif, and Ser729 in the C-terminal hydrophobic motif. Phosphoinositide-dependent kinase-1 (PDK-1) is the upstream kinase that directly phosphorylates the activation loop residue Thr566. This provides a link with the phosphoinositide-3 kinase (PI3-kinase) pathway as the PI3-kinase products, PtdIns(3,4)P2 and PtdIns(3,4,5)P3, bind and recruit PDK-1 to membranes thus leading to phosphorylation of downstream substrates such as PKCε. Phosphorylation of Ser729 in the C-terminal hydrophobic motif depends on the internal catalytic activity of the kinase indicating that it occurs by autophosphorylation rather than an upstream kinase.14 Recent evidence indicates that Ser729 phosphorylation controls intracellular PKCε location and is required for the kinase to achieve mature conformation and to be primed for activation by co-factors.15,16 Further, PKCε phosphorylated at Ser729 accumulates in cells at quiescence.17
Studies attempting to characterize localization of PKCε phosphorylated at Ser729, henceforth referred to as pPKCε, in renal disease models in which cell proliferation and scarring are prevalent features are scarce. In this work
we employed immunohistochemistry methods to assess expression and localization of pPKCε in a rat model of immune-mediated renal inflammation resembling human rapidly progressive glomerulonephritis and characterised by proteinuria and formation of fibrocellular crescents in glomeruli. pPKCε localisation was also examined in a non-inflammatory model of proteinuria resulting from direct, toxin-mediated, glomerular epithelial cell injury.
Materials and Methods
Models of rapidly progressive (crescentic) glomerulonephritis and puromycin aminonucleoside-induced glomerular epithelial cell injury
All animal experiments were performed according to the Institutional Animal Care and Use Committee (IACUC). Male Sprague-Dawley rats weighing 180-200 g were immunized intraperitoneally with 1 mg rabbit IgG emulsified in complete Freund’s adjuvant and given as a total volume of 0.5 mL. Five days after this immunization, animals were injected in the tail vein with a subnephritogenic dose of rabbit immune serum raised against rat particulate GBM as previously described.18 This dose of anti-rat GBM serum (0.15 mL/100 g body weight) was insufficient by itself to cause significant proteinuria when given to rats not preimmunized with rabbit IgG (sub-nephritogenic). The intravenous injection of the anti-rat GBM serum was repeated 24 h after the first injection. The rabbit anti-rat GBM immune serum was heat inactivated and filtered through 1.2 µ filters before each intravenous injection. Control rats were pre-immunized with rabbit IgG in complete Freund’s adjuvant and subsequently given two intravenous injections of non-immune rabbit serum. Studies were performed 15 days following the second injection of anti-rat GBM serum or non-immune rabbit serum. This time point was chosen because it is characterized by proliferation of glomerular epithelial cells (GEC) resulting in crescent formation while scarring is also prominent. On day 14, animals were placed in metabolic cages for urine collection to assess urinary protein and creatinine excretion. Upon completion of this collection, animals were killed and nephrectomized to perform the immunohistochemistry studies described below.
Since GEC injury is a prominent feature in this CGN model, we compared changes in pPKCε expression in GEC with those occurring in a non-inflammatory form of GEC injury resulting from administration of the GEC-specific toxin, puromycin aminonucleoside (PAN). This was administered in doses causing proteinuria the magnitude of which was comparable to that induced by administration of the anti-GBM antibody. A single intraperitoneal injection of PAN (15 mg/100 g body weight) was given to male Sprague-Dawley rats weighing 180-200 g and studies (urine collection and immunohistochemistry) were performed 14 days later. PAN causes proteinuria owing to a direct toxic effect on GEC, which undergo ultrastructural changes resembling those described in human minimal change disease.19
Histology
Rat kidney tissues, were fixed in formalin buffered saline for 24 h, followed by dehydration with graded ethanols (80-90%) for 1 h each. Tissues were then immersed twice in absolute ethanol for 30 min, cleared in xylene for 1 h, and embedded in paraffin at 58°C overnight. Serial 0.5-µm-thick sections were applied to 3’-Aminopropyltriethoxysilane (APES) coated slides and left at 55°C to remove excess paraffin. To perform Periodic Acid Schiff staining,20 sections were further deparaffinized with xylene for 5 min followed by rehydration through a series of graded ethanols (100%, 90%, 80%). Sections were then incubated with 1% (w/v) periodic acid for 10 min, which progressively oxidizes existing tissue aldehydes to carboxylic acid resulting in a strong Schiff’s reaction. Following washing, sections were incubated with Schiffs reagent (Sigma-Aldrich) for 15 min. Harris’ Haematoxylin for 2 min was used as counterstain. Sections were subsequently dehydrated, cleared in xylene for 5 min, and mounted.
Glomerular fibrosis was identified using the Masson-Goldner trichrome stain (Merck A.E., Athens, Greece). Briefly, tissue sections, deparaffinized and rehydrated as described above, were stained with Weigert’s iron hematoxylin (identifies cell nuclei) for 5 min, washed with tap water and decolorized with 1% (v/v) acetic acid. Sections were subsequently placed in a Azophloxine solution for 10 min, rinsed with distilled water and incubated for 1 min with Tungstophosphoric acid Orange G solution. Sections were finally air dried, dehydrated, and mounted. This method stains areas of scarring (collagen) as light green.
Immunohistochemistry
PKCε phosphorylated at Ser729 (pPKCe) was detected in paraffin-embedded tissues. Sections were deparaffinized by two consecutive treatments (5 min each) with xylene. Rehydration was performed with graded ethanols (90%, 80%, and 70%) for 4 min each. Endogenous peroxidase was blocked with 3% H2O2 in methanol for 15 min at room temperature. Antigen retrieval was subsequently performed by boiling the sections with 1x Target Retrieval Solution (Dako Ltd., Athens, Greece) in a steamer for 45 min. The following primary antibodies were used: rabbit anti-human PKC-ε phosphorylated at Ser729 (Santa Cruz Biotechnology, Dallas, TX, USA) diluted at 1:150 (v/v), mouse anti-human Wilms Tumor (WT-1, a marker of glomerular epithelial cells, Dako) diluted at 1:200 (v/v), rabbit polyclonal antibody against a α-smooth muscle Actin synthetic polypeptide, corresponding to the N-terminal of this protein (Abcam, Cambridge, MA, USA), diluted 1:500 (v/v), mouse monoclonal antibody against the rat macrophage marker ED1 (Serotec, Raleigh, NC, USA), and monoclonal mouse anti-human E-cadherin (clone NCH-38, Dako). E-cadherin (Epithelial-cadherin) is a well established adhesion molecule necessary for the maintenance of normal epithelial tissue architecture. Primary antibodies were diluted in REAL™ Antibody Diluent (Dako) and incubated with tissue sections for 30 min at room temperature.
The immunostaining reaction was developed using the REAL™ EnVision™ Detection System Peroxidase/DAB+ Rabbit/Mouse (Dako) to which sections were exposed for 30 min at room temperature. This developing reagent consists of a peroxidase-conjugated polymer backbone, which also carries secondary antibody molecules directed against rabbit and mouse immunoglobulins. Washes were performed using Tris-Buffered Saline-Tween 20 (TBS-T) buffer for 10 min. To detect immunoreactivity on sections, the DAKO REAL DAB+ Chromogen reagent was applied for 10 min. Sections were then counterstained with Hematoxylin and dehydrated using graded ethyl alcohol (80%, 95%, 100%), 3 min each. To completely remove the dehydrating ethanols, sections were immersed in xylene, then mounted and examined. Sections from which the primary antibodies were omitted served as negative controls.
Lectins
To identify the origin of tubules stained positive for pPKCe, we employed lectins that are known to bind to either proximal or non-proximal tubules (see below). The lectins employed were: biotinylated Tetragonolobus purpureas (TP)-derived lectin and biotinylated Arachnis hypogaea (AH)-derived lectin (Vector Lab., Burlingame, CA, USA). Serial (0.5 µ) sections were cut and stained for either pPKCe or biotinylated TP lectin or biotinylated AH lectin. For example, in three sequential 0.5 µ sections, the first was exposed to antibody against PKCe phosphorylated at Ser729, the second to biotinylated TP lectin (diluted at 5 µg/mL) and the third to biotinylated AH lectin (diluted at 1 µg/mL). All lectin incubations were for 60 min at room temperature following which biotinylated Streptavidin, conjugated with Horse Radish Peroxidase (HRP), was applied for 15 min. DAKO REAL DAB+ Chromogen reagent was subsequently applied to achieve colour development and sections were counterstained with hematoxylin, mounted and photographed (Leica DFC 490, Leica Microsystems GmbH, Wetzlar, Germany).
Urine protein and creatinine measurements
Before animal sacrifice, timed urine collections (18 h) were performed by placing animals in metabolic cages with access to food and water ad libitum. Total urine protein and creatinine were measured by established colorimetric methods using commercially available kits (Exocell Inc., Philadelphia, PA, USA) and results were expressed as the ratio of urine protein (Up) to urine creatinine (Uc) concentration. Changes in Up/Uc were expressed as mean ± SEM and comparisons between groups were made using unpaired t-test statistics with a P-value of less than 0.05 indicating statistically significant differences.
Results
In rats with CGN or PAN-induced GEC injury there was heavy proteinuria. Up/Uc values were (n=4): 2.3±0.4 in control, 12.8±2.1 (n=6) in rats with CGN, and 15.3±2.0 in rats with PAN-induced GEC injury (n=4).
In glomeruli of animals with CGN, there was glomerular infiltration by macrophages, periglomerular infiltration by mononuclear cells, and formation of fibrocellular crescents and scarring [Figure 1, panel B (nephritic glomerulus) vs panel A (control glomerulus)]. As expected, in glomeruli of animals that received PAN no apparent changes were detected by light microscopy (not shown). In glomerular cells of control kidneys, pPKC-ε phosphorylated at Ser729 (pPKC-ε) was barely detectable or undetectable (Figure 2A). In animals with CGN, pPKC-ε immunolocalized in glomerular epithelial cells (GEC) (Figure 2B). These cells were identified as GEC on the basis of their positivity for the GEC specific marker WT1 (Figure 2C). In glomeruli with prominent crescent formation, cells within crescents were also positive for pPKC-ε, as shown in Figure 2B. In glomeruli of rats that received PAN and developed proteinuria of comparable severity with that noted in CGN rats, there was no apparent pPKC-ε expression in GEC (Figure 2D). In crescentic glomeruli, there was also loss of E-cadherin compared to non-nephritic glomeruli (Figure 3, panel B vs A) as well as evidence for epithelial-to-mesenchymal cell transformation with cells acquiring a mesenchymal phenotype as identified by expression of α-smooth muscle actin (Figure 3C).
Figure 1.
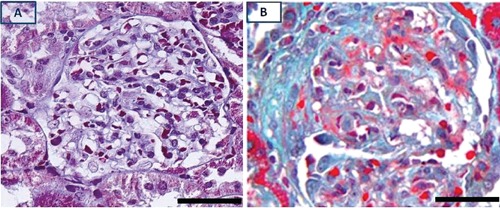
Trichrome stains of control (A) and nephritic glomerulus (B) with fibrocellular crescent. Scarring (collagen) stains as light green. Scale bar: 50 µm.
Figure 2.
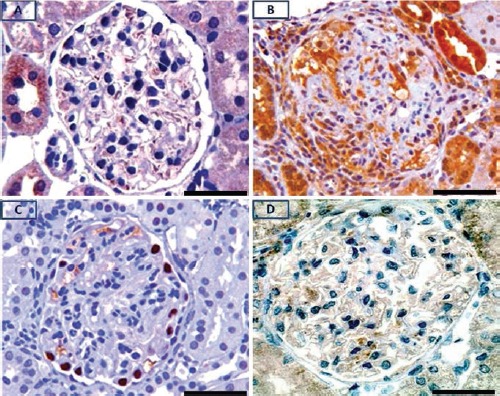
Immunolocalization of pPKCε in glomerulus of control (A), and nephritic animal (B); pPKCε is barely detectable or absent in intrinsic cells of the control glomerulus. pPKCε is intensely expressed in glomerular epithelial cells and within cellular crescent of the nephritic glomerulus (B); immunolocalization of the visceral epithelial cell marker, WT-1 in the nephritic glomerulus (C); lack of pPKCε staining in GEC of a PAN-treated rat (D). Scale bar: 50 μm.
Figure 3.
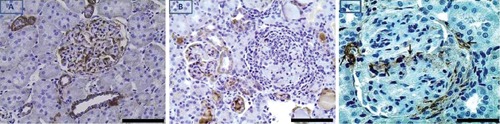
E-cadherin immunohistochemical expression in glomerulus and non-proximal tubules of a control kidney (A) and in glomerulus of a kidney with CGN (B); there is loss of E-cadherin expression in the nephritic glomerulus. (C), crescentic glomerulus with evidence for epithelial-to-mesenchymal (E-to-M) transformation of GEC identified by strong expression of α-smooth muscle actin in cells populating the crescent. Black arrows point to visceral and parietal GEC with E-to-M transformation, and to the vascular pole of the glomerulus. Scale bar: A and B, 100 µm; C, 50 µm.
Since the CGN model employed in this study is macrophage-dependent, we examined whether glomerular localization of infiltrating macrophages (identified as ED1+ cells) was similar to that of pPKC-ε+ cells. As shown in Figure 4, the localization pattern of these two cell types was quite disparate; whereas ED1+ cells distributed throughout the nephritic glomerulus, pPKC-ε+ expression was mainly restricted in GEC. pPKCε also immunolocalized in non-proximal tubules surrounding glomeruli (Figure 2 A,B). A number of these tubules were surrounded by inflammatory cell infiltrates and scarring (Figure 5). The identity of pPKCε positive tubules was further assessed using the biotinylated lectins Arachnis hypogaea (AH) and Tetragonolobus purpureas (TP). AH is specific for distal tubules and collecting ducts,21 while TP stains cells of the Henle’s loop.22 Certain pPKCε positive tubules (Figure 6A) were also positive for the AH lectin (Figure 6B) indicating that they were either distal convoluted or collecting. Another population of pPKCε positive tubules was also positive for the TP lectin as shown in Figure 7. In this figure, two 0.5 µm serial sections were stained for pPKCε and the TP lectin. Shown are tubules that were strongly positive for pPKCε (tubules demarcated by the black arrow heads in panel A), but negative for the TP lectin (same tubules demarcated by the black arrow heads in panel B), indicating they were non-proximal in origin. Also shown, are tubules that were weakly positive for pPKCε (panel A, black arrows) and strongly positive for the TP lectin (panel B, black arrows) indicating their origin as thick segments of Henle’s loops.
Figure 4.
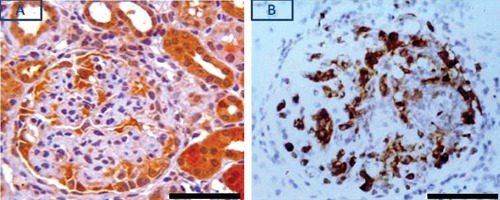
Differential expression of pPKCε (A) and macrophage marker ED1 (B) in nephritic glomerulus: pPKCε expression is mainly restricted in GEC while that of ED1 is observed throughout the glomerulus. Scale bar: 50 µm.
Figure 5.
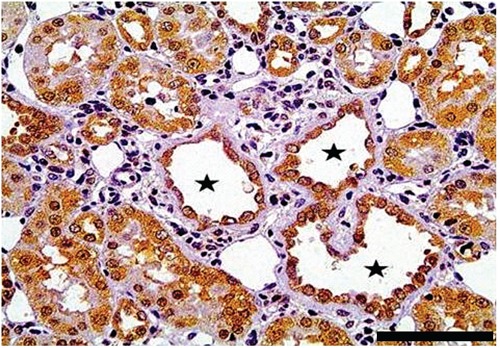
Non-proximal tubules (black stars) that are positive for pPKCε and are surrounded by inflammatory cell infiltrates and scarring. Scale bar: 50 µm
Figure 6.
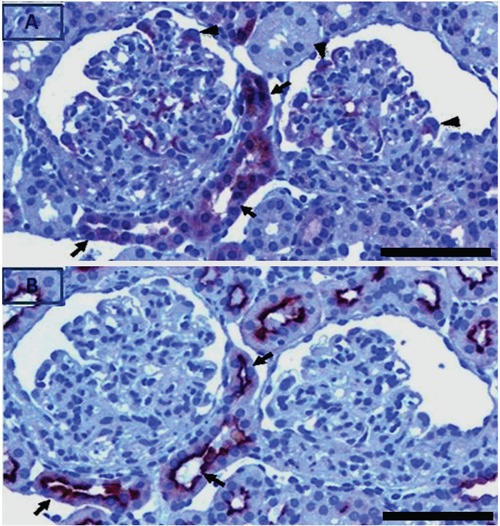
Serial sections of nephritic kidney stained for pPKCε and AH lectin. pPKCε positive tubules (A, black arrows) are also positive for the AH lectin (B, black arrows), which is specific for distal tubules and collecting ducts. In glomeruli, pPKCε localized in GEC (A, black arrow heads). Scale bar: 50 µm.
Figure 7.
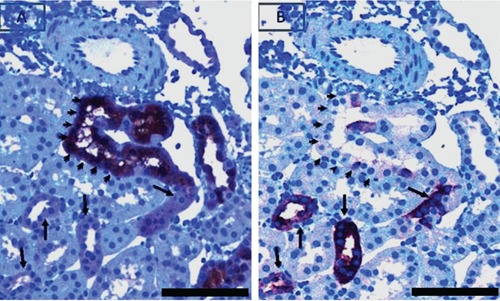
Serial sections of tubules strongly positive for pPKCε (demarcated by the arrow heads in A) but negative for the TP lectin (demarcated by the arrow heads in B). Also shown, are tubules weakly positive for pPKCε (A, black arrows). The same tubules are strongly positive for the TP lectin (B, black arrows). Scale bar: 50 µm.
In a population of non-proximal tubules that were strongly positive for pPKCε, there was heterogeneity in staining of their epithelial cells with positively stained alternating with negatively stained cells (Figure 8 A,B). These tubules were AH lectin positive but TP lectin negative indicating that they were distal or collecting in origin. Further examination of histologic characteristics of these tubules revealed morphology compatible with that of collecting tubules; specifically, they were lined with cuboidal to columnar cells separated by clearly obvious margins and the nuclei of which were closer to the base than apex.
Figure 8.
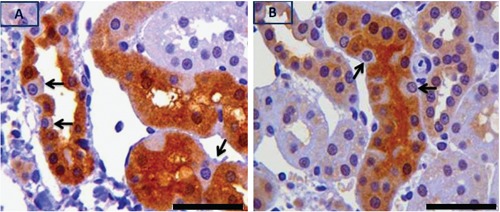
Strongly pPKCε positive tubules in which there is heterogeneity of pPKCε staining amongst epithelial cells (positively alternating with negatively stained cells pointed by black arrows in A and B). These tubules were AH lectin positive and TP lectin negative. Scale bar: 50 µm.
Discussion
In the glomerular inflammation model employed in this study, responses of intrinsic glomerular cells, including proliferation and apoptosis, to pro-inflammatory mediators released by infiltrating macrophages play a crucial role in determining severity of renal dysfunction and the likelihood of progression of inflammation to irreversible stages. In this regard, emphasis has been placed on GEC lesions believed to develop due to effects of pro-inflammatory mediators. GEC are the most vulnerable component of the glomerular filtration barrier owing to their terminally differentiated nature.23 Well characterized GEC lesions include crescent formation,24 scaring and transdifferentiation to a mesenchymal (myofibroblast-type) phenotype.25
Although studies on expression of classical PKCs (α, βI, βII) and novel PKCs (δ, ε) isoenzymes in the normal kidney have been performed,1,26 observations on localization of fully active PKC isoenzymes in lesions such as those described above are scarce. The present study on localization of PKCε phosphorylated at Ser729 in these lesions was prompted by evidence that phosphorylation of this residue is required for the enzyme to assume its mature conformation and to become primed for activation. This may facilitate PKCε dependent processes observed within these lesions. Such processes include: apoptosis,27 which PKCε suppresses,28 cell growth,29 which PKCε promotes,30 and myofibroblast formation associated with scarring,25,31 which PKCε also promotes.32 Phosphorylation of PKCs at the activation loop, turn motif and hydrophobic site in the carboxyl terminal domain primes them for activation by cofactors. The earliest PKCε translation product is an unphosphorylated (immature form) cytoskeleton-associated protein. The initial step for its maturation is a series of ordered phosphorylation steps that prepare the enzyme for catalysis. The first phosphorylation event occurs at the activation loop and at position Thr566 by phosphoinositide-dependent kinase 1 (PDK-1) and functions as a switch by unmasking the entrance of the substrate-binding cavity thus exposing the necessary residues for catalysis.14 This step is followed by a second rapid autophosphorylation event at the turn motif and at position Thr703. Phosphorylation in this position locks the enzyme in a thermally stable catalytic component and in a phosphatase-resistant conformation. The final and third phosphorylation event occurs at the C-terminal hydrophobic motif, which for PKCε is on Ser729. This event can be the result of autophosphorylation or the enzyme can be phosphorylated by its own upstream kinase, particularly PDK-2.14,33 Once the C-terminal serine is phosphorylated, the enzyme assumes its mature conformation. At this point, dephosphorylation of the Ser729 abolishes kinase activity, suggesting that this residue is critical for PKCε activation.34,36 It can, therefore, be proposed that identification of PKCε phosphorylated at Ser729 within tissue lesions would indicate that the kinase has assumed its mature conformation and is primed for activation. Further, the phosphorylation state of Ser729 was shown to control intracellular localization of PKCε in response to cell stimulation.16
Using an antibody against PKCε phosphorylated at Ser729 (pPKCε), the present study demonstrates a robust expression in glomerular epithelial cells (GEC) of animals with crescentic glomerulonephritis and was particularly prominent in cells within crescents. These had characteristics of an epithelial-to-mesenchymal cell transition process. Epithelial to mesenchymal transition (EMT) is typically defined by the acquisition of spindle cell morphology in combination with loss of E-cadherin and upregulation of mesenchymal markers.37-39 Consistent with these observations, cells in nephritic glomeruli had lost E-cadherin expression (Figure 7B) and acquired a mesenchymal phenotype recognized by expression of α-smooth muscle actin (Figure 6). Interestingly, glomerular localization of activated infiltrating macrophages, a prominent feature of the GN model employed, was quite different from that of pPKCε expressing cells (Figure 4). This indicates that infiltrating macrophages is an unlikely cell type in which PKCε phosphorylation at Ser729 occurred. The lack of a robust pPKCε expression in GEC of animals given the GEC-specific toxin PAN and developing comparable degree of proteinuria indicates that effectors of PKCε phosphorylation at Ser729 are at play in immune- but not in non-immune-mediated GEC injury. There are a number of factors that could promote generation of mature (fully phosphorylated) PKCε in GEC and within fibrocellular crescents in immune-mediated glomerular injury. These include several second messengers including DAG and phosphatidylinositol 3,4,5-triphosphate (PIP3) as well as specific fatty acids. In nephritic glomeruli, these messengers are generated by the increased production of cytokines that bind to tyrosine kinase receptors, which transmit signals by recruiting a variety of SH2-domain-containing proteins to multiple phosphorylated tyrosine residues. Key among such cytokines is the platelet-derived growth factor (PDGF) which, in the model of glomerulonephritis employed, immunolocalizes along with its receptor within crescents.40 The PDGF receptor transmits signals by recruiting phospholipase Cγ (PLCγ), phosphatidylinositol 3’-kinase [P13k], Ras GTPaseactivating protein (Ras/GAP), and the Src class kinases.41 In this regard, it was shown that PDGF translocates PKCε from the cytosol to the cell membrane by activating PI3K and PLCγ.42 Reactive oxygen and nitrogen species, shown to be overproduced in the CGN model employed, could also serve as effectors that can activate fully phosphorylated PKC-ε. In this regard, NOX1/NADPH oxidase-derived reactive oxygen species were shown to accelerate translocation of PKCε,43 while hydrogen peroxide (H2O2) and nitric oxide (NO) also activate this kinase.44 Further, NO facilitates PKCε interaction with the specific anchoring protein, RACK2 (receptor for activated C kinase 2), and promotes PKCε translocation and activation.20,45,46
Attempts to elucidate the role of PKCε in fibrosis occurring following tissue injury have yielded conflicting results. In PKCε deficient mice, tubulointerstitial fibrosis (fibronectin and collagen IV) and expansion of mesangial matrix were observed while urine albumin excretion was increased. Interestingly, fibrosis was not observed in other organs, such as liver and lung.47 TGF-β1, phospho-Smad2, and phospho-p38 mitogen-activated protein kinase expression was increased in PKCε deficient mice, suggesting a regulatory role of PKCε on TGF-β1 and its signaling pathway in the kidney.13. These effects of PKCε deficiency were more pronounced in the diabetic kidney and point to a protective role of this kinase. In contrast, pharmacologic inhibition of PKCε activity in a mouse model of hypertension-induced heart failure decreased cardiac parenchymal fibrosis and increased survival.48 Further, a 4-week inhibition of PKCε suppressed chronic inflammation in a heterotopic cardiac transplantation model.49 In addition to its localization in podocytes and crescents of nephritic glomeruli, PKCε phosphorylated at Ser729 also immunolocalized in non-proximal tulules. Using lectin staining these were identified as collecting tubules (positive for the AH lectin) and segments of the Henle’s loop (negative for AH but positive for the TP lectin). Interestingly, a similar preferential localization in non-proximal tubules was previously described for ERK,50 which is a constituent of the PKCε signalling complex.51 The comparable extent of detection of pPKCε in tubules of both control and nephritic animals indicates a constitutive expression which is likely to serve transport function(s). For example, PKCε in rat nephron segments was previously immunolocalized in cortical collecting ducts where it regulates K secretion.52 In our study, pPKCε localization in collecting ducts was heterogeneous with pPKCε positive cells alternating with negative ones (Figure 8). It is likely that the pPKCε positive cells in collecting ducts are of the intercalated type since it was previously shown that such cells express both PKCε and the anion exchanger.52
In summary, the present study identifies the glomerular epithelial cell and fibrocellular crescents as a key site where pPKCε is expressed in experimental crescentic glomerulonephritis. Factors released as a result of the immune/inflammatory process apparently play a role since pPKCε was undetectable in nonimmune mediated GEC injury. Whether the preferential expression of pPKCε in GEC and crescentic lesions facilitates PKCε-dependent processes remains to be determined.
Acknowledgments
This work was submitted as an Abstract at the American Society of Nephrology, November 2010 Annual meeting, Denver, CO, USA.
References
- 1.Pfaff IL, Wagner HJ, Vallon V. Immunolocalization of protein kinase C isoenzymes alpha, beta1 and betaII in rat kidney. J Am Soc Nephrol 1999;10:1861-73 [DOI] [PubMed] [Google Scholar]
- 2.Tipping PG. Crescentic nephritis--is it in your genes? Nephrol Dial Transplant 2008;23:3065-6 [DOI] [PubMed] [Google Scholar]
- 3.Cattell V. Macrophages in acute glomerular inflammation. Kidney Int 1994;45:945-52 [DOI] [PubMed] [Google Scholar]
- 4.Tipping PG, Kitching AR. Glomerulonephritis, Th1 and Th2: what's new? Clin Exp Immunol 2005:142:207-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ganz MB, Abu-Nader R, Saxena R, Grond J. Protein kinase C beta II isoform is up-regulated in human proliferative glomerulonephritis. Exp Nephrol 1997;5:225-32 [PubMed] [Google Scholar]
- 6.Mandal A, Wang Y, Ernsberger P, Kester M. Interleukin-1-induced ether-linked diglycerides inhibit calcium-insensitive protein kinase C isotypes. Implications for growth senescence. J Biol Chem 1997;272:20306-11 [DOI] [PubMed] [Google Scholar]
- 7.Mellor H, Parker PJ. The extended protein kinase C superfamily. Biochem J 1998;332: 281-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Newton AC. Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm. Biochem J 2003;370:361-71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Csukai M, Mochly-Rosen D.Pharmacologic modulation of protein kinase C isozymes: the role of RACKs and subcellular localisation. Pharmacol Res 1999;39 :253-9 [DOI] [PubMed] [Google Scholar]
- 10.Dempsey EC, Newton AC, Mochly-Rosen D, Fields AP, Reyland ME, Insel PA, et al. Protein kinase C isozymes and the regulation of diverse cell responses. Am J Physiol Lung Cell Mol Physiol 2000;279:L429-38 [DOI] [PubMed] [Google Scholar]
- 11.Klein G, Schaefer A, Hilfiker-Kleiner D, Oppermann D, Shukla P, Quint A, et al. Increased collagen deposition and diastolic dysfunction but preserved myocardial hypertrophy after pressure overload in mice lacking PKCepsilon. Circ Res 2005;96: 748-55 [DOI] [PubMed] [Google Scholar]
- 12.Tourkina E, Gooz P, Oates JC, Ludwicka-Bradley A, Silver RM, Hoffman S. Curcumin-induced apoptosis in scleroderma lung fibroblasts: role of protein kinase cepsilon. Am J Respir Cell Mol Biol 2004;31:28-35 [DOI] [PubMed] [Google Scholar]
- 13.Meier M, Menne J, Park JK, Holtz M, Gueler F, Kirsch T, et al. Deletion of protein kinase C-epsilon signaling pathway induces glomerulosclerosis and tubulointerstitial fibrosis in vivo. J Am Soc Nephrol 2007;18:1190-8 [DOI] [PubMed] [Google Scholar]
- 14.Cenni V, Doppler H, Sonnenburg ED, Maraldi N, Newton AC, Toker A. Regulation of novel protein kinase C epsilon by phosphorylation. Biochem J 2002;363:537-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.England K, Rumsby MG. Changes in protein kinase C epsilon phosphorylation status and intracellular localization as 3T3 and 3T6 fibroblasts grow to confluency and quiescence: a role for phosphorylation at ser-729? Biochem J 2000;352:19-26 [PMC free article] [PubMed] [Google Scholar]
- 16.Xu TR, He G, Dobson K, England K, Rumsby M. Phosphorylation at Ser729 specifies a Golgi localisation for protein kinase C epsilon (PKCepsilon) in 3T3 fibroblasts. Cell Signal 2007;19:1986-95 [DOI] [PubMed] [Google Scholar]
- 17.England K, Watson J, Beale G, Warner M, Cross J, Rumsby M. Signalling pathways regulating the dephosphorylation of Ser729 in the hydrophobic domain of protein kinase Cepsilon upon cell passage. J Biol Chem 2001;276:10437-42 [DOI] [PubMed] [Google Scholar]
- 18.Duann P, Lianos EA. GEC-targeted HO-1 expression reduces proteinuria in glomerular immune injury. Am J Physiol Renal Physiol.2009;297:F629-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caulfield JP, Farquhar MG. Loss of anionic sites from the glomerular basement membrane in aminonucleoside nephrosis. Lab Invest 1978;39:505-12 [PubMed] [Google Scholar]
- 20.Balafanova Z, Bolli R, Zhang J, Zheng Y, Pass JM, Bhatnagar A, et al. Nitric oxide (NO) induces nitration of protein kinase Cepsilon (PKCepsilon), facilitating PKCepsilon translocation via enhanced PKCepsilon -RACK2 interactions: a novel mechanism of no-triggered activation of PKCepsilon. J Biol Chem 2002;277:15021-7 [DOI] [PubMed] [Google Scholar]
- 21.Holthofer H. Lectin binding sites in kidney. A comparative study of 14 animal species. J Histochem Cytochem. 1983;31:531-7 [DOI] [PubMed] [Google Scholar]
- 22.Grupp C, John H, Hemprich U, Singer A, Munzel U, Muller GA. Identification of nucleated cells in urine using lectin staining. Am J Kidney Dis 2001;37:84-93 [DOI] [PubMed] [Google Scholar]
- 23.Pavenstadt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev 2003;83:253-307 [DOI] [PubMed] [Google Scholar]
- 24.Le Hir M, Keller C, Eschmann V, Hahnel B, Hosser H, Kriz W. Podocyte bridges between the tuft and Bowman's capsule: an early event in experimental crescentic glomerulonephritis. J Am Soc Nephrol 2001;12:2060-71 [DOI] [PubMed] [Google Scholar]
- 25.Goumenos D, Tsomi K, Iatrou C, Oldroud S, Sungur A, Papaioannides D, et al. Myofibroblasts and the progression of crescentic glomerulonephritis. Nephrol Dial Transplant 1998;13:1652-61 [DOI] [PubMed] [Google Scholar]
- 26.Redling S, Pfaff IL, Leitges M, Vallon V.Immunolocalization of protein kinase C isoenzymes alpha, beta I, beta II, delta, and epsilon in mouse kidney. Am J Physiol Renal Physiol 2004;287:F289-98 [DOI] [PubMed] [Google Scholar]
- 27.Liu D, Nazneen A, Taguchi T, Razzaque MS. Low-dose local kidney irradiation inhibits progression of experimental crescentic nephritis by promoting apoptosis. Am J Nephrol. 2008;28:555-68 [DOI] [PubMed] [Google Scholar]
- 28.Gobbi G, Masselli E, Micheloni C, Nouvenne A, Russo D, Santi P, et al. Hypoxia-induced down-modulation of PKCepsilon promotes trail-mediated apoptosis of tumor cells. Int J Oncol 2010;37: 719-29 [DOI] [PubMed] [Google Scholar]
- 29.Masaki T, Stambe C, Hill PA, Dowling J, Atkins RC, Nikolic-Paterson DJ. Activation of the extracellular-signal regulated protein kinase pathway in human glomerulopathies. J Am Soc Nephrol 2004;15:1835-43 [DOI] [PubMed] [Google Scholar]
- 30.Deuse T, Koyanagi T, Erben RG, Hua X, Velden J, Ikeno F, et al. Sustained inhibition of epsilon protein kinase C inhibits vascular restenosis after balloon injury and stenting. Circulation 2010;122: S170-8 [DOI] [PubMed] [Google Scholar]
- 31.Khan SB, Cook HT, Bhangal G, Smith J, Tam FW, Pusey CD. Antibody blockade of TNF-alpha reduces inflammation and scarring in experimental crescentic glomerulonephritis. Kidney Int 2005;67:1812-20 [DOI] [PubMed] [Google Scholar]
- 32.Leask A, Shi-Wen X, Khan K, Chen Y, Holmes A, Eastwood M, et al. Loss of protein kinase Cepsilon results in impaired cutaneous wound closure and myofibroblast function. J Cell Sci 2008;121:3459-67 [DOI] [PubMed] [Google Scholar]
- 33.Parekh DB, Ziegler W, Parker PJ. Multiple pathways control protein kinase C phosphorylation. Embo J 2000;19:496-503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dutil EM, Toker A, Newton AC. Regulation of conventional protein kinase C isozymes by phosphoinositide-dependent kinase 1 (PDK-1). Curr Biol 1998;8:1366-75 [DOI] [PubMed] [Google Scholar]
- 35.Takahashi M, Mukai H, Oishi K, Isagawa T, Ono Y. Association of immature hypophosphorylated protein kinase cepsilon with an anchoring protein CG-NAP. J Biol Chem 2000;275:34592-6 [DOI] [PubMed] [Google Scholar]
- 36.Waldron RT, Rozengurt E. Protein kinase C phosphorylates protein kinase D activation loop Ser744 and Ser748 and releases autoinhibition by the pleckstrin homology domain. J Biol Chem 2003;278:154-63 [DOI] [PubMed] [Google Scholar]
- 37.Thorner PS, Ho M, Eremina V, Sado Y, Quaggin S. Podocytes contribute to the formation of glomerular crescents. J Am Soc Nephrol 2008;19:495-502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ng YY, Fan JM, Mu W, Nikolic-Paterson DJ, Yang W-C, Huang T-P, et al. Glomerular epithelial-myofibroblast transdifferentiation in the evolution of glomerular crescent formation. Nephrol Dial Transplant 1999;14:2860-72 [DOI] [PubMed] [Google Scholar]
- 39.Fujigaki Y, Sun DF, Fujimoto T, Suzuki T, Goto T, Yonemura K, et al. Mechanisms and kinetics of Bowman's epithelialmyofibroblast transdifferentiation in the formation of glomerular crescents. Nephron 2002;92:203-12 [DOI] [PubMed] [Google Scholar]
- 40.Fujigaki Y, Sun DF, Fujimoto T, Yonemura K, Morioka T, Yaoita E, et al. Cytokines and cell cycle regulation in the fibrous progression of crescent formation in antiglomerular basement membrane nephritis of WKY rats. Virchows Arch 2001;439:35-45 [DOI] [PubMed] [Google Scholar]
- 41.Van Der Geer P, Hunter T, Lindberg RA. Receptor protein-tyrosine kinases and their signal transduction pathways. Annu Rev Cell Biol 1994;10:251-337 [DOI] [PubMed] [Google Scholar]
- 42.Moriya S, Kazlauskas A, Akimoto K, Hirai S, Mizuno K, Takenawa T, et al. Platelet-derived growth factor activates protein kinase C epsilon through redundant and independent signaling pathways involving phospholipase C gamma or phosphatidylinositol 3-kinase. Proc Natl Acad Sci USA,1996;93:151-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ibi M, Matsuno K, Shiba D, Katsuyama M, Iwata K, Kakehi T, et al. Reactive oxygen species derived from NOX1/NADPH oxidase enhance inflammatory pain. J Neurosci 2008;28:9486-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Costa AD, Garlid KD. Intramitochondrial signaling: interactions among mitoKATP, PK Cepsilon ROS, and MPT. Am J Physiol Heart Circ Physiol 2008;295:H874-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koya D, Jirousek MR, Lin YW, Ishii H, Kuboki K, King GL. Characterization of protein kinase C beta isoform activation on the gene expression of transforming growth factor-beta, extracellular matrix components, and prostanoids in the glomeruli of diabetic rats. J Clin Invest 1997;100:115-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xuan YT, Guo Y, Zhu Y, Wang OL, Rokosh G, Bolli R. Endothelial nitric oxide synthase plays an obligatory role in the late phase of ischemic preconditioning by activating the protein kinase C epsilon p44/42 mitogen-activated protein kinase pSer-signal transducers and activators of transcription1/3 pathway. Circulation 2007; 116 :535-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tourkina E, Gooz P, Oates JC, Ludwicka-Bradley A, Silver RM, Hoffman S. Curcumin-induced apoptosis in scleroderma lung fibroblasts: role of protein kinase cepsilon. Am J Respir Cell Mol Biol 2004; 31 :28-35 [DOI] [PubMed] [Google Scholar]
- 48.Inagaki K, Koyanagi T, Berry NC, Sun L, Mochly-Rosen D. Pharmacological inhibition of epsilon-protein kinase C attenuates cardiac fibrosis and dysfunction in hypertension-induced heart failure. Hypertension 2008;51:1565-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koyanagi T, Noguchi K, Ootani A, Inagaki K, Robbins RC, Mochly-Rosen D. Pharmacological inhibition of epsilon PKC suppresses chronic inflammation in murine cardiac transplantation model. J Mol Cell Cardiol 2007;43:517-22 [DOI] [PubMed] [Google Scholar]
- 50.Masaki T, Foti R, Hill PA, Ikezumi Y, Atkins RC, Nikolic-Paterson DJ. Activation of the ERK pathway precedes tubular proliferation in the obstructed rat kidney. Kidney Int 2003;63:1256-64 [DOI] [PubMed] [Google Scholar]
- 51.Ping P, Zhang J, Pierce WM, Jr, Bolli R. Functional proteomic analysis of protein kinase C epsilon signaling complexes in the normal heart and during cardioprotection. Circ Res 2001;88:59-62 [DOI] [PubMed] [Google Scholar]
- 52.Sterling H, Lin DH, Chen YJ, Wei Y, Wang ZJ, Lai J, et al. PKC expression is regulated by dietary K intake and mediates internalization of SK channels in the CCD. Am J Physiol Renal Physiol 2004;286:F1072-8 [DOI] [PMC free article] [PubMed] [Google Scholar]