

Abstract
A dense resident microbial community in the gut, referred as the commensal microbiota, co-evolved with the host, and is essential for many host physiological processes that include enhancement of the intestinal epithelial barrier, development of the immune system, and nutrient acquisition. A major function of the microbiota is protection against colonization by pathogens and overgrowth of indigenous pathobionts that can result from the disruption of the healthy microbial community. The mechanisms that regulate the ability of the microbiota to restrain pathogen growth are complex and include competitive metabolic interactions, localization to intestinal niches, and induction of host immune responses. Pathogens, in turn, have evolved strategies to escape from commensal-mediated colonization resistance. Thus, the interplay between commensals and pathogens or indigenous pathobionts is critical for controlling infection and disease. Understanding pathogen-commensal interactions may lead to new therapeutic approaches against infectious diseases.
Structural composition of the gut microbiota
Vertebrates harbor a densely populated resident microbial community, which consists of bacteria, viruses, and fungi, particularly in mucosal organs, such as the oral cavity and the intestine. This internal microbial community residing within the host is referred as the microbiota1. In healthy individuals, Gram-negative Proteobacteria and Bacteroidetes, and Gram-positive Firmicutes, such as Clostridiales and Lactobacillales, represent the major phyla among intestinal eubacteria, whereas methanogens are the predominant intestinal archaea2. Many of these resident commensals are adapted to the intestinal environment and develop complex ecological networks with other bacteria to acquire nutrients. For example, Lactobacillus species and Eubacterium dolichum lack the ability to synthesize certain amino acids and therefore, acquire these critical molecules from their intestinal habitats 3, 4. Methanogens obtain energy from hydrogen molecules, a waste product of other obligate anaerobes2. Consequently, host-microbial, microbial-microbial as well as microbial-environmental interactions dictate the distribution of individual commensals throughout the gastrointestinal tract.
A critical factor that defines the composition and distribution of the microbiota is the nutrient requirement of individual commensals (Figure 1). Microbes colonize mammalian hosts immediately after birth 5, 6. In neonatal mice, the bacterial composition within the oral cavity and intestinal tract is similar and simply structured; however, after weaning as the diet changes from maternal milk to fiber-rich foods, the bacterial composition dramatically changes 5, 6. The small intestine is rich in mono- and di-saccharides as well as amino acids, which support the growth of certain bacteria particularly Proteobacteria and Lactobacillales (Figure 1). In the distal small intestine including the terminal ileum, simple sugars are absorbed by host cells, so energy sources available for bacterial growth are significantly altered, resulting in changes in bacterial composition. Beyond the ileocecal valve, the vast majority of available carbohydrates are diet- (i.e., plant foods) or host-derived (e.g., mucin, cellular debris, etc.) complex carbohydrates (polysaccharides), which are indigestible by the host. Proteobacteria, such as Escherichia coli, are incapable of digesting polysaccharides, and therefore are unable to utilize complex carbohydrates as an energy source. In contrast, Bacteroides and Clostridiales harbor enzymes that can breakdown host-indigestible polysaccharides, including fibers and mucin, and use them as an energy source. Consequently, the abundance of proteobacteria and Lactobacillales is much lower in the colon, while Bacteroides and Clostridiales are dominant populations within the large intestine 7. Notably, even among bacteria within the same phyla, the ability to scavenge polysaccharides is highly diverse7, suggesting that the polysaccharide content of any diet can greatly affect the relative abundance of bacterial species within Bacteroidetes or Clostridiales. Thus, the distribution of nutrients within the intestine is a major driver of the microbial community structure in the gut. Many studies have shown differences in microbiota composition between healthy individuals and patients with intestinal diseases8-10, which may reflect changes in the availability of host factors and nutrients in the disease state and/or be secondary to the inflammatory response. However, the diversity of microbiota among individuals cannot be simply explained by diet alone. For example, Bifidobacteria, which affect host responses to pathogens, colonize the gut of humans, but not SPF mice. Further ecological analysis of intra-commensal interactions and better characterization of the metabolic activities of individual bacteria are required to understand the diversity of bacteria within the intestine.
Figure 1. Localization of dominant bacterial groups within the intestine.
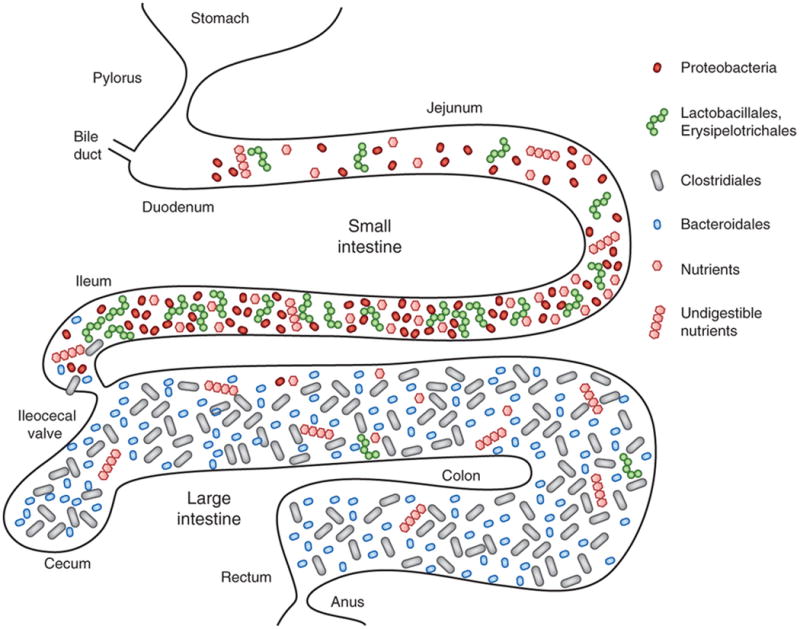
The small intestine is rich in nutrients utilized by both the host and microbe for growth. Proteobacteria (mainly enterobacteria), Lactobacillales and Erysipelotrichales (especially Turicibacter) are dominant in the small intestine. In contrast, the large intestine is poor in such nutrients and therefore harbor much fewer numbers of these bacteria, while Bacteroidetes and Clostridia which can utilize host indigestible fibers as energy sources are enriched.
Protective effect of the microbiota against pathogen infection
Mounting evidence demonstrates that the gut microbiota play a crucial role in host resistance against invading pathogens within the intestine. Consequently, germ-free (GF) and antibiotic-treated mice are more susceptible to various enteric pathogens infection 11-16. How resident microbes prevent pathogen colonization has been studied for many years, and the mechanisms involved largely fall into two categories : 1) Direct interactions between commensals and pathogens, such as competition for shared nutrients and niches, and 2) Commensal-mediated enhancement of host defense mechanisms.
Colonization resistance via direct commensal-pathogen interaction
Both pathogens and commensal bacteria require similar ecological niches to colonize and proliferate in the intestine, and mechanisms to out-compete each other have evolved. Commensal bacteria produce bacteriocins, proteinaceous toxins, that specifically inhibit members of the same or similar bacterial species 17. For example, E. coli produces bacteriocin that directly inhibits the growth of the related pathogen enterohaemorrhagic E. coli (EHEC) 18(Figure 2). Commensals also prevent pathogen infection by altering host environmental conditions (e.g., pH) that are prohibitive for pathogen colonization17. A healthy vaginal flora reduces the pH in the vagina, thereby preventing the colonization of urinary tract pathogens whose optimal pH for growth is neutral 19. Likewise, in the intestine, certain commensal bacteria generate short chain fatty acids (SFCAs), which can alter the local pH to inhibit the growth of certain intestinal pathogens 20, 21(Figure 2). Bifidobacterium blocks colonization of pathogenic E. coli through acidification of its environment12. Similarly, the pH is critical for Bacillus cereus growth and enterotoxin secretion22.
Figure 2. Commensal microbiota prevents colonization by exogenous pathogens and pathobionts.
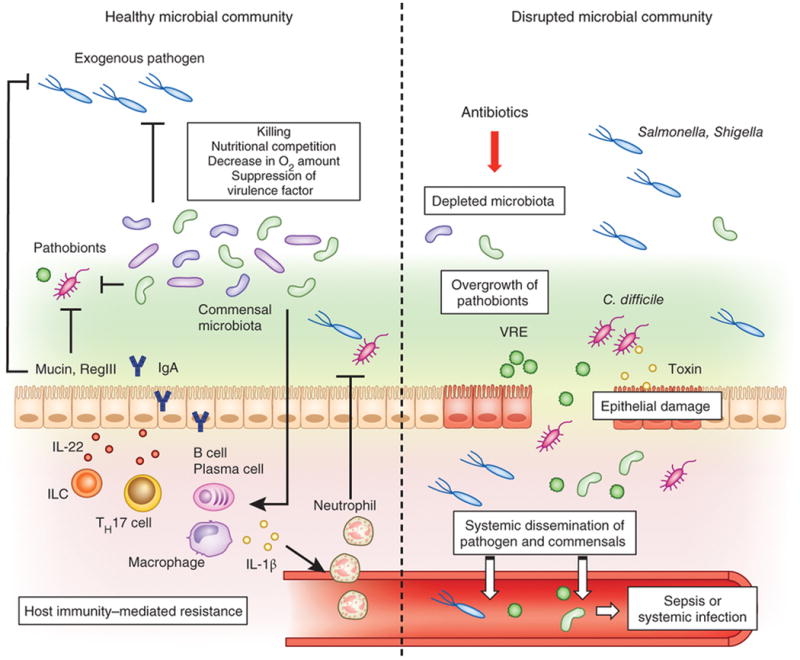
In the healthy gut, the resident bacteria occupy intestinal colonization niches. Commensal microbiota suppresses the proliferation and colonization of incoming enteric pathogens as well as opportunistic pathobionts through multiple mechanisms. Microbiota produces bacteriocins and short-chain fatty acids, which directly inhibit the growth of pathogen and pathobiont. Commensals can also modify virulence factor expression in pathogens by consuming residual oxygen or suppressing growth by their metabolites. Commensal microbiota facilitates host barrier function through up-regulation of the mucus layer, induction of antimicrobial molecules, such as RegIIIβ and γ, and regulating secretion of IgA. Commensal bacteria also prime intestinal macrophages by upregulating pro-IL-1β. Pathogen infection results in the conversion of pro-IL-1β into the enzymatically active mature form of IL-1β, which promotes neutrophil recruitment and pathogen eradication. Antibiotic treatment or other environmental factors that disrupt the commensal microbial community results in diminished colonization resistance against pathogens (e.g. Salmonella, Shigella) and allow the outgrowth of indigenous pathobionts (e.g. Clostridium difficile, vancomycin-resistant Enterococcus) that have the potential to disseminate systemically and induce septic shock and/or systemic organ infection.
An alternative strategy utilized by the indigenous microbial community is the preferential consumption of nutrients required for the growth of competing pathogenic bacteria. For example, commensal E. coli competes with EHEC for organic acids, amino acids, and other nutrients23-26(Figure 2). By consuming common limited resources, the gut microbiota essentially causes the starvation of competing pathogens. Commensal bacteria, through production of specific metabolites, can also affect pathogen virulence to compromise their growth. The SCFA, butyrate, downregulates the expression of several virulence genes including those encoding the Type 3 secretion system (T3SS) proteins in Salmonella enteria Serovar Enteritidis and Typhimurium 27(Figure 2). Moreover, host mucin-derived fucose, which is generated by fucosidase-bearing commensal bacteria such as Bacteroides thetaiotaomicron, modulates the expression of the virulence factor ler, a master regulator of the locus of enterocyte effacement (LEE) genes, in EHEC28(Figure 2). In addition to direct suppression of virulence genes by commensal metabolites, commensals also inhibit pathogen virulence by altering conditions required for virulence activity. For example, ambient oxygen tensions are required for competent secretion of virulence factors by Shigella flexneri 29, and therefore, the consumption of residual oxygen by commensal facultative anaerobes, such as Enterobacteriaceae, may lead to incomplete virulence expression in intestinal lumen (Figure 2).
Indirect mechanisms of colonization resistance by commensals via activation of host immunity
Commensal bacteria also prevent pathogen colonization and infection indirectly by enhancing host defense mechanisms such as functionally promoting mucosal barrier function and enhancing either innate immune responses. The first line of defense against any pathogen invasion is the epithelial barrier. The promotion of epithelial barrier function by commensal bacteria is supported primarily by indirect evidence in which studies have demonstrated that germ free mice and mice deficient in proteins involved in microbial recognition such as Nod2 and the TLR signaling adaptor MyD88 have impaired production of antimicrobial peptides, particularly by Paneth cells of the small intestine 30, 31. Consequently, MyD88-deficient or Paneth-cell deficient mice have impaired epithelial barrier function and increased bacterial translocation of pathogenic bacteria 31, associated with their inability to produce specific antimicrobial peptides (Figure 2). In addition, antimicrobial peptides not only limit enteric infection by their inherent bactericidal activity, but also restrict bacterial colonization. Specifically, mice deficient in MyD88 or RegIIIγ have higher mucosal bacterial loads compared to wildtype littermates with larger numbers of bacteria in direct contact with the surface epithelium within the small intestine 32. Production of RegIIIγ is also upregulated by IL-22, which is induced in group 3 innate lymphoid cells, Th17 cells as well as a certain subset of DCs33-35 by the gut microbiota. IL-22-mediated RegIIIγ production by intestinal epithelium is protective against enteric infection by Citrobacter rodentium, a mouse bacterium that models infection by enteropathogenic E. coli (EPEC) and EHEC33, 36, 37 (Figure 2). In addition to antimicrobial peptide production, bacterial signaling likely through MyD88 also promotes barrier function through the production of secretory IgA that is released by intestinal epithelial cells and functions to bind to microbial antigens, neutralize pathogen activity and prevent infection 38-41. However, since both antimicrobial peptides and IgA are also capable of shaping the gut microbiota 32, 42-44, it remains unclear whether the actual composition of the gut microbiota regulated by these molecules is the main determinant for pathogen resistance.
The gut microbiota, not only promotes mucosal barrier function, but also enhances host immunity to defend against enteric infection. IL-1β is a cytokine typically produced during active infection that is critical for neutrophil recruitment and pathogen eradication. The microbiota have an essential role in the production of homeostatic levels of pro-IL-1β in resident intestinal macrophages that is MyD88-dependent, thereby priming macrophages to respond rapidly to enteric infection by conversion of pro-IL-1β to mature active IL-1β45 (Figure 2). The gut microbiota can also enhance host immunity through MyD88-independent mechanisms. Notably, colonization of GF mice by commensal bacteria induces development of Th17 cells in the intestine, which is important for protection against C. rodentium infection and is MyD88-, TRIF-, and Rip2-independent46.
Disruption of the commensal microbial community leads to outgrowth of pathogens and pathobionts
A consequence of disruption of commensal-mediated colonization resistance is that susceptibility to enteric infection will be markedly increased. Salmonella enterica Serovar Typhimurium (Salmonella) colonize poorly the mouse intestine in the presence of commensal microbiota 13. However, it can proliferate and induce inflammation if the resident bacterial community is disrupted by treatment with antibiotics or if recipient mice have low-complexity microbiota 13, 47. An altered bacterial community structure also may facilitate the overgrowth of potentially harmful subsets of indigenous bacteria within the intestine. In a mouse model, virulent E. coli, whose growth is normally suppressed by commensals, accumulates after antibiotic treatment and can disseminate systemically when the intestinal epithelial barrier is breached by dextran sulphate sodium (DSS), thereby inducing lethal inflammasome activation 48. Similarly, Clostridium difficile, a leading cause of healthcare-associated infectious diarrhea and colitis, 16 typically present at low abundance in the intestine of adult healthy individuals, but with disruption of indigenous bacteria after treatment with broad-spectrum antibiotics as in hospitalized patients, C. difficile can significantly increase in abundance followed by severe intestinal inflammation 16. Like humans, in a mouse model of C. difficile infection, C. difficile can not colonize and induce inflammation in conventional mice, while antibiotic treatment increases the incidence of C. difficile infection. C. difficile does not invade the host systemically, but causes marked epithelial damage through the production of the toxins TcdA and TcdB16, 49(Figure 2). The toxin-mediated epithelial damage can cause systemic dissemination of commensal bacteria which induce lethal septic shock 50(Figure 2). Vancomycin-resistant enterococcus (VRE), which can cause sepsis in immunocompromised patients, is also associated with antibiotic treatment51. Commensal microbiota-mediated induction of antimicrobial peptide RegIIIγ is particularly important for bacterial killing (Figure 2). A role for commensals to regulate RegIIIγ production is supported by the observation that bacterial signaling through MyD88, and more specifically, administration of the bacterial products lipopolysaccharide (LPS) or bacterial flagellin, upregulate RegIIIγ and increase VRE eradication52, 53. However, a recent study suggested that specific bacterial populations within the colon facilitate VRE clearance that occurred independently of host innate immune pathways including MyD8854. Although the mechanism is currently unknown, it may be related to a direct mechanism such as through competition for limited nutrients. It is also possible that both commensal-dependent indirect and direct mechanisms are essential for VRE clearance with antimicrobial production being the primary mechanism within the small intestine, where Paneth cells predominate, and direct inhibition by specific bacteria populations being a primary mechanism within the colon.
The gut microbiota as promoters of enteric infection
Although the commensal microbiota play crucial roles in resistance against enteric pathogen infections, certain pathogens can utilize the microbiota to facilitate their infection. For example, by-products derived from commensal bacteria such as bile salts promote the germination of C. difficile spores 55. In addition to bacterial pathogens, certain enteric viruses can also take advantage of the gut microbiota to promote their replication and transmission. Offspring transmission of mouse mammary tumor virus (MMTV) and replication of poliovirus are significantly reduced in antibiotic-treated or GF mice 56, 57. These enteric viruses directly bind and modify the stimulatory activity of LPS that is secreted by commensal bacteria. MMTV-bound LPS induces the production of the anti-inflammatory cytokine IL-10, which leads to depressed anti-viral immune response by the host and persistent infection of MMTV56. Thus, the microbiota can also potentiate pathogenic infection.
Strategies by which pathogens overcome the colonization resistance by the microbiota
As described above, the microbiota have developed multiple mechanisms to resist pathogen colonization. However, pathogens have evolved strategies to escape these mechanisms. For example, to counteract nutritional competition by commensals, some pathogens have developed alternative nutrient utilization. The pathogen EHEC can utilize galactose, hexuronates, mannose, and ribose as a carbon source for its growth, while commensal E. coli cannot catabolize these sugars25, 58(Figure 3). In addition to altered carbohydrate metabolism, EHEC harbors the EA utilization eut operon in its genome and is capable of utilizing pathogen-specific nutrients in the intestine, such as ethanolamine (EA), which is released into the intestine during epithelial cell turnover25, 59, 60(Figure 3). In contrast, the eut operon is absent in the genome of commensal members of non-pathogenic E. coli thereby prohibiting them from using EA as a nutrient60. Although C. rodentium exhibits a similar carbohydrate metabolic profile as the commensal E. coli61, C. rodentium resides in a distinct niche during replication to escape from the nutritional competition by commensals. Specifically, C. rodentium expresses intimin, a LEE-encoded adhesion molecule, that allows the bacterium to localize to the intestinal epithelial surface where commensal microbiota do not normally reside 61(Figure 3). In addition, some pathogens more efficiently utilize common resources. Iron is an essential resource for the growth of bacteria, and many bacteria produce iron-chelating small molecules, known as siderophores, to acquire ferric iron62. Host cells secrete lipocalin 2 (Lcn2), which blocks the 2,3 dihydroxy benzoate-based siderophore enterobactin (Ent) in E. coli, thereby preventing iron acquisition and proliferation of commensal E. coli. However, some enteric pathogens, such as Salmonella, pathogenic E. coli and Klebsiella pneumoniae, display modified forms of Ent, referred to as salmochelins (also: vary the positioning of hydroxyl groups to the 3,4 positions or use non-catecholate siderophores)63. Lcn2 does not inhibit salmochelin-mediated iron uptake, leading to a growth advantage of the pathogens over the commensals (Figure 3). Thus, enteric pathogens use different strategies to overcome colonization resistance by utilizing pathogen-specific nutritional resources and/or localizing to distinct niches separate from competing commensals.
Figure 3. Pathogen overcome commensal-mediated resistance through multiple strategies.
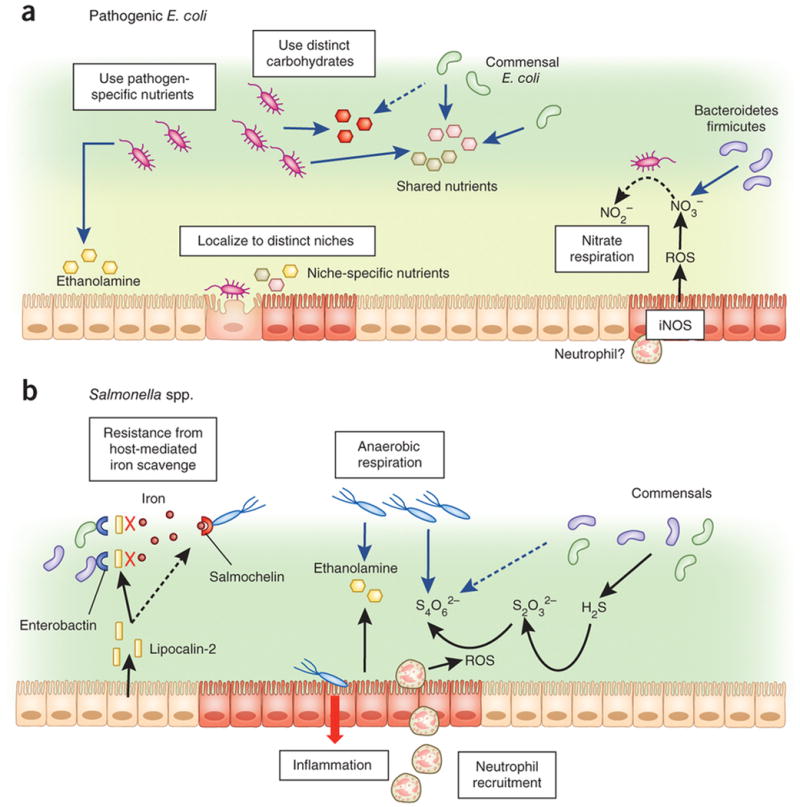
a| Pathogenic Escherichia coli: Pathogenic E. coli is capable of utilizing carbohydrates and other resources, such as ethanolamine distinct from that scavenged by commensals. Pathogenic E. coli can also localize to the intestinal epithelial surface that is devoid of commensal microbiota through expression of adhesion molecules, such as intimin. Pathogen-induced gut inflammation confers a growth advantage to the pathogen through the generation of molecules such as inducible nitric oxide synthase (iNOS) expression by host innate immune cells leading to the release of nitrate (NO3-) that can be utilized as an electron acceptor by E. coli to generate energy through nitrate respiration. Commensal obligate anaerobes, such as Bacteroidetes or Firmicutes lack this ability.
b| Salmonella spp.: Lipocalin-2 derived from host cells block commensal bacterial iron uptake by binding to the bacterial siderophore, enterobactin. Salmonella, however, has a distinct siderophore, salmochelin, for iron uptake, which is not blocked by lipocalin-2. Salmonella-induced gut inflammation also promotes migration of neutrophils that produce reactive oxygen species (ROS), which facilitate conversion of thiosulphate (S2O32-), generated by commensal bacteria, into tetrathionate (S4O62-). Salmonella, but not commensals, is capable of utilizing tetrathionate as an electron acceptor for anaerobic respiration.
Another important strategy utilized by pathogens to acquire a growth advantage over commensals is to promote host inflammation that impedes commensal survival. Most virulent microorganisms express virulence factors that cause intestinal inflammation. Inflammation caused by toxin- or pathogen-mediated diarrhea significantly decreases the number of commensal microbiota in the intestine, and, in turn, increases the chance of colonization/proliferation of incoming pathogens because of less competition64. DSS-induced intestinal inflammation markedly increases the proliferation of C. rodentium in the intestine; however, specific virulence factors are required for optimal colonization and proliferation as the ler mutant C. rodentium, which lacks expression of all LEE-encoded virulence genes is avirulent and fails to acquire a survival advantage from DSS-induced inflammation 61(Figure 3). Recent evidence also demonstrates that Salmonella benefits from intestinal inflammation triggered by the pathogen itself. In the intestine, commensals produce abundant hydrogen sulfide (H2S), and the epithelium converts H2S to thiosulphate (S2O32-) to avoid the H2S-mediated toxic effects on host cells. Infection by Salmonella results in the recruitment of neutrophils that produce reactive oxygen species, resulting in the conversion of S2O32- into tetrathionate (S4O62-)65, 66. Salmonella, but not commensals, contains a gene operon, ttrSR ttrBCA, which allows the utilization of the S4O62-. This tetrathionate respiration provides a growth advantage to Salmonella over commensal microbes within an inflammatory environment67(Figure 3). Moreover, tetrathionate supports anaerobic growth of Salmonella on ethanolamine68(Figure 3). Like Salmonella, pathogenic E. coli including EPEC, EHEC, and C. rodentium, also may benefit from intestinal inflammation. In the inflamed intestine, intestinal epithelium and recruited neutrophils and macrophages that express inducible nitric oxide synthetase (iNOS), upregulate the production of nitrate (NO3-) 69, 70. Obligate anaerobes, such as Bacteroidetes or Firmicutes that are the vast majority of healthy microbial community in the gut, cannot utilize nitrate as an electron acceptor 71. Rather, nitrate reductase-harboring facultive anaerobes, such as E. coli, can utilize NO3- to generate energy for growth, leading to a growth advantage over obligate anaerobes in the inflamed intestine71. Although this mechanism of E. coli overgrowth within the inflamed gut involves commensal-commensal competition, pathogenic E. coli strains, which bear nitrate reductase genes such as narZ in their genome, may use a similar mechanism to acquire a growth advantage over the competitive commensal community (Figure 3). Furthermore, the host inflammatory environment can act as a signal to trigger and enhance virulence factor expression. The human opportunistic pathogen Pseudomonous aeruginosa can bind to interferon-γ through its outer membrane protein OprF, thereby expressing quorum-sensing dependent virulence determinant type I P. aeruginosa (PA-I) lectin72. Thus, pathogens can take advantage of the inflammatory response to promote their growth in host tissues.
Microbiota-targeted therapies for disease treatment
The notion of harnessing the gut microbiota to prevent or fight microbial infection is not new. However, the lack of knowledge about the mechanisms by which commensals regulate colonization resistance against pathogens has hampered progress in the area. Recent mechanistic insight into pathogen-commensal interactions suggest ways to promote the eradication of pathogens. Diarrheagenic Escherichia coli strains, including EPEC and EHEC, cause substantial morbidity and mortality worldwide each year 73. In the C. rodentium model, the capacity to metabolize simple sugars regulates the ability of commensals like E. coli to out-compete the pathogen for energy resources. Thus, administration of commensals metabolically related to EPEC or EHEC or treatment with prebiotics to boost the growth of natural “competitors” may prove effective in the treatment of these enteric diseases. Eradication of EPEC and EHEC by commensals could be further boosted by targeting pathogen LEE virulence at the early phase of infection61. This approach may be effective in eradicating not only enterovirulent E. coli infection, but also other intestinal infections by pathobionts such as C. difficile and VRE. Overgrowth of C. difficile and VRE are leading causes of healthcare-associated infectious diarrhea and colitis 16, 51. There is also evidence that specific bacterial populations within the gut promote C. difficile and VRE clearance54, 74. Although the mechanism is currently unknown, it may be also mediated via a direct mechanism such as through competition between VRE and certain commensals for limited nutrients. Notably, intestinal microbiota transplantation, the infusion of stool microorganisms from a healthy donor, has proved effective in treating recurrent C. difficile infection that is refractory to antibiotic therapy 74-76. However, the variability of the donor commensal populations and the potential presence of hazardous microbes may hamper the use of microbiota transplantation in the clinic. Thus, identification and characterization of the gut commensals that restrain the growth of C. difficile and VRE may lead to the use of single species or defined combinations of protective commensals to treat infections. In addition, understanding the metabolic pathways that are used by gut commensals to prevent the growth of C. difficile and VRE may lead to the development of genetically engineered commensal species with enhanced capacity to limit pathogen colonization.
Conclusion
Recent studies are providing new insight into the mechanisms by which the microbiota regulates the colonization and eradication of pathogens. Particularly revealing have been studies that indicate that the ability of commensals to restrain pathogen growth is dictated by metabolic pathways that control the competition for limited nutrients in the intestine. Furthermore, inflammatory responses have profound effects on the growth of pathogens and certain commensal species. However, the relative contributions of each metabolic pathway and the commensal species involved remain poorly understood. In addition, little is known about how the inflammatory responses affect interactions between pathogens and commensals. There is a delicate balance in microbiota populations in the gut and disruption in this balance leads to dysbiosis and overgrowth of pathobionts leading to pathologic immune responses and disease. The identification and characterization of natural “competitors” that suppress the growth of pathogens and pathobionts may lead to the development of rational approaches to manage intestinal disease. There is also a clear role for host immunity in controlling microbiota populations. However, recent studies have challenged a critical role of innate recognition receptors in determining the composition of the gut microbiota77. Further studies are needed to clarify the mechanism by which the host regulates the microbiota.
Acknowledgments
We thank Eric Martens for critical review of the manuscript. Studies on the microbiota in our laboratory are supported by grants from the National Institutes of Health and the Bill & Melinda Gates Foundation. N. K. is supported by a Postdoctoral Fellowship from Colitis Foundation of America.
Contributor Information
Nobuhiko Kamada, Email: nkamada@umich.edu, Department of Pathology and Comprehensive Cancer Center, University of Michigan, Ann Arbor 48109, USA.
Grace Y. Chen, Email: gchenry@umich.edu, Department of Internal Medicine and Comprehensive Cancer Center, University of Michigan, Ann Arbor 48109, USA.
Naohiro Inohara, Email: ino@umich.edu, Department of Pathology, University of Michigan, Ann Arbor 48109, USA.
Gabriel Núñez, Email: gabriel.nunez@umich.edu, Department of Pathology and Comprehensive Cancer Center, University of Michigan, Ann Arbor 48109, USA.
References
- 1.Hooper LV, Macpherson AJ. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat Rev Immunol. 2010;10:159–169. doi: 10.1038/nri2710. [DOI] [PubMed] [Google Scholar]
- 2.Dridi B, Raoult D, Drancourt M. Archaea as emerging organisms in complex human microbiomes. Anaerobe. 2011;17:56–63. doi: 10.1016/j.anaerobe.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 3.Pridmore RD, et al. The genome sequence of the probiotic intestinal bacterium Lactobacillus johnsonii NCC 533. Proc Natl Acad Sci U S A. 2004;101:2512–2517. doi: 10.1073/pnas.0307327101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Turnbaugh PJ, Backhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe. 2008;3:213–223. doi: 10.1016/j.chom.2008.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matamoros S, Gras-Leguen C, Le Vacon F, Potel G, de La Cochetiere MF. Development of intestinal microbiota in infants and its impact on health. Trends Microbiol. 2013 doi: 10.1016/j.tim.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 6.Hasegawa M, et al. Transitions in oral and intestinal microflora composition and innate immune receptor-dependent stimulation during mouse development. Infect Immun. 2010;78:639–650. doi: 10.1128/IAI.01043-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koropatkin NM, Cameron EA, Martens EC. How glycan metabolism shapes the human gut microbiota. Nat Rev Microbiol. 2012;10:323–335. doi: 10.1038/nrmicro2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Willing B, et al. Twin studies reveal specific imbalances in the mucosa-associated microbiota of patients with ileal Crohn's disease. Inflamm Bowel Dis. 2009;15:653–660. doi: 10.1002/ibd.20783. [DOI] [PubMed] [Google Scholar]
- 9.Li E, et al. Inflammatory bowel diseases phenotype, C. difficile and NOD2 genotype are associated with shifts in human ileum associated microbial composition. PLoS One. 2012;7:e26284. doi: 10.1371/journal.pone.0026284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oh PL, et al. Characterization of the ileal microbiota in rejecting and nonrejecting recipients of small bowel transplants. Am J Transplant. 2012;12:753–762. doi: 10.1111/j.1600-6143.2011.03860.x. [DOI] [PubMed] [Google Scholar]
- 11.Osawa N, Mitsuhashi S. INFECTION OF GERMEREE MICE WITH SHIGELLA FLEXNERI 3A. Jpn J Exp Med. 1964;34:77–80. [PubMed] [Google Scholar]
- 12.Fukuda S, et al. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature. 2011;469:543–547. doi: 10.1038/nature09646. [DOI] [PubMed] [Google Scholar]
- 13.Bohnhoff M, Drake BL, Miller CP. Effect of streptomycin on susceptibility of intestinal tract to experimental Salmonella infection. Proc Soc Exp Biol Med. 1954;86:132–137. doi: 10.3181/00379727-86-21030. [DOI] [PubMed] [Google Scholar]
- 14.Hentges DJ, Freter R. In vivo and in vitro antagonism of intestinal bacteria against Shigella flexneri. I. Correlation between various tests. J Infect Dis. 1962;110:30–37. doi: 10.1093/infdis/110.1.30. [DOI] [PubMed] [Google Scholar]
- 15.Lawley TD, et al. Antibiotic treatment of clostridium difficile carrier mice triggers a supershedder state, spore-mediated transmission, and severe disease in immunocompromised hosts. Infect Immun. 2009;77:3661–3669. doi: 10.1128/IAI.00558-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rupnik M, Wilcox MH, Gerding DN. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat Rev Microbiol. 2009;7:526–536. doi: 10.1038/nrmicro2164. [DOI] [PubMed] [Google Scholar]
- 17.Hammami R, Fernandez B, Lacroix C, Fliss I. Anti-infective properties of bacteriocins: an update. Cell Mol Life Sci. 2012 doi: 10.1007/s00018-012-1202-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schamberger GP, Diez-Gonzalez F. Selection of recently isolated colicinogenic Escherichia coli strains inhibitory to Escherichia coli O157:H7. J Food Prot. 2002;65:1381–1387. doi: 10.4315/0362-028x-65.9.1381. [DOI] [PubMed] [Google Scholar]
- 19.Turovskiy Y, Sutyak Noll K, Chikindas ML. The aetiology of bacterial vaginosis. J Appl Microbiol. 2011;110:1105–1128. doi: 10.1111/j.1365-2672.2011.04977.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cherrington CA, Hinton M, Pearson GR, Chopra I. Short-chain organic acids at ph 5.0 kill Escherichia coli and Salmonella spp. without causing membrane perturbation. J Appl Bacteriol. 1991;70:161–165. doi: 10.1111/j.1365-2672.1991.tb04442.x. [DOI] [PubMed] [Google Scholar]
- 21.Shin R, Suzuki M, Morishita Y. Influence of intestinal anaerobes and organic acids on the growth of enterohaemorrhagic Escherichia coli O157:H7. J Med Microbiol. 2002;51:201–206. doi: 10.1099/0022-1317-51-3-201. [DOI] [PubMed] [Google Scholar]
- 22.Ceuppens S, et al. Enterotoxin production by Bacillus cereus under gastrointestinal conditions and their immunological detection by commercially available kits. Foodborne Pathog Dis. 2012;9:1130–1136. doi: 10.1089/fpd.2012.1230. [DOI] [PubMed] [Google Scholar]
- 23.Momose Y, Hirayama K, Itoh K. Competition for proline between indigenous Escherichia coli and E. coli O157:H7 in gnotobiotic mice associated with infant intestinal microbiota and its contribution to the colonization resistance against E. coli O157:H7. Antonie Van Leeuwenhoek. 2008;94:165–171. doi: 10.1007/s10482-008-9222-6. [DOI] [PubMed] [Google Scholar]
- 24.Momose Y, Hirayama K, Itoh K. Effect of organic acids on inhibition of Escherichia coli O157:H7 colonization in gnotobiotic mice associated with infant intestinal microbiota. Antonie Van Leeuwenhoek. 2008;93:141–149. doi: 10.1007/s10482-007-9188-9. [DOI] [PubMed] [Google Scholar]
- 25.Fabich AJ, et al. Comparison of carbon nutrition for pathogenic and commensal Escherichia coli strains in the mouse intestine. Infect Immun. 2008;76:1143–1152. doi: 10.1128/IAI.01386-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leatham MP, et al. Precolonized human commensal Escherichia coli strains serve as a barrier to E. coli O157:H7 growth in the streptomycin-treated mouse intestine. Infect Immun. 2009;77:2876–2886. doi: 10.1128/IAI.00059-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gantois I, et al. Butyrate specifically down-regulates salmonella pathogenicity island 1 gene expression. Appl Environ Microbiol. 2006;72:946–949. doi: 10.1128/AEM.72.1.946-949.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pacheco AR, et al. Fucose sensing regulates bacterial intestinal colonization. Nature. 2012;492:113–117. doi: 10.1038/nature11623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marteyn B, et al. Modulation of Shigella virulence in response to available oxygen in vivo. Nature. 2010;465:355–358. doi: 10.1038/nature08970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kobayashi KS, et al. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 2005;307:731–734. doi: 10.1126/science.1104911. [DOI] [PubMed] [Google Scholar]
- 31.Vaishnava S, Behrendt CL, Ismail AS, Eckmann L, Hooper LV. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc Natl Acad Sci U S A. 2008;105:20858–20863. doi: 10.1073/pnas.0808723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vaishnava S, et al. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science. 2011;334:255–258. doi: 10.1126/science.1209791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Satoh-Takayama N, et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity. 2008;29:958–970. doi: 10.1016/j.immuni.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 34.Sanos SL, et al. RORgammat and commensal microflora are required for the differentiation of mucosal interleukin 22-producing NKp46+ cells. Nat Immunol. 2009;10:83–91. doi: 10.1038/ni.1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng Y, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- 36.Kiss EA, et al. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science. 2011;334:1561–1565. doi: 10.1126/science.1214914. [DOI] [PubMed] [Google Scholar]
- 37.Qiu J, et al. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity. 2012;36:92–104. doi: 10.1016/j.immuni.2011.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frantz AL, et al. Targeted deletion of MyD88 in intestinal epithelial cells results in compromised antibacterial immunity associated with downregulation of polymeric immunoglobulin receptor, mucin-2, and antibacterial peptides. Mucosal Immunol. 2012;5:501–512. doi: 10.1038/mi.2012.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fagarasan S, Kawamoto S, Kanagawa O, Suzuki K. Adaptive immune regulation in the gut: T cell-dependent and T cell-independent IgA synthesis. Annu Rev Immunol. 2010;28:243–273. doi: 10.1146/annurev-immunol-030409-101314. [DOI] [PubMed] [Google Scholar]
- 40.Suzuki K, et al. The sensing of environmental stimuli by follicular dendritic cells promotes immunoglobulin A generation in the gut. Immunity. 2010;33:71–83. doi: 10.1016/j.immuni.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 41.Strugnell RA, Wijburg OL. The role of secretory antibodies in infection immunity. Nat Rev Microbiol. 2010;8:656–667. doi: 10.1038/nrmicro2384. [DOI] [PubMed] [Google Scholar]
- 42.Petnicki-Ocwieja T, et al. Nod2 is required for the regulation of commensal microbiota in the intestine. Proc Natl Acad Sci U S A. 2009;106:15813–15818. doi: 10.1073/pnas.0907722106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Salzman NH, et al. Enteric defensins are essential regulators of intestinal microbial ecology. Nat Immunol. 2010;11:76–83. doi: 10.1038/ni.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Macpherson AJ, Geuking MB, McCoy KD. Homeland security: IgA immunity at the frontiers of the body. Trends Immunol. 2012;33:160–167. doi: 10.1016/j.it.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 45.Franchi L, et al. NLRC4-driven production of IL-1beta discriminates between pathogenic and commensal bacteria and promotes host intestinal defense. Nat Immunol. 2012;13:449–456. doi: 10.1038/ni.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ivanov II, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Endt K, et al. The microbiota mediates pathogen clearance from the gut lumen after non-typhoidal Salmonella diarrhea. PLoS Pathog. 2010;6:e1001097. doi: 10.1371/journal.ppat.1001097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ayres JS, Trinidad NJ, Vance RE. Lethal inflammasome activation by a multidrug-resistant pathobiont upon antibiotic disruption of the microbiota. Nat Med. 2012;18:799–806. doi: 10.1038/nm.2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ng J, et al. Clostridium difficile toxin-induced inflammation and intestinal injury are mediated by the inflammasome. Gastroenterology. 2010;139:542–552. 552, e541–543. doi: 10.1053/j.gastro.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 50.Hasegawa M, et al. Protective Role of Commensals against Clostridium difficile Infection via an IL-1beta-Mediated Positive-Feedback Loop. J Immunol. 2012 doi: 10.4049/jimmunol.1200821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arias CA, Murray BE. The rise of the Enterococcus: beyond vancomycin resistance. Nat Rev Microbiol. 2012;10:266–278. doi: 10.1038/nrmicro2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brandl K, et al. Vancomycin-resistant enterococci exploit antibiotic-induced innate immune deficits. Nature. 2008;455:804–807. doi: 10.1038/nature07250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kinnebrew MA, et al. Bacterial flagellin stimulates Toll-like receptor 5-dependent defense against vancomycin-resistant Enterococcus infection. J Infect Dis. 2010;201:534–543. doi: 10.1086/650203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ubeda C, et al. Intestinal Microbiota Containing Barnesiella Species Cures Vancomycin-Resistant Enterococcus faecium Colonization. Infect Immun. 2013;81:965–973. doi: 10.1128/IAI.01197-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Giel JL, Sorg JA, Sonenshein AL, Zhu J. Metabolism of bile salts in mice influences spore germination in Clostridium difficile. PLoS One. 2010;5:e8740. doi: 10.1371/journal.pone.0008740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kane M, et al. Successful transmission of a retrovirus depends on the commensal microbiota. Science. 2011;334:245–249. doi: 10.1126/science.1210718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kuss SK, et al. Intestinal microbiota promote enteric virus replication and systemic pathogenesis. Science. 2011;334:249–252. doi: 10.1126/science.1211057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Le Bouguenec C, Schouler C. Sugar metabolism, an additional virulence factor in enterobacteria. Int J Med Microbiol. 2010;301:1–6. doi: 10.1016/j.ijmm.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Perna NT, et al. Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature. 2001;409:529–533. doi: 10.1038/35054089. [DOI] [PubMed] [Google Scholar]
- 60.Bertin Y, et al. Enterohaemorrhagic Escherichia coli gains a competitive advantage by using ethanolamine as a nitrogen source in the bovine intestinal content. Environ Microbiol. 2011;13:365–377. doi: 10.1111/j.1462-2920.2010.02334.x. [DOI] [PubMed] [Google Scholar]
- 61.Kamada N, et al. Regulated virulence controls the ability of a pathogen to compete with the gut microbiota. Science. 2012;336:1325–1329. doi: 10.1126/science.1222195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Crosa JH, Walsh CT. Genetics and assembly line enzymology of siderophore biosynthesis in bacteria. Microbiol Mol Biol Rev. 2002;66:223–249. doi: 10.1128/MMBR.66.2.223-249.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fischbach MA, Lin H, Liu DR, Walsh CT. How pathogenic bacteria evade mammalian sabotage in the battle for iron. Nat Chem Biol. 2006;2:132–138. doi: 10.1038/nchembio771. [DOI] [PubMed] [Google Scholar]
- 64.Lupp C, et al. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe. 2007;2:204. doi: 10.1016/j.chom.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 65.Furne J, Springfield J, Koenig T, DeMaster E, Levitt MD. Oxidation of hydrogen sulfide and methanethiol to thiosulfate by rat tissues: a specialized function of the colonic mucosa. Biochem Pharmacol. 2001;62:255–259. doi: 10.1016/s0006-2952(01)00657-8. [DOI] [PubMed] [Google Scholar]
- 66.Levitt MD, Furne J, Springfield J, Suarez F, DeMaster E. Detoxification of hydrogen sulfide and methanethiol in the cecal mucosa. J Clin Invest. 1999;104:1107–1114. doi: 10.1172/JCI7712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Winter SE, et al. Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature. 2010;467:426–429. doi: 10.1038/nature09415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thiennimitr P, et al. Intestinal inflammation allows Salmonella to use ethanolamine to compete with the microbiota. Proc Natl Acad Sci U S A. 2011;108:17480–17485. doi: 10.1073/pnas.1107857108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kolios G, Valatas V, Ward SG. Nitric oxide in inflammatory bowel disease: a universal messenger in an unsolved puzzle. Immunology. 2004;113:427–437. doi: 10.1111/j.1365-2567.2004.01984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Reinders CA, et al. Rectal nitric oxide and fecal calprotectin in inflammatory bowel disease. Scand J Gastroenterol. 2007;42:1151–1157. doi: 10.1080/00365520701320505. [DOI] [PubMed] [Google Scholar]
- 71.Winter SE, et al. Host-derived nitrate boosts growth of E. coli in the inflamed gut. Science. 2013;339:708–711. doi: 10.1126/science.1232467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wu L, et al. Recognition of host immune activation by Pseudomonas aeruginosa. Science. 2005;309:774–777. doi: 10.1126/science.1112422. [DOI] [PubMed] [Google Scholar]
- 73.Kaper JB, Nataro JP, Mobley HL. Pathogenic Escherichia coli. Nat Rev Microbiol. 2004;2:123–140. doi: 10.1038/nrmicro818. [DOI] [PubMed] [Google Scholar]
- 74.Reeves AE, Koenigsknecht MJ, Bergin IL, Young VB. Suppression of Clostridium difficile in the gastrointestinal tracts of germfree mice inoculated with a murine isolate from the family Lachnospiraceae. Infect Immun. 2012;80:3786–3794. doi: 10.1128/IAI.00647-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.van Nood E, et al. Duodenal infusion of donor feces for recurrent Clostridium difficile. N Engl J Med. 2013;368:407–415. doi: 10.1056/NEJMoa1205037. [DOI] [PubMed] [Google Scholar]
- 76.Petrof EO, et al. Stool substitute transplant therapy for the eradication of Clostridium difficile infection: ‘RePOOPulating’ the gut. Microbiome. 2013;1 doi: 10.1186/2049-2618-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ubeda C, et al. Familial transmission rather than defective innate immunity shapes the distinct intestinal microbiota of TLR-deficient mice. J Exp Med. 2012;209:1445–1456. doi: 10.1084/jem.20120504. [DOI] [PMC free article] [PubMed] [Google Scholar]