

Abstract
Enzymes in the radical SAM (RS) superfamily catalyze a wide variety of reactions through unique radical chemistry. The characteristic markers of the superfamily include a [4Fe–4S] cluster coordinated to the protein via a cysteine triad motif, typically CX3CX2C, with the fourth iron coordinated by S-adenosylmethionine (SAM). The SAM serves as a precursor for a 5′-deoxyadenosyl radical, the central intermediate in nearly all RS enzymes studied to date. The SAM-bound [4Fe–4S] cluster is located within a partial or full triosephosphate isomerase (TIM) barrel where the radical chemistry occurs protected from the surroundings. In addition to the TIM barrel and a RS [4Fe–4S] cluster, many members of the superfamily contain additional domains and/or additional Fe–S clusters. Recently characterized superfamily members are providing new examples of the remarkable range of reactions that can be catalyzed, as well as new structural and mechanistic insights into these fascinating reactions.
Introduction
The radical S-adenosylmethionine (radical SAM, hereafter RS) superfamily of enzymes carry out a wide variety of biological functions including synthesis of cofactors, modification of RNA, DNA repair, and synthesis of antibiotics [1]. There are currently tens of thousands of predicted RS superfamily members spanning the phylogenetic kingdom; however, only a small fraction of these have been characterized. Although the chemistry they catalyze is diverse, the RS enzymes utilize a common mechanism for initiation of catalysis that involves generation of a primary carbon-centered radical intermediate, the 5′-deoxyadenosyl radical (dAdo•), which abstracts a hydrogen atom from the substrate [2–4,5••]. The substrate radical can then undergo radical-mediated and often complex transformations to generate product.
In order to generate the dAdo• radical from SAM, RS enzymes utilize an enzyme-bound [4Fe–4S] cluster. Three of the four irons in the [4Fe–4S] cluster are coordinated by cysteine thiolates (C or Cys) present in a triad motif, generally CX3CX2C although variations exist. The fourth iron of the cluster, often referred to as the unique iron due to its distinct coordination, has no cysteine ligand but rather is coordinated by SAM (Figure 1). The iron–sulfur cluster is active in its reduced [4Fe–4S]+ state, from which it can transfer an electron to the sulfonium of SAM to promote homolytic cleavage of the S–5′C bond of SAM, producing methionine and a 5′-deoxyadenosyl radical intermediate that abstracts a hydrogen atom from substrate (Figure 1) [6]. The highly reactive nature of the dAdo• radical intermediate [7] confers on these enzymes the ability to carry out diverse and difficult chemical transformations requiring abstraction of a hydrogen atom from generally unactivated positions on the substrate. The utilization of this primary carbon radical in catalytic chemistry, however, also requires the RS enzymes to employ exquisite control and protection of the radical intermediate in order to avoid damaging side reactions. Recent insights into the structures of the RS enzymes, and how these structures relate to mechanism and control of reactivity, will be addressed in this review.
Figure 1.
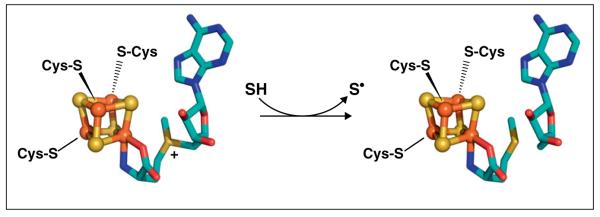
The reductive cleavage of SAM catalyzed by RS enzymes. SAM is coordinated via its amino and carboxyl moieties to the unique iron of a [4Fe–4S] cluster (left). The reduced [4Fe–4S]+ cluster transfers an electron to SAM, thereby promoting homolytic cleavage to generate methionine and a 5′-deoxyadenosyl radical. The 5′-deoxyadenosyl radical abstracts a hydrogen atom from substrate (SH) to produce a substrate radical (S•), methionine, and 5′-deoxyadenosine (right).
Structural insights into the radical SAM superfamily
Binding diverse substrates with a common fold
Several RS enzymes have been structurally characterized; these reveal a common fold consisting of a triosephosphate isomerase (TIM) barrel, which is a full (α/β)8 TIM barrel in some cases (e.g. biotin synthase (BioB) [8], HydE [9], and ThiC [10]) and a partial (α/β)6 TIM barrel in most other cases (Figure 2) [11,12••,13–17,18••]. The size of the TIM barrel inversely correlates with substrate size: enzymes that act on larger substrates tend to have less complete TIM barrels, thus making the barrel opening larger [4,5•]. The glycyl radical generating enzyme pyruvate formate lyase activating enzyme (PFL-AE), for example, is the smallest RS protein structurally characterized to date with little secondary structure outside the partial (α/β)6 TIM barrel (Figure 2a) [11]. The substrate for PFL-AE is among the largest RS substrates known: the 170 kDa homodimeric protein pyruvate formate lyase (PFL). On the basis of the evidence for direct H-atom abstraction from PFL G734 by the dAdo• generated in the PFL-AE active site, it is clear that minimally the glycyl radical domain of PFL must bind within this TIM barrel such that G734 is in close proximity to the [4Fe–4S] cluster and bound SAM in PFL-AE; this is consistent with the presence of an incomplete and splayed TIM barrel that provides a larger binding pocket for substrate [11,19•].
Figure 2.

Crystal structures of representative RS enzymes and Dph2. The Fe–S clusters are shown in rust for iron and yellow for sulfur. SAM molecules (teal carbons) and substrates (purple carbons) are also shown. (a) PFL-AE with SAM and the substrate PFL peptide (PDB ID: 3CB8). (b) SPL with SAM and substrate dinucleoside 5R-SP (PDB ID: 4FHD). (c) BioB with SAM, DTB substrate, and the additional [2Fe–2S] cluster (PDB ID: 1R30). (d) RlmN with SAM (PDB ID: 3RFA). (e) HydE with SAM and additional [2Fe–2S] cluster (PDB ID: 3IIZ). (f) Dph2 with the iron coordinating cysteines in red (PDB ID: 3LZD).
Spore photoproduct (SP) lyase (SPL) is another example of a RS enzyme that acts on a large substrate; in this case, the substrate is the SP (5-thyminyl-5,6-dihydrothymine) thymine dimer contained within double-stranded DNA (dsDNA). A recent structure of SPL reveals that, like PFL-AE, SPL contains a partial (α/β)6 TIM barrel with a wide lateral opening capable of binding large substrates (Figure 2b) [18••,20]. In contrast to PFL-AE and SPL, the RS enzyme BioB acts on a small-molecule substrate, dethiobiotin (DTB), and contains a full (α/β)8 TIM barrel; this full barrel still allows access of the small molecule substrate to the SAM-bound [4Fe–4S] cluster at the active site within the barrel, while also presumably shielding reactive radical intermediates from deleterious side reactions (Figure 2c) [8].
Positioning SAM and substrate for catalysis
In all RS structures to date, the catalytic [4Fe–4S] cluster bound to the characteristic RS cysteine motif resides at the C-terminal end of the TIM barrel, with the unique iron of the cluster pointing into the barrel. This unique iron is coordinated by the amino nitrogen and the carboxylate oxygen of SAM (Figure 1), an interaction first identified by electron nuclear double resonance (ENDOR) spectroscopic studies of PFL-AE [21,22] and subsequently observed in every RS crystal structure in which SAM is bound [5••]. This SAM–cluster interaction is unique to the RS enzymes and provides the key point of contact between the catalytic cluster and SAM for the production of dAdo•. Generally, in the crystal structures solved to date, the active site is exposed to solvent; however, upon binding of substrate, sections of the C-terminal region and the N-terminal region and/or the substrate itself provide a lid for the active site, subsequently blocking off solvent access to the active site [4,23•]. Such closure of the active site cavity during catalysis provides protection for the radical intermediates, including dAdo•, implicated in RS mechanisms. Without such shielding of reaction intermediates, the radicals involved in the catalytic mechanisms of these enzymes might not survive to react with their intended targets due to quenching by solvent. Further, exclusion of solvent would alter the dielectric of the active site, a factor that may be important in modulating the energetics of the catalytic steps [24•].
Similar to other RS enzymes, the [4Fe–4S] cluster in SPL is buried at the top of the barrel, in an environment rich in hydrophobic residues, with SAM bound through the amino and carboxylate groups [18•]. A structure with the dinucleoside 5R-SP bound shows that this substrate binds in proximity to SAM, accompanied by distinct conformational changes that appear to seal off the substrate binding pocket and provide a protected environment for subsequent radical chemistry (Figure 2b) [18••]. The conformational changes occur mainly in the catalytic pocket and the β-hairpin that resides just outside the catalytic pocket. Two residues (Arg304 and Tyr305) in the β-hairpin may help recognize and flip out the SP region from dsDNA through the insertion of the β-hairpin into dsDNA or through interactions with the DNA backbone [18••]. Other residues near the active site change their orientation to allow access of SP to the active site. Another interesting outcome of this structure is the revelation of the structural basis for the previously reported stereospecificity of SPL [20,25], which resides in the steric clashes that would occur when orienting the 5S-SP correctly for H-atom abstraction.
Similar to SPL, the crystal structures of PFL-AE in the presence and absence of a peptide analog of the PFL substrate reveal that binding of the peptide shields the active site from solvent and helps to stabilize and orient SAM in the active site (Figure 2a) [11]. The structures also reveal that binding of substrate induces a large conformational change in loop A of PFL-AE, wherein the loop swings up into the active site and interacts with the substrate to either stabilize and position it for H-atom abstraction or to induce conformational changes in the PFL substrate [11]. Given the evidence for direct H-atom abstraction from G734 of PFL by the dAdo• generated in the PFL-AE active site, together with the observation that G734 is buried in the interior of PFL in its crystal structures [19•,26], the involvement of significant conformational changes for PFL during activation has long been postulated. Recent biochemical and spectroscopic results support such a major conformational change in PFL upon interaction with PFL-AE. Peng et al. showed that in the presence of PFL-AE, PFL favors an open conformation in which the radical domain emerges from its buried position in the interior of PFL [19•]. PFL-AE thus appears to promote a conformational change in PFL that renders its glycyl radical domain accessible for binding in the PFL-AE active site [19•]. The elucidation of the details of this conformational change in PFL, and the detailed mechanism by which PFL-AE promotes this change to allow for glycyl radical formation, awaits further studies.
The SAM–cluster interaction and implications for mechanism
Uncoupled SAM cleavage, in which the dAdo• does not react with substrate but rather is quenched by protein or solvent, is a wasteful and potentially damaging reaction for RS enzymes. One method that RS enzymes use to prevent uncoupled cleavage of SAM is to take advantage of the large difference in redox potential between the [4Fe–4S] cluster and SAM. The reduction potential for SAM is approximately −1.8 V, while the [4Fe–4S] cluster in RS enzymes is only about −450 mV, resulting in a barrier of about 1.4 V or 32 kcal mol−1 [4,27]. Wang and Frey have shown that binding of SAM and substrate to LAM lowers the energy barrier to 9 kcal mol−1, resulting in more favorable conditions for the reductive cleavage of SAM [4,27]. Sulfur K-edge X-ray absorption spectroscopy and density functional theory calculations on PFL-AE have provided evidence for a back-bonding interaction between SAM and the cluster that is increased upon cluster reduction; this interaction is proposed to facilitate electron transfer from the [4Fe–4S]+ cluster to SAM [24•]. Computational results also indicate that reductive cleavage of SAM is sensitive to the dielectric in the active site, and this sensitivity has been proposed to play a role in triggering inner-sphere electron transfer and subsequent SAM cleavage upon substrate binding [24•].
Novel chemistry for radical SAM enzymes
In the last several years, a number of newly characterized RS enzymes have been reported. These RS enzymes carry out novel chemistry and include the C-methyltransferase YtkT involved in the production of the antitumor agent yatakemycin [28], PqqE which is involved in the biosynthesis of pyrroloquinoline quinine (PQQ) [29], HpnP which is involved in the methylation of hopanoids [30], and the methylthiotransferase Cdkal1 [31] which was found to be linked to type 2 diabetes in mice [32]. Other newly identified RS enzymes discussed in more detail below play central roles in antiviral activity [33•,34,35], antibiotic production [36,37•], methylation reactions [12,38•,39,40] and metal cofactor biosynthesis [41–44]. These newly elucidated functions add to the already remarkably diverse chemistry known for the superfamily (see Refs. [2,3,5••,23•]).
Viperin (virus inhibitory protein, endoplasmic reticulum-associated, interferon-inducible) is a mammalian protein that is upregulated in response to viral infections; however, the mechanism for its antiviral activity has yet to be determined [33•,34,35]. The proposed structure of viperin indicates a three-domain protein with a partial (α/β)6 TIM barrel RS domain, a leucine zipper domain possibly for protein folding and anchoring of the protein to the endoplasmic reticulum (ER), and a C-terminal domain which may be involved in substrate recognition or interactions with cofactors [45]. Viperin exhibits enhanced stability upon reconstitution with iron and sulfide [35,46], and UV–vis and electron paramagnetic resonance (EPR) spectroscopic analysis revealed the presence of a [4Fe–4S] cluster typical of the RS superfamily [33•]. Reductive cleavage of SAM was also observed, supporting the hypothesis that viperin is a RS enzyme [33•]. The mode by which RS chemistry aids in the antiviral response, however, is unknown. Viperin has been shown to interact with the enzyme farnesyl pyrophosphate synthase (FPPS) on the cytosolic face of the ER, decreasing its activity and disrupting the formation of lipid rafts, which are involved in budding of a number of viruses including HIV and influenza [33•,47]. While this interaction is an important observation, better understanding of the role of viperin in the antiviral response awaits identification of the reaction(s) it catalyzes.
Recent results demonstrate important roles for RS chemistry in antibiotic biosynthesis. AlbA is a RS enzyme involved in antimicrobial activity; it catalyzes the formation of three thioether bridges on the peptide SboA to produce subtilosin A, a sactibiotic (sulfur-to-α-carbon antibiotic) that has been shown to have antimicrobial activity against bacteria (Figure 3b) [48•,49]. The radical SAM protein NosL is involved in the production of the antibiotic thiopeptide nosiheptide (NOS) [37•]. Thiopeptides are sulfur rich, heterocyclic peptides with a macrocyclic core which includes a nitrogen-containing 6-membered ring central to multiple thiazoles and dehydroamino acids [37••,50]. In most polycylic thiopeptides, the functional side ring formation is independent of the precursor peptide and l-tryptophan provides the variable functional groups [37•]. NosL as well as NocL, which is 78% homologous to NosL and is involved in nocathiacin I (NOC-I) biosynthesis [36], are part of the MIA (3-methyl-2-indolic acid) synthase family that catalyze the conversion of l-tryptophan to MIA through an unusual fragmentation–recombination mechanism (Figure 3C).
Figure 3.

Representative radical SAM reactions. (a) BioB catalyzes the sequential abstraction of hydrogen atoms from C9 and C6 of DTB (left), with insertion of sulfur from the [2Fe–2S] cluster. (b) AlbA incorporates three thioether bonds between three cysteines and the α-C of two phenylalanines and one threonine during the maturation of subtilosin A. Shown is a representative reaction between one cysteine and threonine. (c) NosL and NocL catalyze a fragmentation–recombination reaction of l-Trp to MIA which is incorporated into NOS in the case of NosL or NOC-I in the case of NocL. (d) RlmN and Cfr catalyze the methylation of A2503 of the 23S rRNA utilizing two equivalents of SAM. The sequential methylations catalyzed by these two enzymes can occur in either order.
Methylation of ribosomal RNA (rRNA) usually occurs via an SN2 reaction with the donation of the methyl group from SAM. In the case of the RS enzymes RlmN and Cfr that methylate carbons 2 and 8, respectively, of adenosine 2503 of 23S rRNA, methylation occurs via radical subsequent to transfer of a methyl group to a non-cluster Cys (Figure 3d) [38••,39]. Two equivalents of SAM are needed: one for methylation of the Cys to form mCys, and one for dAdo• production necessary for H-atom abstraction from mCys [38••,39]. In the crystal structure of RlmN, the β7 loop moves to dip into the active site upon the addition of SAM, positioning the catalytic Cys355 closer to the active site for methyl transfer (Figure 2d) [12••]. This Cys355 residue was methylated in the structure, apparently by the first molecule of SAM, suggesting that the second molecule of SAM was bound, awaiting reductive cleavage to initiate H-atom abstraction [12••]. Unlike methylthiotransferases (MTTases) that utilize two [4Fe–4S] clusters for the insertion of a methylthio group into substrate, RlmN and Cfr contain only one [4Fe–4S] cluster bound to the typical RS CX3CX2C motif, and both SAM molecules presumably bind to this same [4Fe–4S] cluster at different steps in the catalytic cycle [12••,38••,39].
Another emerging function for RS enzymes is the biosynthesis of complex metal cofactors including the iron–molybdenum cofactor (FeMo-co) of nitrogenase and the H-cluster of [Fe–Fe]-hydrogenase. NifB is a RS enzyme that inserts the central carbide in an essential step in FeMo-co maturation, with the carbide originating from the methyl group of SAM via novel chemistry [51]. The role of RS chemistry in H-cluster biosynthesis has been partially delineated in recent years [44]. The H-cluster consists of a [4Fe–4S] cluster bridged by a cysteine residue to a 2Fe cluster coordinated by three CO molecules, two CN− ions, and a bridging dithiolate; this cluster is unique to [FeFe]-hydrogenase and is the site where protons are reduced to H2 [42,52,53]. While the [4Fe–4S] cluster portion of the H-cluster is synthesized by the housekeeping Fe–S cluster assembly machinery, the 2Fe subcluster is assembled by three proteins, two of which (HydE and HydG) are RS enzymes [42,43]. HydG has been shown to synthesize the CO and CN− ligands from tyrosine using RS chemistry (Figure 4) [54–56], and HydE is presumed to catalyze formation of the dithiolate ligand, although the substrate and reaction catalyzed remain a mystery. HydG and HydE deliver these synthesized ligands to the GTPase HydF where the 2Fe subcluster is assembled [41,57]. Once constructed, the 2Fe subcluster is transferred to hydrogenase already containing a [4Fe–4S] cluster in order to generate the H-cluster and the active enzyme [52,58,59].
Figure 4.
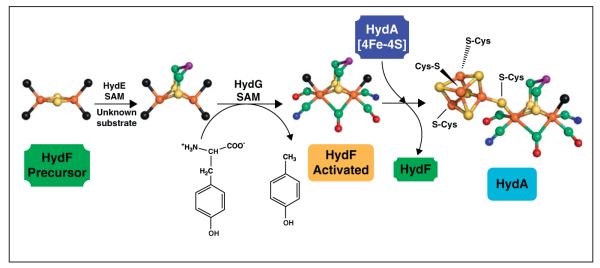
Proposed biosynthesis pathway for HydA H-cluster. HydE uses an unknown substrate to synthesize a dithiolate ligand on a [2Fe–2S] cluster of HydF. HydG catalyzes the decomposition of tyrosine to produce p-cresol, CO, and CN−; the latter two diatomics bind to the H-cluster precursor 2Fe cluster on HydF. HydF then transfers the 2Fe H-cluster precursor to HydA to produce the complete H-cluster and the active hydrogenase. Colors of atoms are as follows: green, carbon; red, oxygen; blue, nitrogen; rust, iron; yellow, sulfur; purple, unknown bridging atom; black, unknown residue coordinating to protein.
Emerging themes in radical SAM chemistry
Multiple Fe–S clusters
All RS enzymes require a [4Fe–4S] cluster in the active site for the binding and reductive cleavage of SAM; however, an emerging theme in the RS field is the presence of a second Fe–S cluster in certain subclasses of RS enzymes. In some cases, the second cluster appears to be a source of sulfur that is inserted into substrate during catalysis, a role first proposed for biotin synthase (BioB) which catalyzes the conversion of DTB to biotin by a sulfur insertion reaction [8,60,61••,62]. BioB contains an additional [2Fe–2S] cluster positioned such that DTB is sandwiched in between SAM and the [2Fe–2S] cluster (Figure 2c) [8]. The [2Fe–2S] cluster was found to undergo reduction concomitant with the formation of a 9-mercaptodethiobiotin (9-MDTB) intermediate; such cluster reduction is consistent with sulfur donation from the cluster thus providing the most recent experimental evidence that the cluster serves as a sulfur source during biotin synthesis (Figure 3a) [60,62]. Lipoate synthase catalyzes a reaction that is quite similar to that of biotin synthase: the insertion of sulfur into an unactivated C–H bond (in this case the C6 and C8 of the octanoyl group) to form the lipoyl cofactor. Lipoate synthase contains two [4Fe–4S] clusters, one of which is the RS active cluster and the other appears to serve as the source of the two sulfides inserted during catalysis [63]. The methylthiotransferases such as RimO [64], MiaB [65], and MtaB [66] also require a sulfur source and contain a second Fe–S cluster; the implication is that the second cluster is the source of the sulfur required in catalysis, but direct biochemical evidence has yet to be reported.
Other potential purposes for auxiliary Fe–S clusters in RS enzymes have also been indicated, including such roles as electron acceptors or anchors for substrates. In the case of BtrN, Grove et al. found that the second [4Fe–4S] cluster cannot be reduced by chemical means, suggesting that this cluster is inaccessible to exogenous reductants [67]. During turnover, however, an EPR signal was observed that is proposed to arise as a result of the second cluster accepting an electron from the RS cluster during catalysis [67]. The second [4Fe–4S] cluster in MoaA binds its substrate 5′-GTP through the N1 of the purine ring to the unique Fe [68]. AlbA also contains a second [4Fe–4S] cluster that is proposed to be an electron acceptor during turnover [48••]. Anaerobic sulfatase-maturating enzymes (anSMEs), on the other hand, contain two additional [4Fe–4S] clusters that are thought to be involved with the reduction of the RS [4Fe–4S] cluster [69]. The anSMEs catalyze the oxidation of cysteine and serine residues of the sulfatase enzymes to produce Cα-formylglycine (FGly) [69]. Mutation studies to knock out the additional clusters resulted in loss of activity and it was proposed that either: first, one of the clusters binds substrate and acts as an electron acceptor while the second cluster transfers an electron from an external electron donor to the RS cluster [70]; or second, in a more recent hypothesis, both clusters act to transfer the electron to the RS cluster and neither cluster coordinates substrate [69].
The [FeFe]-hydrogenase maturation proteins HydE and HydG also contain additional Fe–S clusters. Although spectroscopic studies had indicated the presence of only [4Fe–4S] clusters in HydE, in the crystal structure of HydE there was a [2Fe–2S] cluster in addition to the RS [4Fe–4S] cluster (Figure 2e) [6,9,71]. The [2Fe–2S] cluster of HydE is bound in a site about 20Å from the active site and on the exterior of the barrel separated from the active site by a water-filled cavity, quite different from the [2Fe–2S] cluster in biotin synthase, which was within the barrel and in close proximity to the active site [9]. Furthermore, the cysteine residues that coordinate the [2Fe–2S] cluster in HydE are not conserved across all HydE proteins, suggesting that this second cluster does not play an essential role in catalysis [9]. HydG contains two distinct [4Fe–4S] clusters upon reconstitution with iron and sulfide, and both of these clusters are coordinated by conserved cysteine motifs and have been shown to be essential for HydG activity [55••]. EPR spectroscopic characterization indicates that SAM binds to one of these clusters [55••]. Variants of HydG lacking the ligands for the second cluster were able to produce CN− and p-cresol upon incubation under assay conditions with SAM and tyrosine; however, no CO production was observed, suggesting a role for the second cluster in CO production [53].
Unexpected cysteine motifs
One of the characteristic features of RS enzymes is the cluster-binding CX3CX2C triad motif; however, a number of variations of this cluster-binding motif have now been reported. A CX2CX4C motif was found in 4-amino-5-hydroxymethyl-2-methylpyrimidine phosphate (HMP-P) synthase (ThiC), a RS enzyme that converts 5-aminoimidazole ribonucleotide (AIR) into HMP-P during thiamine biosynthesis [10]. HmdB is a RS enzyme involved in the biosynthesis of the [Fe]-hydrogenase through an undetermined reaction, and exhibits a CX5CX2C motif [72]. Dph2, which is involved in diphthamide biosynthesis, has not been classified as a RS enzyme, although biochemical evidence strongly supports a catalytic mechanism in which a [4Fe–4S] cluster interacts with SAM to generate a 3-amino-3-carboxypropyl (ACP) radical intermediate; such chemistry is clearly analogous to RS reactions [73••,74]. Dph2 exhibits neither the cysteine triad motif nor the typical RS TIM barrel structure (Figure 2f); rather, each of the three cysteines coordinating the Fe–S cluster reside on a separate domain, with over one hundred amino acids separating each cysteine residue [73••,74]. The observation of non-canonical cluster binding motifs in RS enzymes, together with the characterization of RS-like chemistry in a protein that has neither the sequence nor the structural signatures of the superfamily, suggests the likelihood that many additional, as-yet unidentified proteins will ultimately be discovered which catalyze radical reactions using an Fe–S cluster and SAM.
Reductive cleavage of alternate C–S bonds of SAM
RS enzymes have been described as cleaving the S–5′C bond to form methionine and a dAdo• radical intermediate, where dAdo• abstracts a hydrogen from substrate, producing 5′-deoxyadenosine (dAdo) and a substrate radical (Figure 1). Recently, Dph2 and glycerol dehydratase activating enzyme (GDH-AE) were reported to produce alternatively 5′-deoxy-5′-methylthioadenosine (MTA) and a 3-amino-3-carboxypropyl (ACP) radical intermediate, thus implicating the reductive cleavage of an alternate S–C bond of SAM [73••,74,75]. Dph2, as stated previously, is not classified as a RS enzyme but yet it still catalyzes a radical reaction using SAM and an Fe–S cluster [73••]. GDH-AE, on the other hand, is a member of the RS superfamily and is predicted to contain a partial TIM barrel structure similar to PFL-AE [75]. It is unclear why these proteins cleave an alternate S–C bond of SAM although the regioselectivity of the reductive cleavage of SAM has been proposed to be a result of the orientation of SAM with respect to the [4Fe–4S] cluster and substrate [74,76•].
Concluding remarks
The RS superfamily contains an amazing variety of enzymes that carry out diverse and difficult radical reactions that are essential to the metabolic processes in all kingdoms of life. Despite the presence of little sequence homology among superfamily members, these enzymes exhibit a common TIM barrel fold, a common location of a catalytically essential [4Fe–4S] cluster within that barrel, and a mode of binding the [4Fe–4S] cluster through three cysteines to generate a site-differentiated Fe that can be coordinated by SAM. Functional and structural diversity is conferred in some cases by additional domains and by additional Fe–S clusters that can serve a variety of roles in catalysis. New insights into mechanism, as well as newly characterized functions, continue to emerge for this fascinating group of enzymes.
Acknowledgements
Research on RS enzymes in the Broderick Laboratory is supported by the National Institutes of Health (GM54608) and the Department of Energy (DE-FG02-10ER16194). The Astrobiology Biogeocatalysis Research Center is supported by NASA (NNA08C-N85A).
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Sofia HJ, Chen G, Hetzler BG, Reyes-Spindola JF, Miller NE. Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: functional characterization using new analysis and information visualization methods. Nucleic Acids Res. 2001;29:1097–1106. doi: 10.1093/nar/29.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shepard EM, Broderick JB. S-Adenosylmethionine and iron–sulfur clusters in biological radical reactions: the radical SAM superfamily. In: Mander LN, Liu HW, editors. Comprehensive Natural Products. II. Chemistry and Biochemistry. Vol. 8. Elsevier Press; 2010. pp. 625–662. [Google Scholar]
- 3.Frey PA, Hegeman AD, Ruzicka FJ. The radical SAM superfamily. Crit Rev Biochem Mol Biol. 2008;43:63–88. doi: 10.1080/10409230701829169. [DOI] [PubMed] [Google Scholar]
- 4.Duschene KS, Veneziano SE, Silver SC, Broderick JB. Control of radical chemistry in the AdoMet radical enzymes. Curr Opin Chem Biol. 2009;13:74–83. doi: 10.1016/j.cbpa.2009.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5 ••.Vey JL, Drennan CL. Structural insights into radical generation by the radical SAM superfamily. Chem Rev. 2011;111:2487–2506. doi: 10.1021/cr9002616. A review that focuses on the structures of radical SAM enzymes and how each enzyme utilizes their own structural architecture to carry out radical reactions on a wide array of substrates.
- 6.Nicolet Y, Amara P, Mouesca J-M, Fontecilla-Camps JC. Unexpected electron transfer mechanism upon AdoMet cleavage in radical SAM proteins. Proc Natl Acad Sci U S A. 2009;106:14867–14871. doi: 10.1073/pnas.0904385106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hioe J, Zipse H. Radicals in enzymatic catalysis — a thermodynamic perspective. Faraday Discuss. 2010;145:301–313. [Google Scholar]
- 8.Berkovitch F, Nicolet Y, Wan JT, Jarrett JT, Drennan CL. Crystal structure of biotin synthase, an S-adenosylmethionine-dependent radical enzyme. Science. 2004;303:76–79. doi: 10.1126/science.1088493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nicolet Y, Rubach JK, Posewitz MC, Amara P, Mathevon C, Atta M, Fontecave M, Fontecilla-Camps JC. X-ray structure of the [FeFe]-hydrogenase maturase HydE from Thermotoga maritima. J Biol Chem. 2008;283:18861–18872. doi: 10.1074/jbc.M801161200. [DOI] [PubMed] [Google Scholar]
- 10.Chatterjee A, Li Y, Zhang Y, Grove TL, Lee M, Krebs C, Booker SJ, Begley TP, Ealick SE. Reconstitution of ThiC in thiamine pyrimidine biosynthesis expands the radical SAM superfamily. Nat Chem Biol. 2008;4:758–765. doi: 10.1038/nchembio.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vey JL, Yang J, Li M, Broderick WE, Broderick JB, Drennan CL. Structural basis for glycyl radical formation by pyruvate formate-lyase activating enzyme. Proc Natl Acad Sci U S A. 2008;105:16137–16141. doi: 10.1073/pnas.0806640105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12 ••.Boal AK, Grove TL, McLaughlin MI, Yennawar NH, Booker SJ, Rosenzweig AC. Structural basis for methyl transfer by a radical SAM enzyme. Science. 2011;332:1089–1092. doi: 10.1126/science.1205358. The authors reveal the first crystal structure of RlmN where they discovered a single molecule of SAM coordinates of the [4Fe–4S] cluster. The Cys355 residue is methylated and is located proximal to the SAM methyl group which suggests that both equivalents of SAM bind in the same site in the protein during the methylation of rRNA.
- 13.Arragain S, Garcia-Serres R, Blondin G, Douki T, Clemancey M, Latour J-M, Forouhar F, Neely H, Montelione GT, Hunt JF, et al. Post-translational modification of ribosomal proteins: structural and functional characterization of RimO from Thermotoga maritima, a radical S-adenosylmethionine methylthiotransferase. J Biol Chem. 2010;285:5792–5801. doi: 10.1074/jbc.M109.065516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hanzelmann P, Schindelin H. Binding of 5′-GTP to the C-terminal FeS cluster of the radical S-adenosylmethionine enzyme MoaA provides insights into its mechanism. Proc Natl Acad Sci U S A. 2006;103:6829–6834. doi: 10.1073/pnas.0510711103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lepore BW, Ruzicka FJ, Frey PA, Ringe D. The X-ray crystal structure of lysine-2,3-aminomutase from Clostridium subterminale. Proc Natl Acad Sci U S A. 2005;102:13819–13824. doi: 10.1073/pnas.0505726102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Layer G, Moser J, Heinz DW, Jahn D, Schubert W-D. Crystal structure of coproporphyrinogen III oxidase reveals cofactor geometry of radical SAM enzymes. EMBO J. 2003;22:6214–6224. doi: 10.1093/emboj/cdg598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goto-Ito S, Ishii R, Ito T, Shibata R, Fusatomi E, Sekine S-i, Bessho Y, Yokoyama S. Structure of an archaeal TYW1, the enzyme catalyzing the second step of wye-base biosynthesis. Acta Crystallogr Sect D: Biol Crystallogr. 2007;63:1059–1068. doi: 10.1107/S0907444907040668. [DOI] [PubMed] [Google Scholar]
- 18 ••.Benjdia A, Heil K, Barends TRM, Carell T, Schlichting I. Structural insights into recognition and repair of UV-DNA damage by spore photoproduct lyase, a radical SAM enzyme. Nucleic Acids Res. 2012 doi: 10.1093/nar/gks603. http://dx.doi.org/10.1093/nar/gks603. The authors provide the first description of the crystal structure of the radical SAM DNA repair enzyme spore photoproduct lyase, revealing important insights into mechanism.
- 19 •.Peng Y, Veneziano SE, Gillispie GD, Broderick JB. Pyruvate formate-lyase: evidence for an open conformation favored in the presence of its activating enzyme. J Biol Chem. 2010;285:27224–27231. doi: 10.1074/jbc.M109.096875. Using spectroscopic and biochemical approaches, the authors provided evidence that PFL has open and closed conformations that the distribution of the conformations is regulated by the presence of the radical SAM protein PFL-AE.
- 20.Heil K, Kneuttinger AC, Schneider S, Lischke U, Carell T. Crystal structures and repair studies reveal the identity and the base-pairing properties of the UV-induced spore photoproduct DNA lesion. Chem Eur J. 2011;17:9651–9657. doi: 10.1002/chem.201100177. [DOI] [PubMed] [Google Scholar]
- 21.Walsby CJ, Hong W, Broderick WE, Cheek J, Ortillo D, Broderick JB, Hoffman BM. Electron-nuclear double resonance spectroscopic evidence that S-adenosylmethionine binds in contact with the catalytically active [4Fe–4S]+ cluster of pyruvate formate-lyase activating enzyme. J Am Chem Soc. 2002;124:3143–3151. doi: 10.1021/ja012034s. [DOI] [PubMed] [Google Scholar]
- 22.Walsby CJ, Ortillo D, Broderick WE, Broderick JB, Hoffman BM. An anchoring role for FeS clusters: chelation of the amino acid moiety of S-adenosylmethionine to the unique iron site of the [4Fe–4S] cluster of pyruvate formate-lyase activating enzyme. J Am Chem Soc. 2002;124:11270–11271. doi: 10.1021/ja027078v. [DOI] [PubMed] [Google Scholar]
- 23 •.Dowling DP, Vey JL, Croft AK, Drennan CL. Structural diversity in the AdoMet radical enzyme superfamily. Biochim Biophys Acta. 2012;1824:1178–1195. doi: 10.1016/j.bbapap.2012.04.006. This is a recent review of radical SAM enzymes that focuses on the structural aspects of radical SAM enzymes with an analysis of crystal-lographic data as well as biochemical, spectroscopic, and computational studies of specific radical SAM enzymes.
- 24 •.Dey A, Peng Y, Broderick WE, Hedman B, Hodgson KO, Broderick JB, Solomon EI. S K-edge XAS and DFT calculations on SAM dependent pyruvate formate-lyase activating enzyme: nature of interaction between the Fe4S4 cluster and SAM and its role in reactivity. J Am Chem Soc. 2011;133:18656–18662. doi: 10.1021/ja203780t. This paper provides evidence for backbonding interactions between the cluster and SAM that facilitate electron transfer from the cluster to SAM. The results also implicate the importance of the altered dielectric in the active site upon substrate binding as a trigger for the reductive cleavage of SAM.
- 25.Chandra T, Silver SC, Zilinskas E, Shepard EM, Broderick WE, Broderick JB. Spore photoproduct lyase catalyzes specific repair of the 5R but not the 5S spore photoproduct. J Am Chem Soc. 2009;131:2420–2421. doi: 10.1021/ja807375c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Becker A, Fritz-Wolf K, Kabsch W, Knappe J, Schultz S, Volker Wagner AF. Structure and mechanism of the glycyl radical enzyme pyruvate formate-lyase. Nat Struct Biol. 1999;6:969–975. doi: 10.1038/13341. [DOI] [PubMed] [Google Scholar]
- 27.Wang SC, Frey PA. Binding energy in the one-electron reductive cleavage of S-adenosylmethionine in lysine 2,3-aminomutase, a radical SAM enzyme. Biochemistry. 2007;46:12889–12895. doi: 10.1021/bi701745h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang W, Xu H, Li Y, Zhang F, Chen X-Y, He Q-L, Igarashi Y, Tang G-L. Characterization of yatakemycin gene cluster revealing a radical S-adenosylmethionine dependent methyltransferase and highlighting spirocyclopropane biosynthesis. J Am Chem Soc. 2012;134:8831–8840. doi: 10.1021/ja211098r. [DOI] [PubMed] [Google Scholar]
- 29.Wecksler SR, Stoll S, Tran H, Magnusson OT, Wu S-p, King D, Britt RD, Klinman JP. Pyrroloquinoline quinone biogenesis: demonstration that PqqE from Klebsiella pneumoniae is a radical S-adenosyl-l-methionine enzyme. Biochemistry. 2009;48:10151–10161. doi: 10.1021/bi900918b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Welander PV, Coleman ML, Sessions AL, Summons RE, Newman DK. Identification of a methylase required for 2-methylhopanoid production and implications for the interpretation of sedimentary hopanes. Proc Natl Acad Sci U S A. 2010;107:8537–8542. doi: 10.1073/pnas.0912949107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arragain S, Handelman SK, Forouhar F, Wei F-Y, Tomizawa K, Hunt JF, Douki T, Fontecave M, Mulliez E, Atta M. Identification of eukaryotic and prokaryotic methylthiotransferase for biosynthesis of 2-methylthio-N6-threonylcarbamoyladenosine in tRNA. J Biol Chem. 2010;285:28425–28433. doi: 10.1074/jbc.M110.106831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wei F-Y, Suzuki T, Watanabe S, Kimura S, Kaitsuka T, Fujimura A, Matsui H, Atta M, Michiue H, Fontecave M, et al. Deficit of tRNA(Lys) modification by Cdkal1 causes the development of type 2 diabetes in mice. J Clin Invest. 2011;121:3598–3608. doi: 10.1172/JCI58056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33 •.Duschene KS, Broderick JB. The antiviral protein viperin is a radical SAM enzyme. FEBS Lett. 2010;584:1263–1267. doi: 10.1016/j.febslet.2010.02.041. This article provided the first evidence that viperin binds a [4Fe–4S] cluster and is capable of reductively cleaving SAM to produce 5′-deoxyadenosine, classifying it as a radical SAM enzyme.
- 34.Duschene KS, Broderick JB. Viperin: a radical response to viral infection. BioMol Concepts. 2012;3:255–266. doi: 10.1515/bmc-2011-0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haldar S, Paul S, Joshi N, Dasgupta A, Chattopadhyay K. The presence of the iron–sulfur motif is important for the conformational stability of the antiviral protein, viperin. PLoS One. 2012;7:e31797. doi: 10.1371/journal.pone.0031797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Q, Chen D, Lin J, Liao R, Tong W, Xu Z, Liu W. Characterization of NocL involved in thiopeptide nocathiacin I biosynthesis: a [4Fe–4S] cluster and the catalysis of a radical S-adenosylmethionine enzyme. J Biol Chem. 2011;286:21287–21294. doi: 10.1074/jbc.M111.224832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37 ••.Zhang Q, Li Y, Chen D, Yu Y, Duan L, Shen B, Liu W. Radical-mediated enzymatic carbon chain fragmentation–recombination. Nat Chem Biol. 2011;7:154–160. doi: 10.1038/nchembio.512. The authors reported that NosL is a radical SAM enzyme that catalyzes the conversion of l-Trp to MIA through the removal of the Cα-N unit and shifting the carboxylate to the indole ring. NosL works through an unusual fragmentation–recombination reaction that the authors proposed goes through a glycyl radical intermediate.
- 38 ••.Grove TL, Benner JS, Radle MI, Ahlum JH, Landgraf BJ, Krebs C, Booker SJ. A radically different mechanism for S-adenosylmethionine-dependent methyltransferases. Science. 2011;332:604–607. doi: 10.1126/science.1200877. Unlike most SN2 reactions for the methylation of small molecules or macromolecules, the authors found that RlmN and Cfr utilize radical SAM chemistry. These enzymes use a ping–pong mechanism that involves methylation of a conserved Cys residue.
- 39.Grove TL, Radle MI, Krebs C, Booker SJ. Cfr and RlmN contain a single [4Fe–4S] cluster, which directs two distinct reactivities for S-adenosylmethionine: methyl transfer by S(N)2 displacement and radical generation. J Am Chem Soc. 2011;133:19586–19589. doi: 10.1021/ja207327v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yan F, LaMarre JM, Rohrich R, Wiesner J, Jomaa H, Mankin AS, Fujimori DG. RImN and Cfr are radical SAM enzymes involved in methylation of ribosomal RNA. J Am Chem Soc. 2010;132:3953–3964. doi: 10.1021/ja910850y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shepard EM, Boyd ES, Broderick JB, Peters JW. Biosynthesis of complex iron–sulfur enzymes. Curr Opin Chem Biol. 2011;15:319–327. doi: 10.1016/j.cbpa.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 42.Swanson KD, Duffus BR, Beard TE, Peters JW, Broderick JB. Cyanide and carbon monoxide ligand formation in hydrogenase biosynthesis. Eur J Inorg Chem. 2011;2011:935–947. [Google Scholar]
- 43.Duffus BR, Hamilton TL, Shepard EM, Boyd ES, Peters JW, Broderick JB. Radical AdoMet enzymes in complex metal cluster biosynthesis. Biochim Biophys Acta. 2012;1824:1254–1263. doi: 10.1016/j.bbapap.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 44.Peters JW, Broderick JB. Emerging paradigms for complex iron–sulfur cofactor assembly and insertion. Annu Rev Biochem. 2012;81:429–450. doi: 10.1146/annurev-biochem-052610-094911. [DOI] [PubMed] [Google Scholar]
- 45.Jiang D, Guo HT, Xu CX, Chang JH, Gu BH, Wang LJ, Block TM, Guo J-T. Identification of three interferon-inducible cellular enzymes that inhibit the replication of hepatitis C virus. J Virol. 2008;82:1665–1678. doi: 10.1128/JVI.02113-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shaveta G, Shi J, Chow VTK, Song J. Structural characterization reveals that viperin is a radical S-adenosyl-l-methionine (SAM) enzyme. Biochem Biophys Res Commun. 2010;391:1390–1395. doi: 10.1016/j.bbrc.2009.12.070. [DOI] [PubMed] [Google Scholar]
- 47.Wang X, Hinson ER, Cresswell P. The interferon-inducible protein viperin inhibits influenza virus release by perturbing lipid rafts. Cell Host Microbe. 2007;2:96–105. doi: 10.1016/j.chom.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 48 ••.Fluhe L, Knappe TA, Gattner MJ, Schafer A, Burghaus O, Linne U, Marahiel MA. The radical SAM enzyme AlbA catalyzes thioether bond formation in subtilosin A. Nat Chem Biol. 2012;8:350–357. doi: 10.1038/nchembio.798. This article shows that AlbA catalyzes the formation of the three thioether bonds on the peptide SboA to produce subtilosin A through radical SAM chemistry, the first step during subtilosin A production. AlbA contains two clusters: one for SAM cleavage and one for generation of the thioether linkages.
- 49.Zhang Q, Yu Y. Thioether crosslinkages created by a radical SAM enzyme. Chembiochem. 2012;13:1097–1099. doi: 10.1002/cbic.201200196. [DOI] [PubMed] [Google Scholar]
- 50.Yu Y, Duan L, Zhang Q, Liao R, Ding Y, Pan H, Wendt-Pienkowski E, Tang G, Shen B, Liu W. Nosiheptide biosynthesis featuring a unique indole side ring formation on the characteristic thiopeptide framework. ACS Chem Biol. 2009;4:855–864. doi: 10.1021/cb900133x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wiig JA, Hu Y, Lee CC, Ribbe MW. Radical SAM-dependent carbon insertion into the nitrogenase M-cluster. Science. 2012;337:1672–1675. doi: 10.1126/science.1224603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mulder David W, Shepard Eric M, Meuser Jonathan E, Joshi N, King Paul W, Posewitz Matthew C, Broderick Joan B, Peters John W. Insights into [FeFe]-hydrogenase structure, mechanism, and maturation. Structure. 2011;19:1038–1052. doi: 10.1016/j.str.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 53.Nicolet Y, Martin L, Tron C, Fontecilla-Camps JC. A glycyl free radical as the precursor in the synthesis of carbon monoxide and cyanide by the [FeFe]-hydrogenase maturase HydG. FEBS Lett. 2010;584:4197–4202. doi: 10.1016/j.febslet.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 54.Driesener RC, Challand MR, McGlynn SE, Shepard EM, Boyd ES, Broderick JB, Peters JW, Roach PL. [FeFe]-hydrogenase cyanide ligands derived from S-adenosylmethionine-dependent cleavage of tyrosine. Angew Chem Int Ed Engl. 2010;49:1687–1690. doi: 10.1002/anie.200907047. [DOI] [PubMed] [Google Scholar]
- 55 ••.Shepard EM, Duffus BR, McGlynn SE, Challand MR, Swanson KD, Roach PL, Peters JW, Broderick JB. [FeFe]-hydrogenase maturation: HydG-catalyzed synthesis of carbon monoxide. J Am Chem Soc. 2010;132:9247–9249. doi: 10.1021/ja1012273. The authors demonstrated that HydG catalyzes the synthesis of CO from tyrosine.
- 56.Kuchenreuther JM, George SJ, Grady-Smith CS, Cramer SP, Swartz JR. Cell-free H-cluster synthesis and [FeFe] hydrogenase activation: all five CO and CN− ligands derive from tyrosine. PLoS One. 2011;6:e20346. doi: 10.1371/journal.pone.0020346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shepard EM, McGlynn SE, Bueling AL, Grady-Smith C, George SJ, Winslow MA, Cramer SP, Peters JW, Broderick JB. Synthesis of the 2Fe-subcluster of the [FeFe]-hydrogenase H-cluster on the HydF scaffold. Proc Natl Acad Sci U S A. 2010;107:10448–10453. doi: 10.1073/pnas.1001937107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mulder DW, Boyd ES, Sarma R, Lange RK, Endrizzi JA, Broderick JB, Peters JW. Stepwise [FeFe]-hydrogenase H-cluster assembly revealed in the structure of HydA(DeltaEFG) Nature. 2010;465:248–251. doi: 10.1038/nature08993. [DOI] [PubMed] [Google Scholar]
- 59.Mulder DW, Ortillo DO, Gardenghi DJ, Naumov AV, Ruebush SS, Szilagyi RK, Huynh B, Broderick JB, Peters JW. Activation of HydA(Delta EFG) requires a preformed [4Fe–4S] cluster. Biochemistry. 2009;48:6240–6248. doi: 10.1021/bi9000563. [DOI] [PubMed] [Google Scholar]
- 60.Fugate CJ, Jarrett JT. Biotin synthase: insights into radical-mediated carbon–sulfur bond formation. Biochim Biophys Acta. 2012;1824:1213–1222. doi: 10.1016/j.bbapap.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 61 ••.Fugate CJ, Stich TA, Kim EG, Myers WK, Britt RD, Jarrett JT. 9-Mercaptodethiobiotin is generated as a ligand to the [2Fe–2S](+) cluster during the reaction catalyzed by biotin synthase from Escherichia coli. J Am Chem Soc. 2012;134:9042–9045. doi: 10.1021/ja3012963. The authors provide evidence that BioB catalyzes the production of dethiobiotin to biotin through a 9-mercaptodethiobitotin intermediate coordinated to a [2Fe–2S] cluster and that this cluster is the sulfur source for the production of biotin.
- 62.Taylor AM, Stoll S, Britt RD, Jarrett JT. Reduction of the [2Fe–2S] cluster accompanies formation of the intermediate 9-mercaptodethiobiotin in Escherichia coli biotin synthase. Biochemistry. 2011;50:7953–7963. doi: 10.1021/bi201042r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cicchillo RM, Lee K-H, Baleanu-Gogonea C, Nesbitt NM, Krebs C, Booker SJ. Escherichia coli lipoyl synthase binds two distinct [4Fe–4S] clusters per polypeptide. Biochemistry. 2004;43:11770–11781. doi: 10.1021/bi0488505. [DOI] [PubMed] [Google Scholar]
- 64.Lee K-H, Saleh L, Anton BP, Madinger CL, Benner JS, Iwig DF, Roberts RJ, Krebs C, Booker SJ. Characterization of RimO, a new member of the methylthiotransferase subclass of the radical SAM superfamily. Biochemistry. 2009;48:10162–10174. doi: 10.1021/bi900939w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pierrel F, Douki T, Fontecave M, Atta M. MiaB protein is a bifunctional radical-S-adenosylmethionine enzyme involved in thiolation and methylation of tRNA. J Biol Chem. 2004;279:47555–47563. doi: 10.1074/jbc.M408562200. [DOI] [PubMed] [Google Scholar]
- 66.Anton BP, Russell SP, Vertrees J, Kasif S, Raleigh EA, Limbach PA, Roberts RJ. Functional characterization of the YmcB and YqeV tRNA methylthiotransferases of Bacillus subtilis. Nucleic Acids Res. 2010;38:6195–6205. doi: 10.1093/nar/gkq364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Grove TL, Ahlum JH, Sharma P, Krebs C, Booker SJ. A consensus mechanism for radical SAM-dependent dehydrogenation? BtrN contains two [4Fe–4S] clusters. Biochemistry. 2010;49:3783–3785. doi: 10.1021/bi9022126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lees NS, Hanzelmann P, Hernandez HL, Subramanian S, Schindelin H, Johnson MK, Hoffman BM. ENDOR spectroscopy shows that guanine N1 binds to [4Fe–4S] cluster II of the S-adenosylmethionine-dependent enzyme MoaA: mechanistic implications. J Am Chem Soc. 2009;131:9184–9185. doi: 10.1021/ja903978u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Benjdia A, Subramanian S, Leprince J, Vaudry H, Johnson MK, Berteau O. Anaerobic sulfatase-maturating enzyme — a mechanistic link with glycyl radical-activating enzymes? FEBS J. 2010;277:1906–1920. doi: 10.1111/j.1742-4658.2010.07613.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Grove TL, Lee K-h, St Clair J, Krebs C, Booker SJ. In vitro characterization of AtsB, a radical SAM formylglycine-generating enzyme that contains three [4Fe–4S] clusters. Biochemistry. 2008;47:7523–7538. doi: 10.1021/bi8004297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rubach JK, Brazzolotto X, Gaillard J, Fontecave M. Biochemical characterization of the HydE and HydG iron-only hydrogenase maturation enzymes from Thermotoga maritima. FEBS Lett. 2005;579:5055–5060. doi: 10.1016/j.febslet.2005.07.092. [DOI] [PubMed] [Google Scholar]
- 72.McGlynn SE, Boyd ES, Shepard EM, Lange RK, Gerlach R, Broderick JB, Peters JW. Identification and characterization of a novel member of the radical AdoMet enzyme superfamily and implications for the biosynthesis of the Hmd hydrogenase active site cofactor. J Bacteriol. 2010;192:595–598. doi: 10.1128/JB.01125-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73 ••.Zhang Y, Zhu X, Torelli AT, Lee M, Dzikovski B, Koralewski RM, Wang E, Freed J, Krebs C, Ealick SE, et al. Diphthamide biosynthesis requires an organic radical generated by an iron– sulphur enzyme. Nature. 2010;465:891–896. doi: 10.1038/nature09138. Dph2 is not classified as a radical SAM enzyme but contains a [4Fe–4S] cluster that is used to reductively cleave SAM. This article reveals that Dph2 catalyzes the first step of diphthamide biosynthesis through the unusual cleavage of the Cγ-Met–S bond to produce 5′-deoxy-5′-methylthioadenosine and the 3-amino-3-carboxypropyl radical.
- 74.Broderick JB. A radically different enzyme. Nature. 2010;465:877–878. doi: 10.1038/465877a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Demick JM, Lanzilotta WN. Radical SAM activation of the B12-independent glycerol dehydratase results in formation of 5′-deoxy-5′-(methylthio)adenosine and not 5′-deoxyadenosine. Biochemistry. 2011;50:440–442. doi: 10.1021/bi101255e. [DOI] [PubMed] [Google Scholar]
- 76 •.Kampmeier JA. Regioselectivity in the homolytic cleavage of S-adenosylmethionine. Biochemistry. 2010;49:10770–10772. doi: 10.1021/bi101509u. This article explores the differences in the cleavage of SAM by different radical SAM enzymes and suggests that the geometry of the SAM molecule with respect to the [4Fe–4S] cluster determines the cleavage site of SAM.