

Abstract
Sleeping Beauty (SB3) transposon and transposase constitute a DNA plasmid system used for therapeutic human cell genetic engineering. Here we report a comparison of SB100X, a newly developed hyperactive SB transposase, to a previous generation SB11 transposase to achieve stable expression of a CD19-specific chimeric antigen receptor (CAR3) in primary human T cells. The electro-transfer of SB100X expressed from a DNA plasmid or as an introduced mRNA species had superior transposase activity in T cells based on measurement of excision circles released after transposition and emergence of CAR expression on T cells selectively propagated upon CD19+ artificial antigen presenting cells. Given that T cells modified with SB100X and SB11 integrate on average one copy of the CAR transposon in each T-cell genome, the improved transposition mediated by SB100X apparently leads to an augmented founder effect of electroporated T cells with durable integration of CAR. In aggregate, SB100X improves SB transposition in primary human T cells and can be titrated with a SB transposon plasmid to improve the generation of CD19-specific CAR+ T cells.
Keywords: Chimeric antigen receptor, T cells, Sleeping Beauty, transposase, transposon, SB11, SB100X, CD19
Introduction
To overcome immune tolerance, T cells can be genetically modified to express chimeric antigen receptors (CARs) to redirect specificity to tumor-associated antigens (TAA3), such as CD19 (1). These transgenes can be introduced into T cells ex vivo using virus-based vectors and nonviral systems. Viral-based vectors are widely used in research and clinical trials since they provide stable transduction of target cells (2, 3). However, retroviruses' non-random patterns of integration could potentially activate/inactivate oncogenes/tumor suppressor genes leading to autonomous T-cell proliferation (4). Nonviral gene transfer systems based on transposable elements are an alternative approach to introduce desired transgenes. Normally, the target gene is inserted into the genome randomly (5,6). The Sleeping Beauty (SB) transposon system, which inserts at TA dinucleotides repeated arbitrarily across the genome, can be adapted for genetic engineering of T cells (7, 8, 9). This is a result of the stable and efficient integration of an electro-transferred SB transposon by the enzymatic activity of a SB transposase which is typically introduced as a separate DNA plasmid in trans from the DNA plasmid expressing the transposon. SB11 is a hyperactive SB transposase reported to achieve about 100-fold higher integration rates than those achieved by DNA plasmids without transposase activity that use illegitimate recombination to achieve integration (10). Based on the SB11 transposase, we are undertaking a gene therapy clinical trial (IND # 14193) infusing CD19-specific T cells that have been electroporated to introduce SB transposon and transposase to generate CAR+ T cells, which can be selectively propagated on CD19+ artificial antigen presenting cells (aAPC3) (11,12). Using this approach, clinically-sufficient numbers of T cells can be obtained within a few weeks after electroporation of the SB DNA plasmids.
Improvements to the efficiency of transposition may augment our ability to generate CAR+ T cells by reducing the time and associated expense needed to culture clinical-grade genetically modified T cells. Therefore, we investigated the integration efficiency of a CD19-specific CAR transposon, designated CD19RCD28, using a new mutant of SB transposase termed SB100X (13), which had been systematically engineered to have increased enzymatic activity in mammalian cells. Follow-up studies have validated the superiority of SB100X transposase activity in mouse embryonic stem cells and human hematopoietic stem cells (5,14). In preparation for a next-generation trial using the SB system, we compare for the first time the ability of SB11 and SB100X to generate CAR+ T cells from human peripheral blood mononuclear cells (PBMC). Our data reveal that SB100X results in 10 to 100 times more transposition events than SB11 as determined by the excision of CAR transposon from DNA plasmid, which resulted in 3 to 4 times more efficient outgrowth of CD19-specific CAR+ T cells within 28 days after electroporation and with about one copy of CAR transposon per T cell. This apparent increase in enzymatic activity of SB100X is highlighted by our ability to achieve superior outgrowth of CAR+ T cells using just one-tenth the amount of SB100X compared with SB11.
Results
Measuring SB transposition by quantifying excision circle
It has been reported that SB100X results in improved transposition in mouse and human cells (14,19). Therefore, we determined the relative ability of SB100X versus SB11 to mediate a transposition event by Q-PCR adapted to measure DNA fragments (excision circle) (20) that are the expected byproduct produced when CAR transgene (transposon) integrates into T-cell genome, as schematically shown in Figure 1a. The plasmids used to introduce SB transposase in this study are shown in Figure 1b and 1c.
Figure 1. Schematic demonstrating the formation of excision circle and integration of transgene using DNA plasmids from SB system.
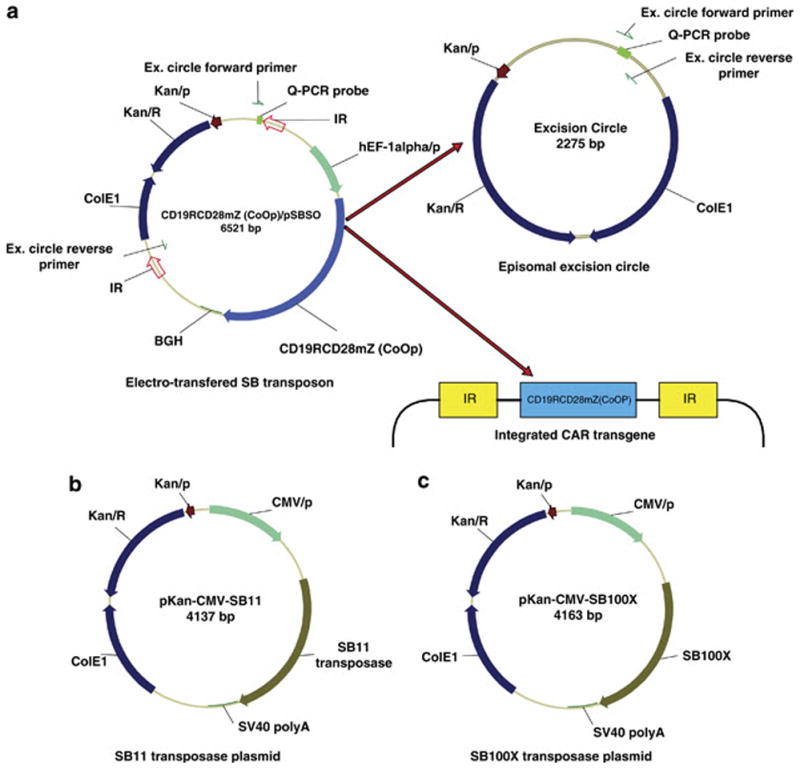
(a) The transgene to be integrated is flanked by two inverted repeats (IR) and mobilized from the CD19RCD28mZ(CoOp)/pSBSO plasmid by a SB transposase. Upon SB transposition, the CAR transposon (CD19RCD28) is inserted into the T-cell genome while the nonintegrated DNA forms an episomal excision circle. The PCR to detect a released excision circle reveals a 77 base pair band, whereas the same primers bound to sites in the CD19RCD28mZ(CoOp)/pSBSO plasmid are 4,298 base pairs apart. (b) Schematic of DNA plasmids expressing SB11 transposase and (c) SB100X transposase.
Abbreviations: BGH: Bovine Growth Hormone polyadenylation signal sequence; CDS: Coding Sequence of gene; CMV/p: Cytomegalovirus promoter; ColE1: Colicin E1 (origin of replication); hEF-1α/p: human elongation factor-1 alpha hybrid promoter; IR: Inverted Repeats; Kan/R: Kanamycin Resistance gene; Kan/P: Kanamycin resistance gene promoter; SV40 poly A: Simian Virus 40 polyadenylation signal sequence.
SB100X transposase improves frequency of transposition
To directly assess the ability of SB100X to improve the frequency of transposition compared with SB11 transposase, we serially measured the formation of non-integrated excision circles by real-time Q-PCR after electroporation of T cells from PBMC. The accumulation of excision circles represents the enzymatic activities of the two SB transposases while measurement of the electro-transferred SB transposon and subsequent expression of CD19RCD28 CAR indicates the overall efficiency of the nonviral gene transfer process. To account for variations in electro-transfer efficiency, we normalized the excision circle data to the amount of DNA transposon recovered after electroporation (excision circle to CD19RCD28 ratio). This allowed us to evaluate the individual SB transposase's enzymatic activities and adjust for possible difference in electroporation efficacy between different samples. Measurement of RPPH1, a subunit of RNase P (21), which is present at one copy on chromosome 14q11.2, was used as internal control in real-time Q-PCR for both quantification of excision circles and transposon DNA species. By varying the relative amounts of DNA plasmids coding for the transposases, SB100X reached peak activity at a concentration of 5 μg/electroporation (Figure 2b). However, at 10 μg/electroporation, SB100X resulted in significant loss of PBMC viability (data not shown). SB11 also showed maximal activity at 5 μg/electroporation (Figure 2a), but the amount of excision circles produced was significantly less than that achieved with SB100X. Overall, SB100X was 10 to 100 times more active compared to SB11. As expected the episomal excision circles are lost to detection as the T cells propagate on the aAPC. To avoid the possibility that the SB transposases could integrate, we assessed whether improved transposition could also be achieved by mRNA coding for SB100X. The two transposases were electro-transferred as mRNA species along with a fixed amount of SB DNA transposon into T cells activated by OKT3. We observed that the introduction of SB100X mRNA at 0.1 μg/electroporation was as active as 10X the amount of SB11. When SB100X mRNA was used at 0.25 μg/electroporation there was a higher transposition activity compared with SB11 at 1 μg/electroporation (Figure 2c). These data again indicate the superior activity of SB100X in primary T cells whether this transposase is expressed from electro-transferred DNA or mRNA.
Figure 2. Evaluation of transposase activity by detecting excision circle formation after SB transposition event.

Excision circle to CD19RCD28 ratio is based upon amount of (a) SB11 and (b) SB100X transposase as detected by Q-PCR at days 1 to 3 after electroporation. The DNA transposon plasmid (CD19RCD28mZ(CoOp)/pSBSO) to express CD19RCD28 CAR was used at 15 μg/electroporation. (c) Excision circle to CD19RCD28 ratio after electro-transfer of mRNA coding for SB100X and SB11, combined with SB DNA transposon plasmid, on day 1 after electro-transfer.
Generation of CAR+ T cells by Sleeping Beauty transposition
Primary human T cells from PBMC were electro-transferred on day 0 of cell culture with SB transposon (CD19RCD28mZ(CoOp)/pSBSO) and one of the two DNA plasmids, pKAN-CMV-SB11 and pKAN-CMV-SB100X, coding for SB11 or SB100X, respectively. The T cells were subsequently propagated for up to 28 days on γ-irradiated CD19+ aAPC added every 7 days in presence of soluble recombinant IL-2 cytokine (Figure 3a). Our initial approach to generating clinical-grade CAR+ T cells uses 5 μg of pKAN-CMV-SB11 along with 15 μg of CD19RCD28mZ(CoOp)/pSBSO in the electroporation of 2X107 PBMC, therefore, this was used as a starting point to assess the ability of SB100X to improve the rate of transposition and subsequent outgrowth of CAR+ T cells. However, when we electroporated T cells with the DNA plasmid coding for SB100X at 5 μg/electroporation with 15 μg/electroporation of the DNA plasmid coding for CAR transposon, this accentuated cell death the day after electro-transfer as demonstrated by Trypan blue staining and failure to propagate CAR+ T cells. The electro-transfer of DNA plasmid coding for SB100X at lower amounts did not compromise cell viability as the genetically modified T cells could be propagated on aAPC. Indeed, upon reducing the concentration of the DNA plasmid coding for this transposase to 0.1 μg/electroporation and 0.5 μg/electroporation, SB100X successfully integrated the transposon to support the outgrowth of CAR+ T cells. When using 10-fold less (0.5 μg/electroporation) than the input concentration of SB11 (5 μg/electroporation), we calculate that SB100X was about 3.6 times more efficient than SB11 in generating CAR+ cells as assessed at day 28 of co-culture with CD19+ aAPC. Indeed, even 0.1 μg/electroporation of the DNA plasmid coding for SB100X was almost as efficient as 5 μg/electroporation of SB11 when the number of CAR+ T cells were counted at 28 days of tissue culture (Figure 3b and 3c). Expression of the CAR on electroporated and propagated T cells was documented by flow cytometry (Figure 3d). Thus, transposition mediated by SB100X results in improved outgrowth of CAR+ T cells.
Figure 3. Selective outgrowth of CD19-specifc CAR+ T cells after transposition with SB100X versus SB11 transposases.
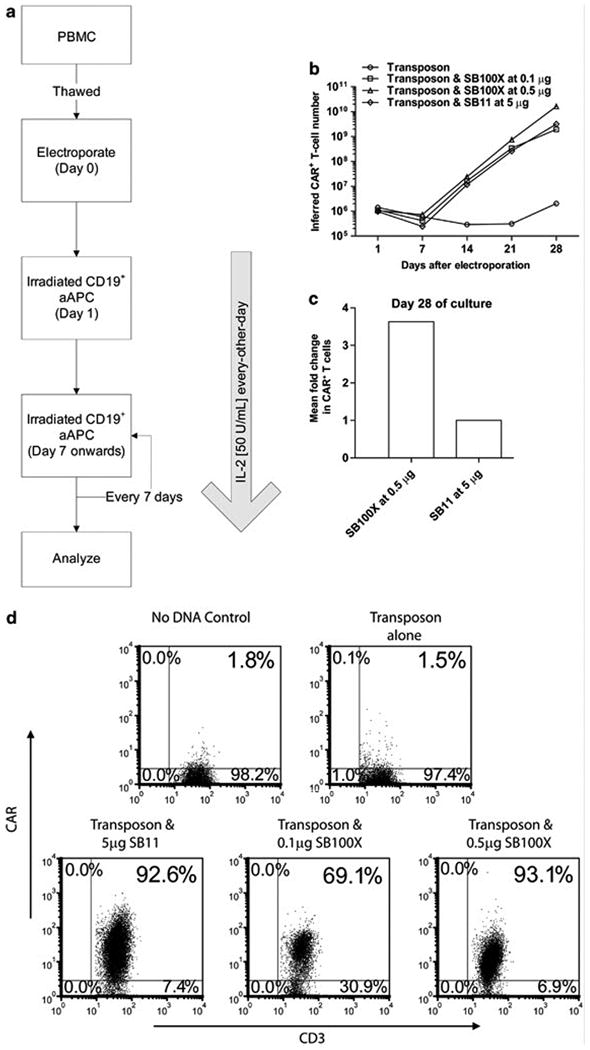
(a) Schematic outlining co-culture process to generate CD19-specifc CAR+ T cells. 105 CAR+ T cells from PBMC were stimulated with γ-irradiated CD19+ aAPC (clone #4) every 7 days at a 1:2 (CAR+ : aAPC) ratio in presence of soluble IL-2. (b) Kinetics of CAR+ T-cell numeric expansion by repetitive co-culture with CD19+ aAPC. T cells were electroporated on Day 0 with SB DNA plasmid transposon expressing CD19RCD28 and graded doses of DNA plasmids expressing SB100X or SB11. (c) Fold change in number of CAR+ T cells at day 28 of co-culture on aAPC from two independent experiments. (d) Expression of CAR (CD19RCD28) on CD3+ T cells by flow cytometry at day 28 of culture.
CAR+ T cells can be generated using SB100X mRNA
Since SB transposase coded by mRNA species was capable of accomplishing SB transposition, we determined if CAR+ T cells could be selectively propagated on aAPC after electro-transfer of DNA plasmid coding for CD19RCD28 and mRNA coding for SB100X or SB11. We adapted our propagation method to generate CAR+ T cells per Figure 4a so that T cells were pre-activated with OKT3 to improve uptake of and expression from mRNA. As shown in Figure 4b, 0.25 μg/electroporation of SB100X and 1 μg/electroporation of SB11 successfully produced CAR+ T cells that could be propagated on CD19+ aAPC. The superiority of the SB100X transposase to support the outgrowth of CAR+ T cells was apparently not due to differences in integrity of the mRNA (Figure 4c).
Figure 4. CAR+ T cells generated by electro-transfer of mRNA coding for SB transposases and DNA plasmid coding for CD19RCD28 CAR transposon.
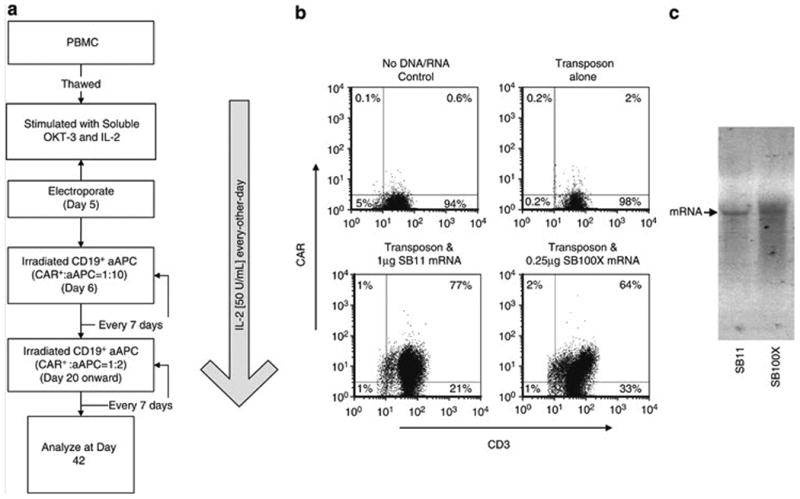
When using transposase mRNA to generate CAR+ T cells, the SB100X mRNA was used at 0.1 μg/electroporation, 0.25 μg/electroporation, and 1 μg/electroporation while the SB11 mRNA concentration was at 1 μg/electroporation. The DNA plasmid expressing Cd19RCD28 was used at 10 μg/electroporation. (a) Schematic used to generate CAR+ T cells. (b) Expression of CAR on numerically expanded CD3+ T cells at day 42 of co-culture with aAPC. (c) Integrity of mRNA after in vitro transcription used to express SB11 and SB100X.
Transposition using SB100X and SB11 result in comparable number of integration events per T cell
Given that SB100X gives rise to greater a greater number of transposition events compared with SB11, we investigated whether this enzyme resulted in multiple integration events. To evaluate the number of integration events per electroporated and propagated T cell, we measured the copy number of CD19RCD28 transpose relative to the copy number of RNase P gene by real-time Q-PCR. The calculated transgene copy per T cell is 0.95 ± 0.068 gene (Mean ± Standard Error Mean, SEM) for 0.5 μg/electroporation SB100X and 0.80 ± 0.033 (Mean ± SEM) for 5 μg/electroporation SB11, respectively (Figure 5a). This difference is not statistically significant (P=0.1846). These data revealed that the number of integrated copies of CAR is approximately 1 per T-cell genome upon transposition with both SB100X and SB11.
Figure 5. Measurement of number of copies of integrated CAR transgene and detection of SB transposases.
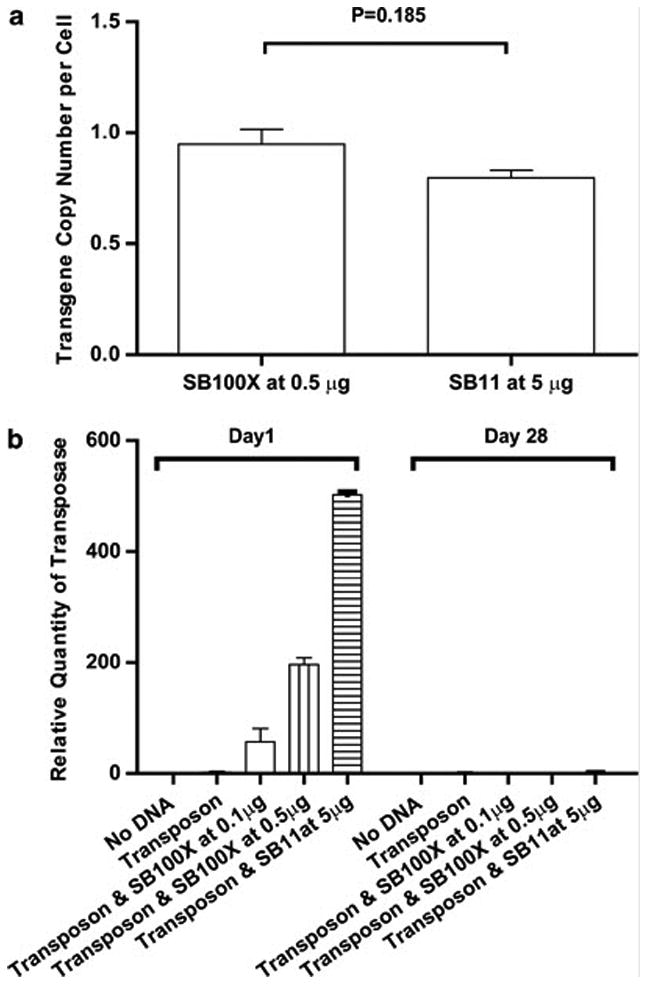
(a) Copy number of CAR transgene, normalized to RNase P, at day 28 of co-culture on CD19+ aAPC after SB transposition with SB100X or SB11. Q-PCR using CAR-specific primers revealed CD19RCD28 copy numbers at 0.95 and 0.80 transgene per T cell after electro-transfer of 0.5 μg DNA plasmid coding for SB100X and 5 μg DNA plasmid coding for SB11, respectively. There was no statistical difference in the copy number of integrated CAR transgenes. (b) Measurement by Q-PCR using transposase-specific primers at Day 1 (day after electroporation) and at Day 28 of co-culture of genetically modified T cells on CD19+ aAPC. Transposon was added at 15 μg/electroporation along with graded doses of DNA coding for SB100X and SB11 transposases.
SB100X DNA cannot be detected in electroporated and propagated T cells
To establish that SB100X or SB11 DNA is not present in propagated T cells, we developed a Q-PCR assay to reveal integrated transposase plasmid. Our data demonstrated that after 28 days of in vitro culture, the SB100X transposase, as well as that of SB11, are absent in the cultured cells (Figure 5b).
CAR+ T cells generated by SB transposition with SB100X exhibit redirected killings for CD19+ tumor cells
The electroporated and propagated T cells generated using SB11 and SB100X transposases were evaluated for their ability to be activated for effector functioning in a CAR-dependent manner. We demonstrated by chromium release assay (CRA) that both sets of T cells exhibited redirected killings for CD19+ target (Figure 6a and 6b).
Figure 6. Killing of tumor cells by genetically modified T cells.

Lysis by CRA of CD19+ tumor cells (Daudi, NAML-6 and genetically modified U251T) compared with CD19neg parental U251T cells by T cells genetically modified with (a) SB11 and (b) SB100X generated from T cells electroporated with DNA plasmids.
Discussion
We and others have used transposon and transposase systems to improve integration efficiency of DNA plasmids expressing immunoreceptors (8,9,22,23,17). Building upon these data, we have adapted the SB system for human application (11) to use the SB11 transposase to integrate CAR into TA dinucleotide repeats across the genomes of populations of human T cells.
The development of SB100X raised the possibility that we could use this new SB transposase to improve the integration frequency of CAR transposon in T cells. We were able to demonstrate that SB100X had up to 100 times higher rates of transposition compared to SB11. This is attributed to improved enzymatic activity, but the improved transposition efficiency may also be due to different translational efficiencies difference as well as post-translational modifications. The efficiency of transposition was dependent on the amount of electro-transferred transposase as concentrations of 5 to 10 μg/electroporation of SB100X led to cell death. The reasons for this toxicity are not known. However, it is known that overexpression of transposase can lead to inhibition of SB transposition (6,16). This highlights the need to titrate the input concentrations between the transposon and the transposase. By reducing the amount of SB100X relative to SB11, we were able to demonstrate its superior activity.
The ability of SB100X to augment the selective propagation of CAR+ T cells on CD19+ aAPC raised the possibility that this transposase led to the insertion of multiple copies of the transposon per cell. Indeed, multiple integrations have been observed after SB100X-mediated transposition in other cells (6,13). However, this was disproved when we found that on average, there was approximately 1 copy of the CAR transgene per T cell which was similar to the integration efficacy with SB11. The lower number of integrants per genome compared with the published reports of SB100X activity may be due to an intrinsic property of the T-cell genome, genotoxicity leading to loss of viability of T cells carrying multiple copies of the transposon, and/or a reflection of the selective pressure provided by the aAPC to selectively propagate CAR+ T cells bearing just one copy of the transposon.
That SB100X has intrinsically improved transposase activity is revealed by the release and detection of more excision circles when SB100X was used compared with SB11. Presumably, the release of the excision circles from a population of electroporated T cells is correlated with the stable integration of the CAR transgene, which is measured per T cell. Thus, it appears that the superior enzymatic activity of SB100X results in an improved efficiency of transposon integration and a beneficial founder effect leading to the subsequent improved outgrowth of CAR+ T cells upon in vitro propagation on aAPC. This finding pertains to the safety of SB100X for, as with SB11, it does not lead to multiple insertions of the CAR transgene when T cells are electroporated and co-cultured on aAPC.
The increased enzymatic activity of SB100X was also evident when we could reduce the amount of transposase DNA plasmid needed to accomplish the integration of CAR into T cells. Indeed, we could achieve superior numeric expansion of CAR+ T cells on CD19+ aAPC using 0.5 μg/electroporation of SB100X compared with 10 times as much SB11. This also has implications for improved safety of SB system in human trials as a decrease in the amount of SB transposase delivered by DNA plasmid presumably decreases the chance of integration of the transposase into the genome and the potential for re-mobilization of the inserted transposon. When we evaluated for the presence of integrated plasmid expressing SB100X, we did not detect a signal by Q-PCR which curtails the possibility of a re-hopping event after SB mediated transposition. However, it is possible that DNA for SB transposase were present at a level below limit of detection. To exclude the possibility that SB100X can integrate into the T-cell genome we demonstrated that electro-transfer of mRNA species coding for this integrase could mediate transposition and further, that the efficiency of integration was again higher than SB11. Previously, it has been shown that SB11 transposase coding by mRNA can mediate transposition (24). Furthermore, viral vectors have been used to deliver SB transposase to improve the pattern of integration of an integrase-deficient lentivirus to achieve a more random pattern of integration than can be achieved with lentiviral integrase (25, 26).
We are currently undertaking the first clinical application of the SB system, which has successfully received an Investigational New Drug Application from the Food and Drug Administration, to electrotransfer the SB11 transposase to express CD19RCD28 transposon in autologous T cells for infusion in patients with B-lineage lymphoma. With the development of SB100X, our data demonstrate the SB100X may be a desirable alternative transposase to the use of SB11 for use in clinical trials to adoptively transfer CAR+ T cells.
Materials and Methods
Plasmids
The SB transposon contains the codon optimized (CoOp) second-generation CD19RCD28 CAR, specific for human CD19, flanked by the SB inverted repeats. This CAR has been previously described (15). In brief, the ampicillin resistance gene (AmpR) and origin of replication from the plasmid CoOpCD19RCD28/pT-MNDU3 was replaced with a DNA fragment encoding the kanamycin resistance gene (KanR) and origin of replication (ColE1) from the pEK Vector (8). The human elongation factor-1α (hEF-1α) promoter from pVitro4 vector (InvivoGen, San Diego, CA) was swapped with MNDU3 promoter to generate the DAN plasmid CD19RCD28mZ(CoOp)/pSBSO (Figure 1a). The pKan-CMV-SB11 DNA plasmid (Figure 1b), coding for the SB11 transposase was constructed by digesting pCMV-SB11(kindly provided by Dr. Perry Hackett, University of Minnesota) (16) with PvuII, harvesting the fragment coding for SB11 transposase and ligating to the AseI and PacI fragment, which contained the Kanamycin-resistant gene and ColE1 origin of replication from pEK vector. pKan-CMV-SB100X (Figure 1c), coding for SB100X transposase was built by digesting both the pKan-CMV-SB11 plasmid and pCMV(CAT)T7-SB100X plasmid (13) with AvaI and PsiI and then annealing the fragment coding for SB100X with the backbone of pKan-CMV-SB11 plasmid. All DNA plasmids were purified using Qiagen (Valencia, CA) endotoxin-free reagents. The integrity of the DNA plasmids coding SB100X and SB11 transposase was assessed by Experion automatic electrophoresis station (Bio-Rad, CA).
Cells
After obtaining consent, PBMC from healthy donors were isolated by density gradient centrifugation over Ficoll-Paque-Plus (Pharmacia Biotech, Pistacaway, NJ) and stored in liquid nitrogen. K562 were transduced with lentivirus to express CD64, CD86, CD137L and membrane-bound IL-15 and were cloned (clone #4, kindly provided by Dr. Carl June, University of Pennsylvania). The construction of CD19+-K562 aAPC was previously reported (17). The CD19+ Daudi, (catalog number CCL-213; ATCC, VA) and NALM-6 (catalog number ACC128; DSMZ, Germany) were cultured in RPMI 1640 with 10% FCS. U251T glioblastoma cell line (a gift from Dr. Waldemar Debinski, Wake Forest University, NC) was genetically modified with the ΔCD19/pSBSO vector and stable transfectants expressed truncated CD19 (18). Both, parental CD19neg U251T and CD19+ U251T cells were cultured in Dulbecco's modified Eagle's medium (Hyclone, Logan, UT), supplemented with 2 mM Glutamax-1 (Gibco-Invitrogen, Carlsbad, CA) and 10% heat-inactivated fetal calf serum (FCS).
Generation of CAR+ T cells by transposition with SB DNA plasmids coding for CAR and transposases
On day 0, PBMC were thawed at 37°C, washed once with phenol-free RPMI 1640 and rested for 2 hours at 37°C. 2x107 PBMC/cuvette were resuspended in Human T Cell Nucleofector buffer (Lonza Inc., Basel, Switzerland) along with 15 μg CD19RCD28mZ(CoOp)/pSBSO plasmid coding for CD19RCD28 CAR and graded doses of DNA plasmids coding for SB100X or SB11. After electroporation with Nucleofector II (Lonza Inc.) using program U14, the cells were washed once with phenol-free RPMI 1640 and resuspended in 1 mL of phenol free RPMI 1640 (without FCS) and cultured in a 12-well plate at 37°C for 4 hours. Then 1 mL of phenol-free RPMI 1640 with 20% of FCS was added. The next day, 3X105 cells were harvested for DNA extraction and immunophenotyping. The remaining cells were stimulated by 1:2 (CAR+T cells : aAPC) weekly addition of γ-irradiated (100 Gy) CD19+ aAPC (clone #4) for 28 days of co-culture. 50 IU/mL recombinant human interleukin 2 (rhIL-2; Chiron) was added every-other-day beginning at Day 1.
In vitro transcription of SB100X or SB11 mRNA
20 μg of the SB transposase plasmids were digested with SpeI and purified with Qiaquick gel extraction kit (Qiagen, CA) and the DNA concentration was measured. 10 μg of the linearized plasmid DNA was used to synthesize mRNA with T7 RiboMAX Express Large Scale RNA Production System (Promega, WI). A PolyA tail was added to the newly synthesized mRNA molecule with Poly(A) Tailing Kit (Ambion,TX). After quantification the mRNA was analyzed by gel electrophoresis, aliquoted, and stored in Nalgene cryogenic vials (Thermo Fisher Scientific, MA) at -80°C for future use.
Generation of CAR+ T cells by transposition with SB DNA plasmid coding CAR and mRNA coding for transposases
PBMC were resuspended in RPMI 1640 with 5% heat-inactivated AB human serum (Invitrogene, CA) and 50 ng/mL of OKT3 monoclonal antibody (eBioscience, CA) and 50 U/mL of IL-2, which was re-added to the culture every-other-day. After 5 days, 107 cells/cuvette were electroporated using 10 μg DNA plasmid (CD19RCD28mZ(CoOp)/pSBSO) coding for CD19RCD28 and graded amounts of SB transposase mRNA. The propagation of the T cells was achieved using 1:10 (CAR+ T cells to aAPC) ratio of γ-irradiated CD19+ aAPC for first 2 weeks and 1:2 ratio thereafter. aAPC clone #4 were added every 7 days and IL-2 every-other-day beginning at day 1.
Flow cytometry
Cells first incubated with 1:200 diluted APC-labeled goat anti-human IgG Fc (Cat. #12-0569-42, Jackson ImmunoResearch Laboratories Inc, PA) were then washed once and stained with 1:50 anti-CD3-FITC (Cat. #349201, BD Biosciences, CA) plus 1:50 anti-CD56-PE (Cat. # 12-0569, eBiosciences, CA). After staining, all cells were resuspended in 100 μL FACS buffer and live/dead cells were differentiated upon the addition of Propidium Iodide (PI, Catalog number P4864; Sigma-Aldrich, St. Louis, MO) then analyzed with flow cytometry (FACSCalibur, BD Biosciences, CA).
Real-time Quantitative PCR
All of the primers, probes, and TaqMan Gene Expression Master Mix were purchased from Applied Biosystem (Foster City, CA). The Q-PCR reactions were performed in a Steponeplus Real-time PCR system (Applied Biosystem, CA) with TaqMan Real-time Quantitative PCR technique as recommended. To measure excision circles, the forward primer (5′-TCCCAGTCACGACGTTGTAAAA-3′) and probe (5′-CCAGTGAATTCGAGCTC-3′) bound 5′ of the CAR cassette and the reverse primer (5′-CGTTGGCCGATTCATTATCG-3′) bound 3′ of the CAR cassette in the SB transposon DNA plasmid CD19RCD28mZ(CoOp)/pSBSO as illustrated in Figure 1a. A positive PCR reaction reveals a 77 base pair band that is generated only after the CAR transposon was excised. To measure CD19RCD28 CAR, the primer sequences are: forward primer 5′-CAGCGACGGCAGCTTCTT-3′; reverse primer 5′-TGCATCACGGAGCTAAA-3′; probe 5′-AGAGCCGGTGGCAGG-3′. To measure SB transposases, a common primer and probe set were designed to target the plasmid backbone shared by pKan-CMV-SB100X and pKan-CMV-SB11. These sequences are: forward primer 5′-AAGGCCAGGAACCGTAAAAAG-3′; reverse primer 5′-GGCGGAGCCTATGGAAAAA-3′; and probe 5′-CCGCGTTGCTGGC-3′. RNase P primer and probe set of TaqMan RNase P Control Reagents Kit (Cat. #4316844, Applied Biosystem, CA) were used as Q-PCR internal control.
Analysis of Q-PCR results
An RNase P CT vs. Cell number standard curve (Curve A, Figure 1s a) was achieved by a serial dilution (200 ng, 20 ng and 2 ng) of genomic DNA from a genetically modified Jurkat cell clone bearing one copy of CAR transgene based on Southern blotting (Maiti et al., in preparation). Since one cell has about 7 pg of DNA, 200 ng total DNA is equivalent to approximately 28,570 cells. CT represents the threshold circle where the Q-PCR was deemed positive. With Curve A we calculated the cell number from corresponding RNase P CT value. In parallel, a CD19RCD28 transgene CT vs. transgene number curve (Curve B, Figure 1s b) was generated to compute the number of integrated transgenes. Twenty nanogram of sample genomic DNA from genetically modified and propagated primary T cells could then be analyzed by Q-PCR. The RNase P CT was used to calculate cell numbers with Curve A and the integrated transgene copy number was deduced from transgene CT with Curve B. The copy numbers per cell were calculated with the following formula:
TgN is the number of integrated transgenes. Cell N represents the cell number deduced from RNase P CT. Quantification of excision circle was achieved with comparative CT method provided by the Steponeplus realtime PCR system (Applied Biosystem).
Chromium Release Assay
The redirected specificity of the genetically modified T cells was determined by chromium release assay (CRA) using 51Cr-labeled Daudi, NALM-6 and U251T cells as targets. The T cells (effectors) were harvested 28 days following stimulation with aAPC, washed, and plated in V-bottom microtiter plates (Costar, Cambridge, MA) in triplicate at 105, 5 × 104, 2.5 × 104, 1.25 × 104 cells/well and with 5 × 103 target cells. After incubation at 37°C for 4 hours followed by centrifugation, 50 μL aliquots of cell-free supernatant were harvested and counted with Topcount NXT (PerkinElmer, MA). The percent of specific cytolysis was calculated from the release of 51Cr as follows:
Control wells contained target cells incubated in media. The maximal 51Cr was determined by measuring the 51Cr content released by target cells lysed with 1% Triton100.
Statistical analysis
The Student t test was employed to determine statistical significance and p < 0.05 was considered significant. Where applicable the data are reported as an average + standard deviation (SD). Electroporations were performed on two different donors while Q-PCR reactions were carried out in triplicate.
Supplementary Material
Figure 1s. Curves used in calculation of transgene copy number per cell. The Q-PCR reactions of standards for curve and samples are running in the same 96-plate. R2 is the Coefficient of determination. (a) Curve A: Cell number versus RNase P threshold circle value. (b) Curve B: Transgene number versus actual CD19RCD28 threshold circle value.
Acknowledgments
We thank Dr. Perry Hackett University of Minnesota for Sleeping Beauty system and Dr. Carl June from University of Pennsylvania for assistance generating the K562-aAPC. We are grateful to the Flow Cytometry Core Laboratory at MDACC.
Footnotes
Supported by CCSG Grant (CA16672), RO1 (CA124782, CA120956, CA141303), R21 (CA116127), DOD (PR064229), The Alex's Lemonade Stand Foundation, The Burroughs Wellcome Fund, The Gillson Longenbaugh Foundation, The Ladies Leukemia League, The Leukemia and Lymphoma Society, The Lymphoma Research Foundation, The Miller Foundation, The National Foundation for Cancer Research, The Pediatric Cancer Research Foundation, The Scales Family, The William Lawrence and Blanche Hughes Foundation.
Abbreviations used in this paper: SB, Sleeping Beauty; CAR, chimeric antigen receptor; TAA, tumor-associated antigen; aAPC, artificial antigen presentation cell.
Conflict of Interest: The authors have no competing financial interests in relation to the work described.
References
- 1.Numbenjapon T, Serrano LM, Chang WC, Forman SJ, Jensen MC, Cooper LJ. Antigen-independent and antigen-dependent methods to numerically expand CD19-specific CD8+ T cells. Exp Hematol. 2005;35(7):1083–1090. doi: 10.1016/j.exphem.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 2.Tan PH, Tan PL, George AJ, Chan CL. Gene therapy for transplantation with viral vectors--how much of the promise has been realised? Expert Opin Biol Ther. 2006;6:759–772. doi: 10.1517/14712598.6.8.759. [DOI] [PubMed] [Google Scholar]
- 3.Ciuffi A. Mechanisms governing lentivirus integration site selection. Curr Gene Ther. 2008;8:419–429. doi: 10.2174/156652308786848021. [DOI] [PubMed] [Google Scholar]
- 4.Kaiser J. Gene therapy. Seeking the cause of induced leukemias in X-SCID trial. Science. 2003;299:495. doi: 10.1126/science.299.5606.495. [DOI] [PubMed] [Google Scholar]
- 5.Liang Q, Kong J, Stalker J, Bradley A. Chromosomal mobilization and reintegration of Sleeping Beauty and piggyBac transposons. Genesis. 2009;47:404–408. doi: 10.1002/dvg.20508. [DOI] [PubMed] [Google Scholar]
- 6.Grabundzija I, Irgang M, Mátés L, Belay E, Matrai J, Gogol-Döring A, Ivics Z, et al. Comparative analysis of transposable element vector systems in human cells. Mol Ther. 2010;18:1200–1209. doi: 10.1038/mt.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang X, Wilber AC, Bao L, Tuong D, Tolar J, Zhou X, et al. Stable gene transfer and expression in human primary T cells by the Sleeping Beauty transposon system. Blood. 2006;107:483–491. doi: 10.1182/blood-2005-05-2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singh H, Manuri PR, Olivares S, Dara N, Dawson MJ, Cooper LJ, et al. Redirecting specificity of T-cell populations for CD19 using the Sleeping Beauty system. Cancer Res. 2008;68:2961–2971. doi: 10.1158/0008-5472.CAN-07-5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peng PD, Cohen CJ, Yang S, Hsu C, Jones S, Morgan RA, et al. Efficient nonviral Sleeping Beauty transposon-based TCR gene transfer to peripheral blood lymphocytes confers antigen-specific antitumor reactivity. Gene Ther. 2009;16:1042–1049. doi: 10.1038/gt.2009.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Geurts AM, Yang Y, Clark KJ, Liu G, Cui Z, Hackett PB, et al. Gene transfer into genomes of human cells by the Sleeping Beauty transposon system. Mol Ther. 2003;8:108–117. doi: 10.1016/s1525-0016(03)00099-6. [DOI] [PubMed] [Google Scholar]
- 11.Williams DA. Sleeping Beauty vector system moves toward human trials in the United States. Mol Ther. 2008;16:1515–1516. doi: 10.1038/mt.2008.169. [DOI] [PubMed] [Google Scholar]
- 12.Hackett PB, Largaespada DA, Cooper LJ. A transposon and transposase system for human application. Mol Ther. 2010;18:674–683. doi: 10.1038/mt.2010.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mátés L, Chuah MK, Belay E, Jerchow B, Manoj N, Izsvák Z, et al. Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nat Genet. 2009;41:753–761. doi: 10.1038/ng.343. [DOI] [PubMed] [Google Scholar]
- 14.Xue X, Huang X, Nodland SE, Mátés L, Ma L, Zhou X, et al. Stable gene transfer and expression in cord blood-derived CD34+ hematopoietic stem and progenitor cells by a hyperactive Sleeping Beauty transposon system. Blood. 2009;114:1319–1330. doi: 10.1182/blood-2009-03-210005. [DOI] [PubMed] [Google Scholar]
- 15.Kowolik CM, Topp MS, Gonzalez S, Pfeiffer T, Cooper LJ, et al. CD28 costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Res. 2006;66:10995–11004. doi: 10.1158/0008-5472.CAN-06-0160. [DOI] [PubMed] [Google Scholar]
- 16.Geurts AM, Yang Y, Clark KJ, Liu G, Cui Z, Hackett PB, et al. Gene transfer into genomes of human cells by the Sleeping Beauty transposon system. Mol Ther. 2003;8:108–117. doi: 10.1016/s1525-0016(03)00099-6. [DOI] [PubMed] [Google Scholar]
- 17.Manuri PV, Wilson MH, Maiti SN, Mi T, Singh H, Cooper LJ, et al. piggyBac transposon/transposase system to generate CD19-specific T cells for treatment of B-lineage malignancies. Hum Gene Ther. 2010;21:427–437. doi: 10.1089/hum.2009.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Serrano LM, Pfeiffer T, Olivares S, Numbenjapon T, Bennitt J, Cooper LJ, et al. Differentiation of naive cord-blood T cells into CD19-specific cytolytic effectors for posttransplantation adoptive immunotherapy. Blood. 2006;107:2643–2652. doi: 10.1182/blood-2005-09-3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Izsvák Z, Chuah MK, Vandendriessche T, Ivics Z. Efficient stable gene transfer into human cells by the Sleeping Beauty transposon vectors. Methods. 2009;49:287–297. doi: 10.1016/j.ymeth.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 20.Liu G, Aronovich EL, Cui Z, Whitley CB, Hackett PB. Excision of Sleeping Beauty transposons: parameters and applications to gene therapy. J Gene Med. 2004;6:574–583. doi: 10.1002/jgm.486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Szilagyi A, Blasko B, Szilassy D, Fust G, Sasvari-Szekely M, Ronai Z. Real-time PCR quantification of human complement C4A and C4B genes. BMC Genet. 2006;7:1. doi: 10.1186/1471-2156-7-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakazawa Y, Huye LE, Dotti G, Foster AE, Vera JF, Wilson MH, et al. Optimization of the PiggyBac transposon system for the sustained genetic modification of human T lymphocytes. J Immunother. 2009;32:826–836. doi: 10.1097/CJI.0b013e3181ad762b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang X, Guo H, Kang J, Choi S, Zhou TC, Zhou X, et al. Sleeping Beauty transposon-mediated engineering of human primary T cells for therapy of CD19+ lymphoid malignancies. Mol Ther. 2008;16:580–589. doi: 10.1038/sj.mt.6300404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilber A, Frandsen JL, Geurts JL, Largaespada DA, Hackett PB, McIvor RS. RNA as a source of transposase for Sleeping Beauty-mediated gene insertion and expression in somatic cells and tissues. Mol Ther. 2006;13:625–630. doi: 10.1016/j.ymthe.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 25.Staunstrup NH, Moldt B, Mátés L, Villesen P, Jakobsen M, Ivics Z, Mikkelsen JG, et al. Hybrid lentivirus-transposon vectors with a random integration profile in human cells. Mol Ther. 2009;17:1205–1214. doi: 10.1038/mt.2009.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vink CA, Gaspar HB, Gabriel R, Schmidt M, McIvor RS, Qasim W, et al. Sleeping beauty transposition from nonintegrating lentivirus. Mol Ther. 2009;17:1197–1204. doi: 10.1038/mt.2009.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure 1s. Curves used in calculation of transgene copy number per cell. The Q-PCR reactions of standards for curve and samples are running in the same 96-plate. R2 is the Coefficient of determination. (a) Curve A: Cell number versus RNase P threshold circle value. (b) Curve B: Transgene number versus actual CD19RCD28 threshold circle value.