

Abstract
We report here on the building-up of a database of information related to 386 cases of Incontinentia Pigmenti collected in a thirteen-year activity (2000–2013) at our centre of expertise. The database has been constructed on the basis of a continuous collection of patients (27.6/year), the majority diagnosed as sporadic cases (75.6%). This activity has generated a rich source of information for future research studies by integrating molecular/clinical data with scientific knowledge. We describe the content, architecture and future utility of this collection of data on IP to offer comprehensive anonymous information to the international scientific community.
Keywords: Incontinentia pigmenti, Genomic disorder, Neuroectodermal disorder, Molecular diagnosis, Registry, Database
Introduction
Incontinentia pigmenti (IP; OMIM#308300) is a rare multisystemic genomic disorder with an estimated prevalence at birth of 0.7/100,000 [1]. IP is X-linked and usually lethal in males, and affecting the skin, but also other neuroectodermal tissues, in females. The skin lesions are the first clinical manifestations that appear, starting in the neonatal period with a vesiculobullous eruption (Stage I) and following a three stage evolution varying in duration from months to years, namely a verrucous stage (Stage II), a hyperpigmented stage (Stage III), and finally a hypopigmented stage (Stage IV) usually continuing throughout life [2,3]. Such skin defects, that follows Blaschko lines, are always present in IP and are therefore considered the main diagnostic criteria for IP according to Landy and Donnai (1993) [2]. The severity of the disease is related to the presence of neurological and/or ocular impairment [4]. Overall, the prevalence of functional Central Nervous System (CNS) manifestations is approximately 30% [5,6] ranging from a single-seizure episode to severe motor and intellectual disability [7]. Ophthalmologic abnormalities are present in approximately 20%–37% of IP patients [5,6,8]. IP is due to a mutation of the X-linked IKBKG/NEMO gene (Inhibitor of Kappa polypeptide gene enhancer in B-cells, Kinase Gamma/Nuclear Factor κB, Essential Modulator, GenBank NM_003639.3, OMIM#300248). Most cases have a recurrent deletion (IKBKGdel or NEMOdel4-10), removing exons 4–10 of the IKBKG/NEMO gene. Non recurrent genomic rearrangements in the IP locus and point mutations in the IKBKG/NEMO coding region have also been reported [9-11]. IKBKG/NEMO encodes for NEMO/IKKγ a regulatory subunit of the Inhibitor of the kappaB (IκB) Kinase (IKK) complex required for the canonical NF-κB pathway activation involved in many fundamental physiological and pathological functions [12,13]. Most IP female patients present with a skewed X-inactivation. The X-chromosome linked IKBKG/NEMO mutation causes an unbalanced X-inactivation in female IP patients [14], as in other X-linked diseases [15,16], because the absence of the NEMO/IKKγ protein makes the IP cells more sensitive to apoptosis [9]. In males, the extensive apoptosis is responsible for their early fetal lethality [17]. Occasionally, male patients with IP have been reported. They have shown the characteristic skin lesions observed in females and presented a postzygotic mosaicism for the IKBKG/NEMO exons4-10 gene deletion [18]. IP has also been diagnosed in males with a 47,XXY karyotype (Klinefelter syndrome) [19]. The large heterogeneity of defects, the severe clinical presentations, and the wide spectrum of IKBKG/NEMO alterations [7,11,14,20] makes the selection of homogeneous groups of patients difficult, precluding any therapeutic approaches. Indeed, despite the considerable progress that has been made in detailing the basic pathology of the IP disorder, the gap between research and clinical care has remained wide. Moreover, the paucity of patients collected at each single diagnostic centre makes an overall epidemiological report difficult. The integration of scattered resources may be crucial for the success of future scientific accomplishments.
Methods
Here, we report the setting up of a central data repository relating to a cohort of IP patients, the data having been collected in a 13-year-long experience (2000–2013) at our Italian centre of expertise for the molecular diagnosis of IP [21]. The IP patients included in our study have been selected on the basis of the Landy and Donnai (1993) [2] diagnostic criteria, and they also meet the most recently updated IP criteria [22]. We have constructed the first platform for the integration of molecular and clinical data on IP patients. Our sample comprises 386 patients (261 from Italian, 105 from European and 20 from non-European clinical centers), with an annual average of 27.6 new cases of IP diagnosed per year (Figure 1).
Figure 1.
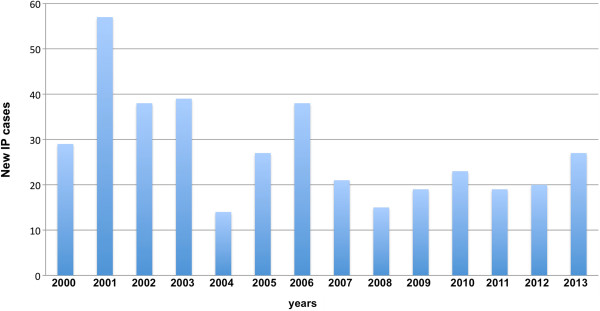
Annual distribution of the IP samples that have been received by the IGB centre for molecular diagnosis.
All the clinical information has been obtained for each patient through their completion of a clinical IP questionnaire developed by the Incontinentia Pigmenti International Foundation (IPIF, [http://www.ipif.org/ip_consortium.html], further extended by the France Incontinentia Pigmenti association (FIP, [http://incontinentia-pigmenti.fr/]) and by the Italian ASSociation of Incontinentia Pigmenti (IPASSI, [http://www.incontinentiapigmenti.it/]). A clinical IP questionnaire is available upon request from these organizations. We have integrated the clinical data with molecular diagnosis results for the IKBKG/NEMO alteration, by way of a well-standardized protocol [10,14,23]. The technical development of the register has involved significant preparatory work consisting in the building up of an in-house electronic database which is comprehensive and permissive, and which has a flexible structure able to register in an anonymous form the pool data from the patients. We have assigned one record to each IP sample, registered with a pseudonymous code. Each record has three domains: the pedigree, the clinical and the genetic domain. The Web domain will be available at link [http://www.igb.cnr.it/ipgb]. The data are not accessible to everyone but only to authorized users through the use of a protected password. A data-mining interface has been developed to ensure maximum flexibility so that users can perform any search they want using the “search” button placed in the homepage after the “log in”. It is possible to perform multiple searches at once. To make the database permissive and flexible the first page contains only the three domains set.
Results
The pedigree and clinical information are available for 308 IP cases, while the genetic data are available for 193 samples, respectively (Table 1). The pedigree domain contains more detailed entries accommodating the family data, for example the presence of an IP mother, sister, or grandmother, indicating the inheritance of the disease. We have registered 233 sporadic cases (75.6%) and 72 familial cases (23.4%) in our IP cohort.
Table 1.
IP data registry
| Number of cases | Percentage of cases | |
|---|---|---|
|
Database information
|
386 records |
|
| IP female samples |
349 |
90.4 |
| IP male samples |
37 |
9.6 |
|
Pedigree domain
|
308 available |
|
| Sporadic cases |
233 |
75.6 |
| Familial cases |
72 |
23.4 |
|
Clinical domain*
|
308 available |
|
| Skin defects |
308 |
100 |
| CNS defects |
97 |
31.5 |
| Ophthalmologic defects |
94 |
30.5 |
| Teeth defects |
134 |
43.5 |
| Hair defects |
82 |
26.6 |
| Fingernail defects |
45 |
14.6 |
| Developmental evolution |
30 |
9.7 |
|
Genetic domain
|
193 available |
|
|
NEMOdel4-10 |
145 |
75.1 |
|
IKBKG/NEMO point mutation |
32 |
22.1 |
| IP locus rearrangement |
7 |
3.6 |
| No known alteration found | 9 | 4.7 |
*IP patients can have more than one defect affecting different organ systems.
The clinical domain consists of seven clinical items, one for each aspect of the phenotype presentation: “Skin defects”, “CNS defects”, “Ophthalmologic defects”, “Teeth defects”, “Hair defects”, “Fingernail defects” and “Developmental evolution”. A drop-down menu has been assigned to each item that, in most cases, contains details about all the specific alterations affecting the tissue, and, in addition, an open space for the annotation of novel clinical features. For example, the item “Skin defects” contains a drop-down menu indicating the stage of the IP skin abnormality, the age of onset, the type of alteration, and the region of the body in which the alteration is present. We report that the most frequent first symptoms leading to diagnosis, typically skin alterations (Stage I), appear before the first year in 99% of cases. The second stage and the third stage are reported within the first year in 96.6% and in 82.8% of cases, respectively. After this date the fourth stage is generally present (Table 2). The specific frequency of each IP specific neuroectodermal defect observed in our cohort is shown in Table 1. CNS abnormalities were present in 31.5% (Table 1). In 17 cases, these were diagnosed by magnetic resonance imaging (Table 3).
Table 2.
IP skin clinical data
| |
Skin alteration age of onset |
|||
|---|---|---|---|---|
| Skin defects | IP cases | <1° month | 1° month-1° year | >1° year |
| Stage I |
183 |
160(87.4%) |
21(11.5%) |
2(1.1%) |
| Stage II |
90 |
38(42.2%) |
49(54.4%) |
3(3.3%) |
| Stage III |
87 |
18(27.6%) |
58(55.2%) |
11(17.1%) |
| Stage IV | 81 | 0 | 0 | 81(100%) |
Table 3.
IP clinical data
| Type of defect | Number of cases** | Percentage of cases |
|---|---|---|
|
CNS defects
|
97
|
|
| Seizures |
39 |
40.2 |
| Mental retardation |
29 |
29.9 |
| Spastic paresis |
16 |
16.5 |
| Cerebral atrophy |
13 |
13.4 |
| Microcephaly |
11 |
11.3 |
| Hydrocephaly |
5 |
5.1 |
| Ischemic strokes* |
5 |
5.1 |
| White matter alterations* |
4 |
4.1 |
| Arachnoid cysts* |
3 |
3.1 |
| Cortico-subcortical atrophy* |
3 |
3.1 |
| Brain morphological alterations* |
2 |
2.1 |
|
Teeth defects
|
134
|
|
| Delayed primary dentition |
46 |
34.3 |
| Cone/peg shaped teeth |
30 |
22.3 |
| Delayed permanent dentition |
30 |
22.3 |
| Teeth dystrophy |
23 |
17.2 |
| Impactions |
23 |
17.2 |
|
Ophthalmologic defects
|
94
|
|
| Vision defects |
16 |
17 |
| Retinopathy |
15 |
15.9 |
| Retinal detachment |
8 |
8.5 |
| Microphthalmia |
6 |
6.4 |
| Retinal neuropathy |
6 |
6.4 |
| Retinal vascular visorders |
5 |
5.3 |
|
Hair defects
|
82
|
|
| Alopecia |
8 |
9.7 |
| Hypertrichosis |
3 |
3.6 |
|
Fingernail defects
|
45
|
|
| Nail dystrophy |
29 |
64 |
|
Developmental evolution
|
30
|
|
| Recurrent infections |
36 |
11.7 |
| Syndactyly of fingers or toes | 4 | 1.3 |
*Data obtained by Magnetic Resonance Imaging analysis in 17 IP cases.
**IP patients can have more than one defect affecting different organ systems.
The genetic domain of the database contains four items: “NEMOdel4-10”, “IKBKG/NEMO point mutation”, “IP locus rearrangement”, and “No known alteration found” (Table 1). The mutation names comply with the accepted guidelines proposed by the Human Genome Variation Society [http://www.hgvs.org/mutnomen] [24]. The genetic domain reveals that 75.1% of patients have the NEMOdel4-10 deletion, 22.1% have an IKBKG/NEMO point mutation, 3.6% have an extended deletion in the IKBKG/NEMO locus, and 4.7% have no known alteration in the IP locus (Table 1).
Finally, in each domain (pedigree, clinical, and genetic) an additional item, named “Supplementary Information”, records in the database any supplementary data from the patient and his/her family when these are available (examples: presence of miscarriages; presence of nonpathogenic alterations in IP locus[25] such as deletions or point mutations in the NEMO pseudogene).
Discussion
The building up of this database represents the first detailed integrated clinical/molecular diagnostic platform on IP patients, the largest collection of an IP cohort in Italy and to the best of our knowledge the first presented to the scientific community worldwide. Thus, this phenotype and genotype database related to IP acts as a unique attempt to improve patient care and healthcare planning since it collects together information that would otherwise be scattered. Finally, we strongly believe that the use of the database is a powerful tool to facilitate the selection of biological samples and/or the enrolment of patients for the organization of appropriate clinical trials. Physicians wishing to include patients in the IP database may contact the Authors: Francesca Fusco and/or Matilde Valeria Ursini by email (incontinentia.pigmenti@igb.cnr.it).
Abbreviations
IP: Incontinentia Pigmenti; OMIM: Online Mendelian Inheritance in Man; CNS: Central Nervous System; IKBKG: Inhibitor of Kappa polypeptide gene enhancer in B-cells, Kinase gamma; NEMO: Nuclear factor κB Essential MOdulator; IKKγ: Inhibitor of Kappa Kinase gamma; IκB: Inhibitor of NF-κB; IKK: IκB Kinase; NF-κB: Nuclear Factor-κB; IPIF: Incontinentia Pigmenti International Foundation; FIP: France Incontinentia Pigmenti association; IPASSI: Italian Incontinentia Pigmenti ASSociation.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
FF: drafted the main part and created the final version of the manuscript, and designed the clinical and scientific content of the database, MP: implemented the content of the database, MIC: contributed to the content of the database, AP: contributed to the content of the database, EE: contributed to the content of the database, PM: contributed to the content of the database, MBL: participated in the design of the database- architecture and the data security concept of the registry, MVU: contributed to the content design of the database, and reviewed and revised the manuscript. All authors read and approved the final manuscript.
Contributor Information
Francesca Fusco, Email: francesca.fusco@igb.cnr.it.
Mariateresa Paciolla, Email: paciolla@igb.cnr.it.
Matilde Immacolata Conte, Email: mconte@igb.cnr.it.
Alessandra Pescatore, Email: pescatore@igb.cnr.it.
Elio Esposito, Email: espositoe@igb.cnr.it.
Peppino Mirabelli, Email: pemirabelli@gmail.com.
Maria Brigida Lioi, Email: maria.lioi@unibas.it.
Matilde Valeria Ursini, Email: matildevaleria.ursini@igb.cnr.it.
Acknowledgments
We are grateful to the patients, their families and physicians, the association France Incontinentia Pigmenti (FIP, [http://incontinentia-pigmenti.fr/]) the Italian Incontinentia Pigmenti ASSociation (IPASSI, [http://www.incontinentiapigmenti.it/]), DHITECH, Progetto Formazione PON n°01-02342 for the fellowship to M.I.C and the Basilicata Innovazione [http://www.basilicatainnovazione.it] for supporting M.P.
References
- Orphanet Report Serie. Prevalence of rare diseases: bibliographic data Rare Diseases collection. 2013. Listed in alphabetical order of disease or group of diseases, http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_alphabetical_list.pdf.
- Landy SJ, Donnai D. Incontinentia pigmenti (Bloch-Sulzberger syndrome) J Med Genet. 1993;30:53–59. doi: 10.1136/jmg.30.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuerle A, Ursini MV. In: GeneReviews® [Internet] Pagon RA, Adam MP, Ardinger HH, Bird TD, Dolan CR, Fong CT, Smith RJH, Stephens K, Seattle WA, editor. Seattle: University of Washington; 2010. Incontinentia pigmenti; pp. 1993–2014. http://www.ncbi.nlm.nih.gov/books/NBK1472. [Google Scholar]
- Goldberg MF. The skin is not the predominant problem in incontinentia pigmenti. Arch Dermatol. 2004;140:748–750. doi: 10.1001/archderm.140.6.748. [DOI] [PubMed] [Google Scholar]
- Meuwissen MEC, Mancini GMS. Neurological findings in incontinentia pigmenti: a review. Eur J Med Genet. 2012;55:323–331. doi: 10.1016/j.ejmg.2012.04.007. [DOI] [PubMed] [Google Scholar]
- Minić S, Trpinac D, Obradović M. Systematic review of central nervous system anomalies in incontinentia pigmenti. Orphanet J Rare Dis. 2013;8:25–35. doi: 10.1186/1750-1172-8-25. doi:doi:10.1186/1750-1172-8-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzamiglio MR, Piccardi L, Bianchini F, Canzano L, Palermo L, Fusco F, D'Antuono G, Gelmini C, Garavelli L, Ursini MV. Incontinentia pigmenti: learning disabilities are a fundamental hallmark of the disease. PLoS One. 2014;9:e87771. doi: 10.1371/journal.pone.0087771. doi:10.1371/journal.pone.0087771. eCollection 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadj-Rabia S, Froidevaux D, Bodak N, Hamel-Teillac D, Smahi A, Touil Y, Fraitag S, de Prost Y, Bodemer C. Clinical study of 40 cases of incontinentia pigmenti. Arch Dermatol. 2003;139:1163–1170. doi: 10.1001/archderm.139.9.1163. [DOI] [PubMed] [Google Scholar]
- Smahi A, Courtois G, Vabres P, Yamaoka S, Heuertz S, Munnich A, Israël A, Heiss NS, Klauck SM, Kioschis P, Wiemann S, Poustka A, Esposito T, Bardaro T, Gianfrancesco F, Ciccodicola A, D'Urso M, Woffendin H, Jakins T, Donnai D, Stewart H, Kenwrick SJ, Aradhya S, Yamagata T, Levy M, Lewis RA, Nelson DL. International Incontinentia Pigmenti (IP) Consortium. Genomic rearrangement in NEMO impairs NF-κB activation and is a cause of incontinentia pigmenti. Nature. 2000;405:466–472. doi: 10.1038/35013114. [DOI] [PubMed] [Google Scholar]
- Fusco F, Paciolla M, Napolitano F, Pescatore A, D'Addario I, Bal E, Lioi MB, Smahi A, Miano MG, Ursini MV. Genomic architecture at the Incontinentia Pigmenti locus favours de novo pathological alleles through different mechanisms. Hum Mol Genet. 2012;21:1260–1271. doi: 10.1093/hmg/ddr556. [DOI] [PubMed] [Google Scholar]
- Conte MI, Pescatore A, Paciolla M, Esposito E, Miano MG, Lioi MB, McAleer MA, Giardino G, Pignata C, Irvine AD, Scheuerle AE, Royer G, Hadj-Rabia S, Bodemer C, Bonnefont JP, Munnich A, Smahi A, Steffann J, Fusco F, Ursini MV. Insight into IKBKG/NEMO locus: report of new mutations and complex genomic rearrangements leading to incontinentia pigmenti disease. Hum Mutat. 2014;35:165–177. doi: 10.1002/humu.22483. [DOI] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Signaling to NF-κB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- Nelson DL. NEMO, NFκB signaling and incontinentia pigmenti. Curr Opin Genet Dev. 2006;16:282–288. doi: 10.1016/j.gde.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Fusco F, Bardaro T, Fimiani G, Mercadante V, Miano MG, Falco G, Israël A, Courtois G, D'Urso M, Ursini MV. Molecular analysis of the genetic defect in a large cohort of IP patients and identification of novel NEMO mutations interfering with NF-κB activation. Hum Mol Genet. 2004;13:1763–1773. doi: 10.1093/hmg/ddh192. [DOI] [PubMed] [Google Scholar]
- Migeon BR. Non-random X chromosome inactivation in mammalian cells. Cytogenet Cell Genet. 1998;80:142–148. doi: 10.1159/000014971. [DOI] [PubMed] [Google Scholar]
- Desai V, Donsante A, Swoboda KJ, Martensen M, Thompson J, Kaler SG. Favorably skewed X-inactivation accounts for neurological sparing in female carriers of Menkes disease. Clin Genet. 2011;79:176–182. doi: 10.1111/j.1399-0004.2010.01451.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtois G, Smahi A, Israël A. NEMO/IKKγ: linking NF-κB to human disease. Trends Mol Med. 2001;7:427–430. doi: 10.1016/S1471-4914(01)02154-2. [DOI] [PubMed] [Google Scholar]
- Fusco F, Fimiani G, Tadini G, Michele D, Ursini MV. Clinical diagnosis of incontinentia pigmenti in a cohort of male patients. J Am Acad Dermatol. 2007;56:264–267. doi: 10.1016/j.jaad.2006.09.019. [DOI] [PubMed] [Google Scholar]
- Kenwrick S, Woffendin H, Jakins T, Shuttleworth SG, Mayer E, Greenhalgh L, Whittaker J, Rugolotto S, Bardaro T, Esposito T, D'Urso M, Soli F, Turco A, Smahi A, Hamel-Teillac D, Lyonnet S, Bonnefont JP, Munnich A, Aradhya S, Kashork CD, Shaffer LG, Nelson DL, Levy M, Lewis RA. International IP Consortium. Survival of male patients with incontinentia pigmenti carrying a lethal mutation can be explained by somatic mosaicism or Klinefelter syndrome. Am J Hum Genet. 2001;69:1210–1217. doi: 10.1086/324591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusco F, Pescatore A, Bal E, Ghoul A, Paciolla M, Lioi MB, D'Urso M, Rabia SH, Bodemer C, Bonnefont JP, Munnich A, Miano MG, Smahi A, Ursini MV. Alterations of the IKBKG locus and diseases: an update and a report of 13 novel mutations. Hum Mutat. 2008;29:595–604. doi: 10.1002/humu.20739. [DOI] [PubMed] [Google Scholar]
- Fusco F, Pescatore A, Steffann J, Royer G, Bonnefont JP, Ursini MV. Clinical utility gene card for: Incontinentia Pigmenti. Eur J Hum Genet. 2013;21 doi: 10.1038/ejhg.2012.227. doi:10.1038/ejhg.2012.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minić S, Trpinac D, Obradović M. Incontinentia pigmenti diagnostic criteria update. Clin Genet. 2014;85:536–542. doi: 10.1111/cge.12223. [DOI] [PubMed] [Google Scholar]
- Bardaro T, Falco G, Sparago A, Mercadante V, Gean Molins E, Tarantino E, Ursini MV, D'Urso M. Two cases of misinterpretation of molecular results in incontinentia pigmenti, and a PCR-based method to discriminate NEMO/IKKγ gene deletion. Hum Mutat. 2003;21:8–11. doi: 10.1002/humu.10150. [DOI] [PubMed] [Google Scholar]
- den Dunnen JT, Antonarakis SE. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat. 2000;15:7–15. doi: 10.1002/(SICI)1098-1004(200001)15:1<7::AID-HUMU4>3.0.CO;2-N. Erratum in: Hum Mutat 2002, 20:403. [DOI] [PubMed] [Google Scholar]
- Ursini MV, Conte MI, Pescatore A, Miano MG, Fusco F. Molecular Genetics of Incontinentia Pigmenti. Chichester: eLS. John Wiley & Sons, Ltd; 2012. doi:10.1002/9780470015902.a0024332. [Google Scholar]