

Abstract
Dental caries is a common infectious disease associated with acidogenic and aciduric bacteria, including Streptococcus mutans. Organisms that cause cavities form recalcitrant biofilms, generate acids from dietary sugars and tolerate acid end products. It has recently been recognized that micro-organisms can produce functional amyloids that are integral to biofilm development. We now show that the S. mutans cell-surface-localized adhesin P1 (antigen I/II, PAc) is an amyloid-forming protein. This conclusion is based on the defining properties of amyloids, including binding by the amyloidophilic dyes Congo red (CR) and Thioflavin T (ThT), visualization of amyloid fibres by transmission electron microscopy and the green birefringent properties of CR-stained protein aggregates when viewed under cross-polarized light. We provide evidence that amyloid is present in human dental plaque and is produced by both laboratory strains and clinical isolates of S. mutans. We provide further evidence that amyloid formation is not limited to P1, since bacterial colonies without this adhesin demonstrate residual green birefringence. However, S. mutans lacking sortase, the transpeptidase enzyme that mediates the covalent linkage of its substrates to the cell-wall peptidoglycan, including P1 and five other proteins, is not birefringent when stained with CR and does not form biofilms. Biofilm formation is inhibited when S. mutans is cultured in the presence of known inhibitors of amyloid fibrillization, including CR, Thioflavin S and epigallocatechin-3-gallate, which also inhibited ThT uptake by S. mutans extracellular proteins. Taken together, these results indicate that S. mutans is an amyloid-forming organism and suggest that amyloidogenesis contributes to biofilm formation by this oral microbe.
Introduction
Amyloid represents a fibrous cross β-sheet quaternary structure comprised of ordered aggregates of peptides or proteins that demonstrate common biophysical properties (Nilsson, 2004). While amyloid formation has been extensively studied in the context of pathological states, for example Alzheimer’s disease, the concept of functional amyloid has only recently emerged (Epstein & Chapman, 2008; Fowler et al., 2007; Gebbink et al., 2005; Maury, 2009a; Otzen & Nielsen, 2008). In bacteria, amyloid formation may be the rule rather than the exception (Otzen & Nielsen, 2008). There are now numerous examples of surface-localized microbial proteins that aggregate and assemble into functional amyloid fibrils.
Amyloid fibres have a tensile strength comparable to steel (Smith et al., 2006). The unique physical and morphological properties of amyloid and the fact that it can be formed in a regulated manner suggest that it probably represents a common quaternary structure that is widespread in biology. Therefore, it does not always represent a misfolded protein structure, but instead represents a low-energy quaternary structure that can occur in the context of function or disease (Fowler et al., 2007). Amyloid is a non-covalent oligomer of extended intermolecular hydrogen-bonded β-sheets that self-assemble to form fibres ~5–13 nm in diameter, ranging in length from tens of nanometers to micrometers. Amyloidogenic proteins can be large or small, structural or catalytic, abundant or sparse (Fowler et al., 2007). The cross β-sheet structure appears to be highly conserved and to represent a primitive structure that can be generated by many polypeptide sequences. It has been hypothesized that amyloid has existed for as long as proteins have (Chernoff, 2004) and that under extreme earth conditions ~3.9 billion years ago, β-sheet molecular structures were the first self-propagating and information-processing biomolecules (Maury, 2009b).
Streptococcus mutans is an organism that is well adapted to a biofilm lifestyle. Research has now demonstrated a high prevalence of amyloids within natural biofilms, and confirmed the production of extracellular amyloid by environmental isolates within the phyla Bacteriodetes, Firmicutes and Actinobacteria and within the class Gammaproteobacteria (Larsen et al., 2008, 2007). The list of examples of well-characterized, amyloid-forming proteins/systems is growing. Escherichia coli, Salmonella typhimurium and other members of the family Enterobacteriaceae produce aggregative surface fibres called curli or tafi (Barnhart & Chapman, 2006; Chapman et al., 2002; Römling et al., 1998) that are involved in adhesion and biofilm formation (Saldaña et al., 2009; Zogaj et al., 2003). Known amyloid-forming pathogens now also include Mycobacterium tuberculosis (Alteri et al., 2007), Klebsiella pneumoniae (Bieler et al., 2005), Staphylococcus aureus (Schwartz et al., 2012) and Candida albicans (Garcia et al., 2011; Otoo et al., 2008; Ramsook et al., 2010; Rauceo et al., 2004). Plant pathogens include members of the genera Xanthomonas, Erwinia and Pseudomonas (Epstein & Chapman, 2008). The hydrophobins expressed by most fungi are amyloidogenic and are involved in fungal coat formation and the modulation of adhesion and surface tension (Butko et al., 2001; de Vocht et al., 2000; Kwan et al., 2006; Mackay et al., 2001). The chaplins, secreted hydrophobic proteins produced by the soil organism Streptomyces coelicolor, are also amyloidogenic (Sawyer et al., 2011). Bacillus subtilis produces matrix-encased, agar-surface and floating biofilms that contain both amyloid and exopolysaccharide (Romero et al., 2010), with an accessory protein now implicated in the assembly of the amyloid fibres and their linkage to the cell wall (Romero et al., 2011). The Pseudomonas fluorescens group also produces amyloid that can confer a biofilm phenotype (Dueholm et al., 2010).
The adhesin P1 of S. mutans (molecular mass ~185 kDa) exists in a fibrillar layer on the cell surface (Ayakawa et al., 1987) and contributes to the organism’s cariogenicity (Crowley et al., 1999). It mediates interactions with salivary constituents, host matrix proteins and other oral bacteria (Brady et al., 2010). The protein has repetitive discontinuous alanine (A)- and proline (P)-rich tandem repeats that flank a variable (V)-region (Brady et al., 1991). P1 is targeted and attached to the cell wall by the sortase transpeptidase (Ton-That et al., 2004) and has an unusual tertiary structure (Larson et al., 2010). The A-region adopts a long α-helix that intertwines with the P-repeat polyproline II helix to form a long narrow stalk. The intervening V region comprises a β-sandwich arranged in two sheets (Troffer-Charlier et al., 2002), and the C terminus of P1 has three structurally related β-sandwich domains (Larson et al., 2011; Nylander et al., 2011). Hence, there are two globular domains within P1 comprised of β-sheet structure and that lie on either end of an extended stalk. The propensity of P1 to aggregate during handling and the identification of amyloid-promoting sequences by multiple computational algorithmic programs led us to examine P1 further for amyloid-like properties. Our current studies show that P1 is amyloidogenic, that additional S. mutans proteins also demonstrate this property, that amyloidogenic proteins are produced by S. mutans laboratory strains and clinical isolates, that amyloid is detectable in human dental plaque and that S. mutans biofilm formation is inhibited by known inhibitors of amyloid fibrillization.
Methods
Bacterial strains.
S. mutans strains used in this study included NG8, its isogenic mutant PC3370 that is devoid of the spaP gene encoding P1 (Crowley et al., 1999) and clinical isolates 3svf1, 1svf1, 5ST1 and 81D2, which were kindly provided by Dr Robert Burne, University of Florida. A sortase-negative strain was generated by allelic exchange mutagenesis. Primers used are listed in Table S1, available in the online version of this paper. A DNA fragment encompassing srtA plus 5′ and 3′ flanking sequences was amplified by PCR using genomic DNA from NG8 as the template. The erythromycin gene (ermAM) with a ribosome-binding site was amplified by PCR from the plasmid template pVA838 (Macrina et al., 1982), with restriction sites NheI and StuI incorporated in the primers. Each fragment was ligated to pCR2.1-TOPO T/A (Life Technologies), followed by restriction digestion and directional cloning to replace srtA with ermAM. The resulting plasmid was linearized, gel-purifed and used to naturally transform S. mutans NG8. Transformants were selected on 5 µg erythromycin ml−1. Integrants were confirmed by PCR.
Collection of human dental plaque.
Whole-pooled supragingival human dental plaque was collected from all smooth surfaces of the dentition using sterile periodontal curettes. Plaque was transferred to and dispersed in sterile microcentrifuge tubes containing 10 mM sodium phosphate buffer, pH 7.0, and was immediately transported on ice to the laboratory for analysis. Samples were collected under the auspices of the University of Florida Institutional Review Board (Protocol no. 665-2008). S. mutans was isolated from plaque by streaking onto selective media, TSY20B (Schaeken et al., 1986), containing 20 % sucrose and bacitracin (0.1 unit ml−1) in a trypticase-soy and yeast extract agar base with incubation for 48 h at 37 °C in an anaerobic GasPak jar (BBL). Raised, rough colonies with glucan puddles were isolated and confirmed by using the API 20 Strep (bioMérieux) system and PCR (Sato et al., 2003).
Expression and purification of P1 polypeptides.
Full-length P1 (CG14) was expressed as a recombinant, six histidine (his)-tagged protein (Brady et al., 1998). Subclones encoding P1 fragments were generated by PCR amplification of spaP DNA, using S. mutans NG8 genomic DNA (A3VP1, aa 386–874) or plasmid pDC20 (Brady et al., 1998) (P3C, aa 921–1486; NA1, aa 39–308) as the template. Primers used to generate the truncated polypeptides are listed in Table S1. A3VP1 was expressed as a six his-tagged polypeptide with pET30(c) (EMD Millipore) as the vector and E. coli TOP10 (Life Technologies) as the host. P3C and NA1 were dual-tagged with a cleavable maltose-binding protein (MBP) on the N terminus and a non-cleavable six-his tag on the C terminus, and they were expressed using pMAL-p2X (New England BioLabs) as the vector and E. coli TOP10 as the host. Recombinant polypeptides were purified using TALON metal affinity resin (Clontech) and amylose affinity resin (New England BioLabs) chromatography, as appropriate, according to the manufacturer’s instructions. NA1 and P3C fusion proteins were cleaved with Factor Xa (New England BioLabs) to remove the MBP moiety. A stable breakdown product of P3C that retains the C-terminal his-tag was present during purification and was determined by N-terminal sequencing to begin at aa 1000. This corresponds to the C terminus of P1, characterized by X-ray crystallography (Larson et al., 2011), and it was therefore included in our analyses, in addition to P3C. The homogeneity of each purified P1 polypeptide was confirmed by SDS-PAGE and Western blotting. Protein concentrations were determined by using the bicinchoninic acid assay with BSA as the standard. Fig. S1 is a schematic representation of the polypeptides utilized in this study.
Preparation of S. mutans extracellular proteins.
S. mutans strain NG8 and its spaP- and srtA-negative derivatives were grown in a chemically defined medium (Palmer et al., 2012) and cultured overnight at 37 °C. Cells were removed by centrifugation at 10 000 g for 30 min and the culture supernatant was filtered through a 0.2 µm Nalgene filter. Proteins present in the culture supernatant were harvested by ammonium sulfate precipitation (60 % saturation), followed by extensive dialysis in PBS, pH 7.2.
Congo red (CR) and Thioflavin T (ThT) binding.
Two hundred microlitres of full-length P1 or fragments thereof (5 µM) in distilled water was stirred at 4 °C for 2 days in an Eppendorf tube on a Fisher Scientific Isotemp stir plate at the highest setting, using a 10×2.5 mm micro stir bar and these served as the aggregated samples. Unstirred samples were also analysed. CR (0.7 mg ml−1 in 5 mM potassium phosphate, 150 mM NaCl, pH 7.4; Sigma-Aldrich) was filtered through a 0.2 µm syringe filter prior to use and spectra were obtained between 400 and 700 nm with an AVIV spectrophotometer (Lakewood). Ten microlitres of stirred or non-stirred protein was added, mixed well and incubated at room temperature for 30 min before the spectra were obtained. BSA (Sigma-Aldrich) served as a negative control. A solution without CR was used as a blank. In the ThT-binding assay a stock 8 mg ml−1 ThT phosphate solution (10 mM phosphate, 150 mM NaCl, pH 7.0) was filtered through a 0.2 µm filter. Samples contained 14 mM ThT and 5 µM P1 polypeptide or BSA in a quartz cuvette with 3 mm excitation and emission path lengths. The fluorescence intensity of ThT was measured at 25 °C with excitation at 442 nm. Emission intensities were collected from 460 to 600 nm with a 1 s integration time by a Photon Technology International Fluorimeter equipped with an LPS-220B lamp power supply, MD-5020 motor drive, BryteBox and water bath. The absorbance at the excitation wavelength was less than 0.1 to prevent inner filter effects. Inhibition of amyloid fibrillization by the green tea polyphenol epigallocatechin-3-gallate (EGCG) was also evaluated by measuring the ThT fluorescence spectrum. S. mutans NG8 extracellular proteins were stirred at 4 °C for 48 h with 1 mM, 100 µM, 10 µM or 1 µM EGCG (Sigma-Aldrich; catalogue no. E4143) dissolved in sterile PBS solution. A protein sample stirred in the absence of EGCG served as the positive control. Following stirring in the cold, ThT was added to a final concentration of 2 µM. Samples were mixed and stored in the dark for 30 min at room temperature and then excited at 442 nm. Fluorescence emission spectra were measured from 450 to 700 nm in a quartz cuvette with a 0.2 cm path length, using a Shimadzu RF-5301 PC Spectrofluorophotometer. A ThT-only sample served as the negative control for background fluorescence. The assay was performed twice, with triplicate samples in each assay. Statistically significant differences between samples with and without inhibitor were determined by Student’s t-test.
Transmission electron microscopy (TEM).
One microlitre of sonicated, stirred full-length P1 (2 mg ml−1) was placed on a carbon-coated 400-mesh nickel grid and the excess solution was dried using filter paper. The dried sample was stained with 2 % uranyl acetate, excess liquid was removed and the sample was air-dried at room temperature. The images were examined using an Hitachi H-700 electron microscope at ×50 000 magnification.
Detection of green birefringence.
CR solution was prepared by dissolving 2 g NaCl in 20 ml deionized H2O and by suspending 0.5 g CR in 80 ml 100 % ethanol; these two solutions were combined, filtered and stored at room temperature as a stock solution. To test amyloid formation by bacterial cells growing on a surface, an S. mutans colony was scraped off a Todd–Hewitt agar plate that had been incubated overnight at 37 °C and held at 4 °C ≥48 h prior to the removal of colonies. An E. coli strain expressing curli (MC4100) and a temperature-sensitive, curli-negative mutant (LSR11, MC4100 :: ΔcsgBACcsgDEFG) (Epstein et al., 2009) (kindly provided by Matthew Chapman, University of Michigan) were grown for 48 h at 26 °C on YESCA agar and they served as positive and negative controls, respectively. Bacterial colonies were suspended in 15 µl deionized H2O on a cleaned microscope slide, then mixed with 5 µl CR solution. Human dental plaque samples were treated in a similar manner as the cells that were scraped from a plate. Control slides were prepared with CR solution only and with bacterial suspensions without CR. To test amyloid formation by proteins, either purified P1 polypeptides or S. mutans extracellular protein preparations were adjusted to ~0.5 mg ml−1 in 50 mM NaH2PO4, 300 mM NaCl, pH 8.0 and stirred for 48 h at 4 °C to induce aggregation. BSA was used as a negative control. Ten microlitres of protein solution was mixed with 5 µl, 1 : 50 dilution, CR stock solution and visualized as wet mounts. Birefringence was evaluated under polarized light with an inverted, computer-controlled Leica DMIRB microscope equipped with a motorized XY stage (Piezo Physic Instrument, crystal focus − 100 micron maximum travel). Wet mounts were viewed with a ×63 Leica plan-apo oil, 1.32 NA, objective with transmitted light, without the neutral density filter or the Walliston prism in place. The field diaphragm was two thirds closed and the polarizer was moved at a 90° angle to observe the birefringence of the samples. Matching images were viewed with white light only, without polarization.
A defining property of amyloid fibrils is resistance to proteinase K digestion and disaggregation of fibrils by formic acid. Aggregated S. mutans extracellular proteins were treated with formic acid (88 %, certified aldehyde free; Fisher Scientific), according to the protocol indicated on the Abcam website for the formic acid extraction of insoluble amyloid beta from mouse brain (http://www.abcam.com/index.html?pageconfig=resource&rid=10730), or proteolysed. Aggregated proteins were also treated with proteinase K (Worthington Biochemical Corporation) as described previously for E. coli curli by Shewmaker et al. (2009), prior to staining with CR as described above.
Inhibition of biofilm formation.
Mid-exponential phase cultures of S. mutans strains were grown in Todd–Hewitt broth with 0.3 % yeast extract, and 100 µl washed cell suspension was adjusted to OD600 0.1 in chemically defined medium containing 0.5 % glucose (Bouvet et al., 1981), with or without an inhibitor and this mixture was used to inoculate wells of a 96-well microtitre plate. Compounds were tested for interference of S. mutans biofilm formation at concentrations known to interfere with fibrillization of disease-associated amyloids and included CR (10 µM) (Heiser et al., 2000; Kuner et al., 2000), Thioflavin S (ThS) (100 µM) (Sharp et al., 2009) and EGCG (1 mM) (DaSilva et al., 2010; Ehrnhoefer et al., 2008; Grelle et al., 2011; Lopez del Amo et al., 2012). The CR stock solution was prepared as described above, under the detection of green birefringence. ThS and EGCG stocks were prepared as described above, under CR and ThT uptake. Corresponding diluents were used as controls for the non-inhibitor comparisons. After incubation for 48 h at 37 °C in an anaerobic chamber, wells were washed and biofilms were stained with 50 µl 0.1 % crystal violet solution for 15 min. After washing, plates were dried and the optical density was read at OD595 in an iMark microplate reader (Bio-Rad Laboratories). Eight replicates were performed for each experimental condition. Statistically significant differences for each strain, with and without inhibitor, were determined by Student’s t-test. To visualize the green birefringence of S. mutans biofilm-grown cells, 50 µl CR solution was used to resuspend the contents of two biofilm wells for each strain and then 10 µl was transferred to a clean glass slide and imaged.
Results
Uptake of amyloidophilic dyes and visualization of fibrillization by the S. mutans adhesin P1
Amyloid formation typically follows a time profile that includes a lag phase, during which there is no detectable amyloid. The lag phase represents the time necessary to build up a fibril nucleus that, once formed, acts as a template for the deposition of additional protein molecules to form the mature fibril (Harper & Lansbury, 1997). In in vitro assays, fibrillization is routinely triggered by mechanical agitation to increase the probability of the intermolecular collisions necessary to form the initial nucleus. We used two assays to assess the potential amyloid formation by S. mutans proteins. First, to test if P1 is amyloidogenic, the binding of CR to P1 polypeptides that had been stirred in the cold for 48 h to initiate fibrillization was evaluated (Fig. 1a). As stated earlier, two regions within P1 contain significant β structure. One is within the region A3VP1 (Larson et al., 2010) and the other is in the C terminus region (Larson et al., 2011). Compared with CR alone, the spectra of the stirred CG14, A3VP1, P3C and C terminus samples demonstrated dye binding and a shift in the CR absorbance maxima that is characteristic of amyloid fibrils (Klunk et al., 1999). In comparison, the spectra of the BSA-negative control and NA1 samples were more similar to that of CR alone. The N-terminal P1 polypeptide NA1 is not predicted to have a β structure and would therefore not be expected to be amyloidogenic. When unstirred samples were evaluated (Fig. 1b), there was no evidence of CR binding to A3VP1; however, the C-terminal polypeptides still showed evidence of dye binding and a spectral shift. This suggests differences in the nucleation events required for amyloid formation by these two amyloidogenic regions within P1. In the case of unstirred samples, the spectra of CG14, which contains both amyloidogenic domains, were similar to those of P3C and the C terminus.
Fig. 1.
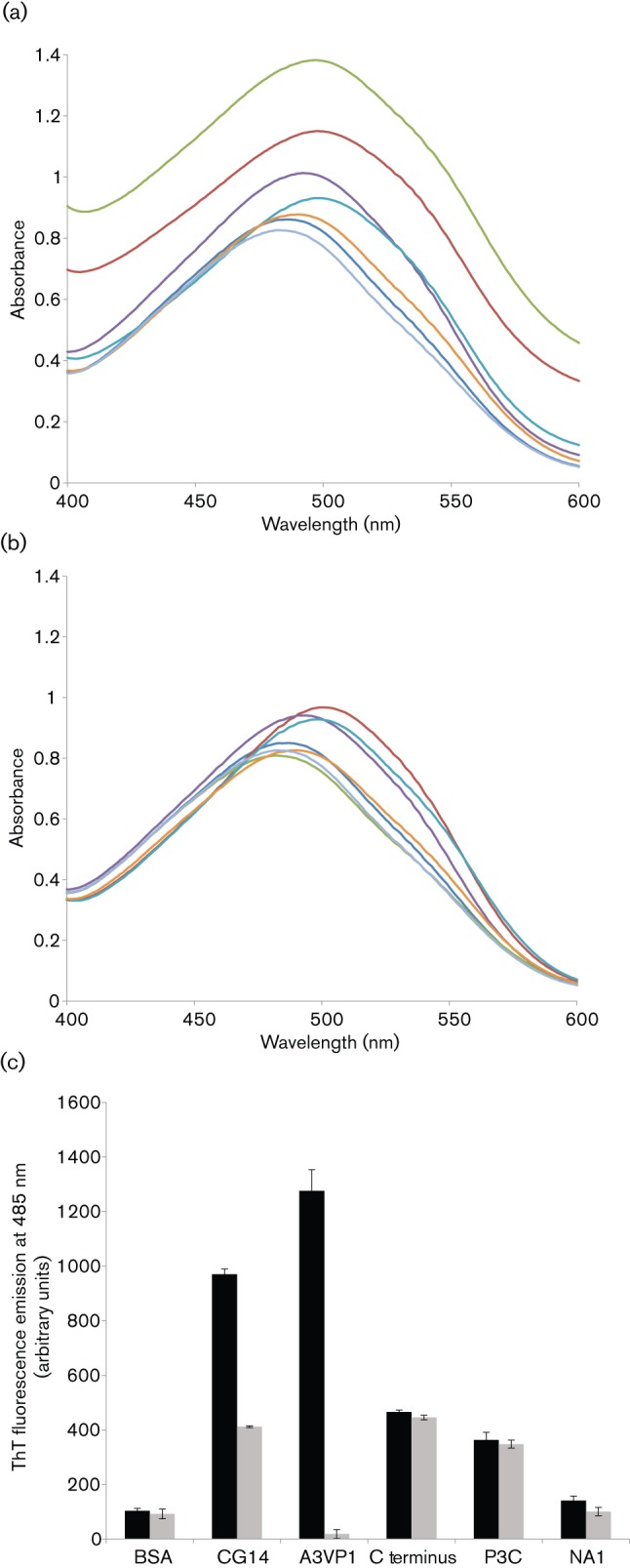
Binding of amyloidophilic dyes to purified recombinant P1 polypeptides. All samples were tested at 5 µM. (a) Absorbance spectra of CR alone and in the presence of aggregated (stirred) samples. (b) Absorbance spectra of CR alone and in the presence of unstirred samples. Dark blue, BSA; red, CG14; green, A3VP1; purple, C terminus; aqua, P3C; orange, NA1; light blue, CR alone. (c) ThT-dependent fluorescence of stirred and unstirred samples. ThT fluorescence emission at 485 nm, following excitation at 442 nm, is shown. P1 alone did not contribute to the fluorescence signal. No change in ThT fluorescence intensity was observed in the absence of P1. Black bars, stirred; grey bars, not stirred. Error bars are sem.
The ability of full-length P1 and the truncated β structure-containing fragments to interact with CR led us to test the polypeptides for ThT-dependent fluorescence, a more specific indicator of amyloid fibrillization (Nilsson, 2004). A pronounced shift in fluorescence intensity was observed for the stirred CG14, A3VP1, P3C and C terminus polypeptides (Fig. 1c). In addition, similar to the CR assay, ThT binding was also observed in the unstirred samples for those polypeptides that contained the C terminus. There was no background fluorescence associated with P1 in the absence of ThT, and no spectral shift in ThT fluorescence was observed in the absence of P1. Again, minimal dye binding was observed for the BSA and NA1 negative controls.
In light of the positive results in both the CR and ThT assays, we confirmed by using TEM that P1 forms amyloid fibrils (Fig. 2). A mesh of amyloid fibrils was clearly visible (shown in three fields) and was similar in appearance to published TEM images of other bacterial amyloids (Romero et al., 2010; Wang & Chapman, 2008).
Fig. 2.
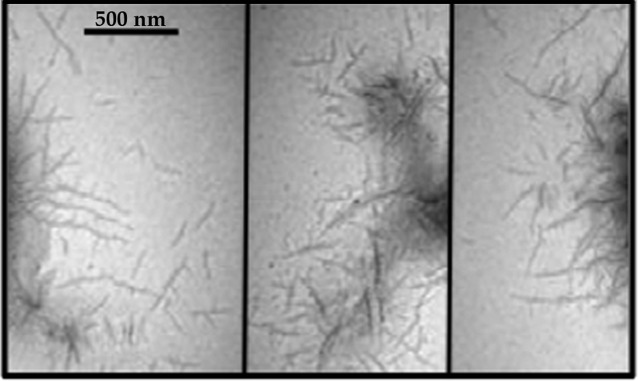
TEM image of aggregated P1 stained with uranyl acetate. Three different fields are shown. Bar, 500 nm.
Green birefringence of S. mutans, S. mutans proteins and dental plaque following CR staining
Based on the dye-binding properties and the ability of P1 to form fibrils as shown by TEM, we also looked for CR-associated green birefringence of P1. Simple CR staining alone is not definitive for amyloid, but when viewed with polarized light through crossed filters, congophilic amyloid foci cause yellowish apple-green birefringence (Howie et al., 2008). This technique is commonly used to visualize amyloid plaques in situ in post-mortem tissue samples from patients with cerebral amyloid pathologies (Jin et al., 2003). As expected, the aggregated S. mutans P1 sample, but not the BSA control, demonstrated green birefringence (Fig. 3a).
Fig. 3.

Green birefringence, characteristic of amyloid formation, was visualized following staining with CR. (a) Green birefringence of aggregated P1 viewed under cross-polarized light. BSA was used as a negative control. (b) Green birefringence of wild-type S. mutans cells grown on an agar surface compared with similarly grown mutants lacking P1 or sortase transpeptidase. Colonies of a curli-expressing strain of E. coli and its corresponding curli-negative mutant were used as positive and negative controls, respectively. The images were taken using cross-polarized (top panels) and non-polarized (bottom panels) light. Bars, 3 µm (a) and 30 µm (b).
Next, we looked for evidence of green birefringence associated with S. mutans cells (Fig. 3b). As positive and negative controls, we utilized an E. coli strain that produces curli amyloid fibres and a corresponding curli-negative mutant strain, respectively (Epstein et al., 2009). No green birefringence was observed when planktonic bacteria present in broth culture were stained with CR (not shown). However, when cells were grown on the surface of agar plates prior to CR staining and viewed under cross-polarized light (top panels), as opposed to non-polarized light (bottom panels), yellowish-green birefringence typical of amyloid was observed for the wild-type strain, as well as for the P1-deficient mutant. This suggests that P1 is not the only amyloidogenic protein produced by S. mutans. In contrast, green birefringence was not detected for an srtA mutant devoid of the sortase transpeptidase enzyme responsible for coupling P1 and other surface proteins to the cell wall. This suggests a possible nucleation event at the cell surface reminiscent of fibrillization of the E. coli CsgA/CsgB curli proteins (Hammer et al., 2007). Green birefringence following CR staining was observed only after plates had been stored for at least 48 h, consistent with a lag phase for amyloid induction or accumulation of a critical mass of amyloid material required for visualization. E. coli colonies of a curli-deficient mutant are white on CR agar indicator plates, whereas the wild-type strain is red (Zhou et al., 2012). All strains and species of streptococci we have tested produce red colonies to some degree on CR plates, including the S. mutans sortase mutant, probably due to the dye-binding behaviour of polysaccharide moieties. Because CR can bind other repetitive structures like cellulose in biological samples, it is important to confirm results, as we have done, with purified protein and to establish amyloid formation by multiple methods, including the ThT uptake and green birefringence assays, and particularly the definitive identification of fibrils by TEM.
To further test whether the wild-type S. mutans and mutant strains were capable of producing amyloidogenic material, extracellular proteins shed into bacterial culture supernatants were tested for green birefringence. Following mechanical agitation to initiate amyloid fibrillization, samples from the wild-type, as well as the P1- and sortase-deficient strains, demonstrated this property following staining with CR (Fig. 4a). The detection of amyloidogenic proteins in the culture supernatant of the sortase-negative strain would be expected, since this strain would still produce the proteins, but just not be able to attach them to the cell surface. As further confirmation that the observed green birefringent material was amyloid, the classical test to demonstrate formic acid dissolution and protease resistance was employed (Shewmaker et al., 2009), as a defining property of amyloid fibrils is resistance to proteinase K digestion and disaggregation of fibrils by formic acid. As would be predicted for a bacterial amyloid, CR-associated green birefringence of S. mutans extracellular proteins was not affected by treatment with proteinase K, but was completely destroyed following treatment with formic acid (Fig. 4b). Resistance to pepsin and trypsin was also observed (not shown).
Fig. 4.

(a) Green birefringence of S. mutans extracellular proteins derived from wild-type strain NG8 and P1- and SrtA-deficient mutants. (b) Amyloid material derived from S. mutans NG8 culture supernatant treated with formic acid or proteinase K. (c) Green birefringence of human dental plaque following staining with CR. Bars, 30 µm (a and c) and 3 µm (b).
Finally, to determine that amyloid fibrillization is not an in vitro artefact and occurs in vivo within the prevailing environment of the oral cavity, dental plaque samples were collected from human subjects and immediately tested for green birefringence. Thus far, we have tested samples from three adults (Fig. 4c). One was caries active (patient 1) and the other two were caries non-active. Green birefringent material indicative of amyloid was present in all three samples. S. mutans was isolated from patients 1 and 3. The production of amyloidogenic proteins by these clinical isolates was confirmed by testing extracellular proteins from their culture supernatants for green birefringence as well (not shown). Given the reported prevalence of amyloid within environmental biofilms (Larsen et al., 2007), its detection in a caries-non-active adult, such as patient 2, from whom S. mutans was not isolated, would also be predicted.
S. mutans biofilm formation is affected by inhibitors of amyloid fibrillization
There have been numerous reports of small polyphenol molecules inhibiting the formation of different amyloid fibrillar assemblies in vitro, as well as associated toxicities in vivo (Porat et al., 2006). We chose four compounds that have been documented to inhibit the formation of fibrillar aggregates associated with amyloidopathies and assessed their effects on biofilm formation by S. mutans. These included CR, because it has commonly been used as an inhibitor of fibril formation with dozens of reports in the literature, ThT and ThS, since both have widely been reported to interact with amyloid fibrils, yet they differ in their ability to inhibit protein–protein interactions (Sharp et al., 2009), and EGCG that has been widely reported to attenuate amyloid-beta (Aβ) fibrillogenesis (DaSilva et al., 2010). Aβ is an amyloidogenic peptide associated with Alzheimer’s disease.
When evaluated at concentrations known to interfere with fibrillization of disease-associated amyloids, CR (Fig. 5a), ThS (Fig. 5b) and EGCG (Fig. 5c) all interfered with S. mutans biofilm formation. To ensure that the observed effects were not due to an artefact of a laboratory strain, we included four clinical isolates in our assays. With the exception of CR treatment of strain NG8, all the inhibitors significantly prevented biofilm formation by all the S. mutans strains tested. Nevertheless, CR was effective against the P1-deficient mutant PC3370, suggesting that its target is a molecule other than P1. Although ThT also impeded biofilm formation by all strains, it inhibited bacterial growth as well (not shown), so its effect on biofilm formation per se could not be assessed. At the concentrations used, the other inhibitors did not affect the growth of any S. mutans strains. The sortase-negative mutant was not included in the biofilm inhibition experiments, since we found it to be severely impaired in biofilm formation, demonstrating >80 % less cell mass than the parent strain.
Fig. 5.

S. mutans biofilm formation in the absence (black bars) and presence (grey bars) of known inhibitors of amyloid fibrillization, CR (10 µM) (a), ThS (100 µM) (b) and EGCG (1 mM) (c). Strains 3svf1, 1svf1, 5ST1 and 81D2 are clinical isolates. NG8 was used as the parent to generate the P1-deficient isogenic mutant PC3370 (Crowley et al., 1999). Error bars represent sem. In each case there was a significant decrease in biofilm formation in the presence of inhibitor at the P≤0.0001 level, except NG8 plus ThS (P≤0.0015) and 1svf1 plus CR (P≤0.0085). There was no significant difference between NG8 with CR and NG8 without CR at the P≤0.05 level.
Given the effects of the amyloid inhibitors on S. mutans biofilm formation, we looked for evidence of green birefringent material within the biofilms. When biofilm-grown cells were stained with CR and then dislodged from the wells, green birefringent material indicative of amyloid was observed for both the wild-type and P1-deficient strains (Fig. 6). Because of its pronounced biofilm defect, the sortase mutant could not be evaluated in this way.
Fig. 6.
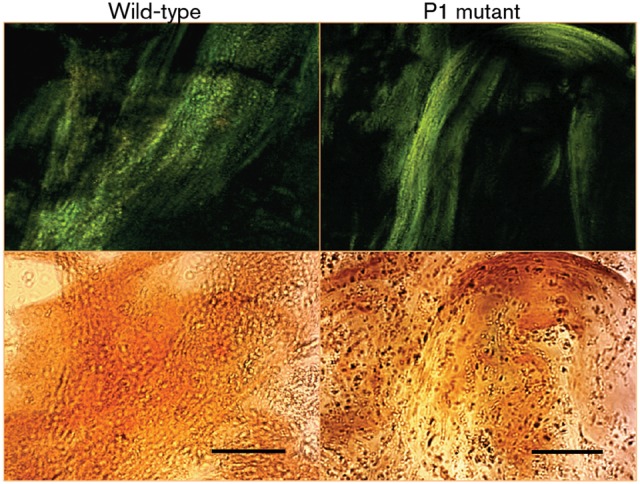
Green birefringence of biofilm-grown cells of S. mutans wild-type and P1-deficient mutant strains, following staining with CR. The images were taken using cross-polarized (top panels) or non-polarized (bottom panels) light. Bars, 30 µm.
EGCG inhibits amyloid fibrillization by S. mutans extracellular proteins
The mechanism of inhibition of amyloid fibrillization by polyphenols has often been attributed to their anti-oxidative properties, but in vitro inhibition of amyloid formation often does not correlate with oxidative conditions (Porat et al., 2006). It has now been reported that structural properties of the polyphenol compounds can mediate their inhibitory effect. Given its pronounced inhibitory effect on S. mutans biofilm formation, we wished to test whether the green tea polyphenol EGCG also had an effect on amyloid formation by S. mutans extracellular proteins, as they are able to interact with ThT, following mechanical agitation to induce fibrillization. CR and ThS could not be tested in this assay because of their interfering effects on ThT fluorescence emission spectra. The pronounced increase in the fluorescence intensity of ThT, following excitation at 442 nm, in the presence of fibrillated S. mutans proteins was notably diminished by 1 mM EGCG (Fig. 7a). EGCG had no effect on ThT fluorescence in the absence of P1. We further showed that the inhibitory effect is dose-dependent, with significant inhibition of ThT uptake by fibrillated S. mutans proteins, and this effect is observed down to 10 µM EGCG (Fig. 7b).
Fig. 7.
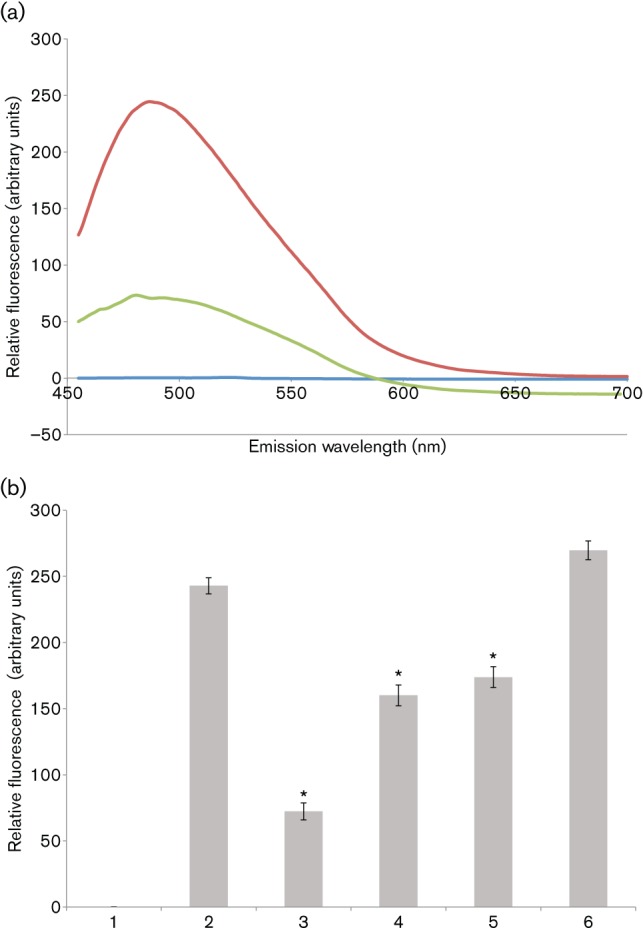
EGCG inhibits amyloid fibrillization by S. mutans extracellular proteins. (a) ThT fluorescence emission spectra, following excitation at 442 nm. S. mutans extracellular proteins were stirred in the cold to induce amyloid fibrillization in the presence (green) and absence (red) of 1 mM EGCG and ThT fluorescence emission was monitored from 450–700 nm. Background fluorescence was evaluated in the absence of added protein (blue). (b) Inhibition of amyloid fibrillization tested over a range of EGCG concentrations. ThT fluorescence emission at 485 nm, following excitation at 442 nm, is shown. Bars: 1, ThT-only control; 2, no EGCG; 3, 1 mM EGCG (P≤0.001); 4, 100 µM EGCG (P≤0.0011); 5, 10 µM EGCG (P≤0.0024); 6, 1 µM EGCG. Error bars represent sem. Significant differences between samples with and without inhibitor, indicated above in parentheses, are denoted with asterisks.
Discussion
S. mutans is particularly effective at colonizing hard tissues of the human oral cavity and its adherence is mediated by sucrose-dependent and -independent mechanisms (Koga et al., 1986; Nobbs et al., 2009). In the absence of sucrose, its wall-associated fibrillar lectin, known variously as antigen I/II, PAc or P1, facilitates attachment to the acquired pellicle on teeth. This multifunctional adhesin mediates interactions with salivary constituents, host cell matrix proteins such as fibronectin, fibrinogen and collagen, and other oral bacteria (Beg et al., 2002; Jenkinson & Demuth, 1997; Lamont et al., 2002). Antigen I/II family proteins are present on most oral streptococci as well as on some strains of group A and group B streptococci. These proteins all contain repetitive A-rich and P-rich regions, and therefore appear to share the common architectural feature of an extended stalk. However, most antigenic, structural and functional differences map to the V and C-terminal domains of the proteins (Brady et al., 2010). Cross-reactive antibodies map in the vicinity of the stalk, whereas less cross-reactivity is observed in the globular regions where unique functional attributes have been localized (McArthur et al., 2007; Robinette et al., 2011). It is not yet known whether other antigen I/II family molecules are capable of amyloid fibrillization. The presence of β-sheet structures in the globular V and C-terminal domains of P1 suggests a role for these two regions in amyloid formation. Indeed, we observed both CR and ThT binding to full-length P1, as well as to truncated polypeptides that contained either of these two regions, but not to an N-terminal polypeptide that is not predicted to adopt a β structure. The extended stalk formed by the A- and P-rich regions is reminiscent of a collagen structure, raising the intriguing possibility that fibril formation may involve formation of a hybrid ultrastructure comprised of both amyloid-like β-sheet assemblies and collagen-like α-polyproline type II helices. Not all P1 is covalently linked to the peptidoglycan. It is readily extracted from the cell surface with SDS or mechanical shaking in phosphate buffer, and is also readily found in the culture medium. Curiously, a similar pattern of binding of two different anti-P1 monoclonal antibodies to the outer surface of the P1 fibrillar layer on S. mutans was observed previously by immunogold electron microscopy (Ayakawa et al., 1987), but in light of the solved crystal structures, these antibodies map to opposite ends of the A–P stalk (Robinette et al., 2011). This suggests that all P1 present on the cell surface or in polymerized fibrils may not be oriented in the same direction. This is supported by additive ELISAs in which purified V and C-terminal domains can interact to increase recognition by anti-P1 monoclonal antibodies that were originally made using whole-cell walls as the immunogen (not shown). Now that the tertiary structure of P1 has been largely solved, understanding the ultrastructure of the adhesin as it exists on the cell surface and as a component of amyloid fibrils will probably lead to further insight into its structure/function relationships.
P1 is not essential for S. mutans biofilm formation (Ahn et al., 2008) and as described in this report, other cell-surface-localized amyloidogenic proteins probably contribute to this process. While much of dental caries microbiology has focused on S. mutans as a single agent, it is appreciated that caries pathology is associated with common attributes of microbial populations within the lesions, in which S. mutans might even be absent (Takahashi & Nyvad, 2008). Elucidating general characteristics of caries pathogens could provide additional promising targets for novel caries control methods and opportunities for disrupting cariogenic biofilms (Burne et al., 2009). The recognition of the role of functional amyloid in microbial biofilm formation and maintenance is the basis of a growing literature topic. Indeed, the practicality of this information and the ability to interfere with pathogenic E. coli biofilm formation using small molecule inhibitors that target amyloid biogenesis have been demonstrated (Cegelski et al., 2009; Hett & Hung, 2009). In addition, biofilm formation can be prevented and existing biofilms can be broken down by a bacterial factor identified in Bacillus subtilis, which is comprised of a mixture of d-amino acids that act at nanomolar concentrations to cause the release of amyloid fibres that link cells within the biofilm together (Kolodkin-Gal et al., 2010). The d-amino acids were incorporated into the peptide side chain of peptidoglycan in place of the terminal d-alanine (Lam et al., 2009). This suggests a close association between functional amyloid fibrils and the bacterial cell wall. Sortase cleaves cell-surface-localized substrates of Gram-positive organisms at a consensus motif near their C-termini, followed by linkage to the cell-wall peptidoglycan. Three of the eight chaplin proteins (ChpA–C) of Streptomyces coelicolor are sortase substrates. Chaplins are a class of hydrophobic proteins that spontaneously self-assemble into amyloid fibrils and mediate attachment to hydrophobic surfaces and penetration at the air–liquid interface (Claessen et al., 2003; de Jong et al., 2009). Interestingly, P1 is also a sortase substrate (Nobbs et al., 2007) and increases the hydrophobicity of cells to which it is attached (Lee et al., 1989). Although a P1-deficient mutant grown on an agar surface still demonstrated the green birefringence characteristic of amyloid fibrils, following staining with CR, the S. mutans srtA mutant lost this property. In addition, the srtA mutant does not form biofilms, even though amyloidogenic material is still detectable in its culture supernatants. P1 is one of six sortase substrates expressed by S. mutans. Evaluating the amyloidogenic potential of other sortase substrates will be the basis of future work. In addition, it is possible that, as observed in the Streptomyces coelicolor chaplin system (Claessen et al., 2003), proteins other than sortase substrates may contribute to amyloidogenesis by S. mutans.
The current study is the first report, to the best of our knowledge, of amyloidogenesis by an oral bacterium and within human dental plaque. We have also shown that several known inhibitors of amyloid formation interfere with biofilm formation of wild-type S. mutans, as well as a P1-deficient mutant, again suggesting that P1 is not the only amyloid-forming protein in this organism. Given the propensity of oral pathogens to exist in biofilm communities, functional amyloid probably represents a common biological property that will be pivotal to understanding mechanisms of pathogenesis and identifying potential targets for therapeutic intervention. Dozens of different small polyphenol molecules inhibit the formation of different amyloid fibrillar assemblies in vitro and their associated toxicities in vivo (Porat et al., 2006). Numerous inhibitors, including CR, are composed of at least two phenolic rings, with two to six atom linkers and a minimum number of three OH groups on the aromatic rings. It has been suggested that such structural similarities imply 3D conformations that facilitate the non-covalent interaction with β-sheet structures, and that the interaction occurs only when the amyloidogenic protein transforms into the assembly conformation. More recently, however, in a study of Alzheimer's disease Aβ peptide, it was reported that small molecule inhibitors segregate into distinct classes that preferentially inhibit oligomerization, fibrillization or both (Necula et al., 2007). Although the mechanism of small polyphenol molecules to impede fibrillar assemblies has been attributed to their anti-oxidative properties, it is now believed to be more likely that their structural properties are responsible (Porat et al., 2006). This is important to keep in mind as reports emerge regarding anti-cariogenic therapeutic properties of naturally occurring polyphenol compounds.
Based on the growing list of newly identified microbial amyloid-like surface structures and the large array of phylogenetically distant bacteria that express them, such systems have probably been selected and refined by evolution. For example, FapC of P. fluorescens contains repeat motifs and conserved Asn/Gln consensus residues similar to curli, tafi and the prion and spider silk amyloid proteins (Dueholm et al., 2010). However, the functional amyloid hydrophobins and chaplins of fungi and actinobacterial streptomyces, and the TasA protein of B. subtilis do not contain either repeat motifs or Asn/Gln consensus residues. Thus, there must be multiple mechanisms to promote and stabilize microbial amyloids and our results reiterate the importance of dissecting the unique structural aspects of the proteins that relate to fibrillization in any given system. Importantly, it also suggests that, based on unique individual aspects, targeted disruption of amyloidogenesis by particular microbes will be feasible (Cegelski et al., 2009), but only by having a firm command of the amyloidogenic proteins produced and an understanding of unique aspects of their assembly and ultrastructure. Differential effects of specific inhibitors on particular amyloid assemblies could explain the disparity between S. mutans strain NG8 and its P1-deficient derivative with regard to CR inhibition of biofilm formation. This report represents, what we believe to be, the first description of amyloid formation within streptococci and its apparent association with biofilm formation. A recent report regarding Staphylococcus aureus also links functional amyloid to biofilms. In this organism small peptides called phenol-soluble modulins (PSMs) form amyloid fibrils that promote biofilm integrity (Schwartz et al., 2012). Mutants lacking PSMs were susceptible to biofilm disassembly by matrix-degrading enzymes and mechanical stress.
In summary, there is a growing realization that amyloid formation is a directed, widespread, functional process that contributes to the biology of numerous micro-organisms, with particular relevance for adhesion and biofilm formation. We have now demonstrated amyloid formation by the cariogenic pathogen S. mutans, which is not surprising considering its biofilm niche. In addition to the contribution of amyloids to virulence by facilitating the adhesion, biofilm formation and invasion of pathogens, microbial amyloids have been postulated to contribute to systemic diseases, including Alzheimer’s and Parkinson’s, possibly by seeding amyloid formation in the brain (Broxmeyer, 2002; Díaz-Corrales et al., 2004; MacDonald, 2006; Miklossy et al., 2006). Injection of curli fibrils into mice led to an accelerated formation of the disease-associated amyloid protein AA (Lundmark et al., 2005) and amyloid fibrils added to the drinking water of mice decreased the lag time to the onset of disease in an amyloidosis model (Lundmark et al., 2002). The association of oral infectious disease with systemic disease is well recognized and it has been reported that newly diagnosed Alzheimer’s patients have a significantly higher level of active dental caries than individuals with other dementia diagnoses or those without dementia (Ellefsen et al., 2008). While the evaluation of a potential link between S. mutans and neurological disease is beyond the scope of the current study, these intriguing, suggested associations increase the imperative to understand basic fundamental information about amyloid formation by a ubiquitous oral pathogen.
Acknowledgements
The authors recognize Arnold Bleiweis for his insight and creative discussions at the inception of this project. This work was supported by National Institute of Health/National Institute of Dental and Craniofacial Research grants R01DE08007 and R01DE21789 to L. J. B and grants SC1GM81756 and R01GM098616 to P. N. L.
Abbreviations:
- A
alanine
- Aβ
amyloid-beta
- CR
Congo red
- EGCG
epigallocatechin-3-gallate
- P
proline
- TEM
transmission electron microscopy
- ThT
Thioflavin T
- ThS
Thioflavin S
- V
variable
Footnotes
A supplementary figure and a supplementary table are available with the online version of this paper.
References
- Ahn S. J., Ahn S. J., Wen Z. T., Brady L. J., Burne R. A. (2008). Characteristics of biofilm formation by Streptococcus mutans in the presence of saliva. Infect Immun 76, 4259–4268. 10.1128/IAI.00422-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alteri C. J., Xicohténcatl-Cortes J., Hess S., Caballero-Olín G., Girón J. A., Friedman R. L. (2007). Mycobacterium tuberculosis produces pili during human infection. Proc Natl Acad Sci U S A 104, 5145–5150. 10.1073/pnas.0602304104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayakawa G. Y., Boushell L. W., Crowley P. J., Erdos G. W., McArthur W. P., Bleiweis A. S. (1987). Isolation and characterization of monoclonal antibodies specific for antigen P1, a major surface protein of mutans streptococci. Infect Immun 55, 2759–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnhart M. M., Chapman M. R. (2006). Curli biogenesis and function. Annu Rev Microbiol 60, 131–147. 10.1146/annurev.micro.60.080805.142106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beg A. M., Jones M. N., Miller-Torbert T., Holt R. G. (2002). Binding of Streptococcus mutans to extracellular matrix molecules and fibrinogen. Biochem Biophys Res Commun 298, 75–79. 10.1016/S0006-291X(02)02390-2 [DOI] [PubMed] [Google Scholar]
- Bieler S., Estrada L., Lagos R., Baeza M., Castilla J., Soto C. (2005). Amyloid formation modulates the biological activity of a bacterial protein. J Biol Chem 280, 26880–26885. 10.1074/jbc.M502031200 [DOI] [PubMed] [Google Scholar]
- Bouvet A., van de Rijn I., McCarty M. (1981). Nutritionally variant streptococci from patients with endocarditis: growth parameters in a semisynthetic medium and demonstration of a chromophore. J Bacteriol 146, 1075–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady L. J., Crowley P. J., Ma J. K., Kelly C., Lee S. F., Lehner T., Bleiweis A. S. (1991). Restriction fragment length polymorphisms and sequence variation within the spaP gene of Streptococcus mutans serotype c isolates. Infect Immun 59, 1803–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady L. J., Cvitkovitch D. G., Geric C. M., Addison M. N., Joyce J. C., Crowley P. J., Bleiweis A. S. (1998). Deletion of the central proline-rich repeat domain results in altered antigenicity and lack of surface expression of the Streptococcus mutans P1 adhesin molecule. Infect Immun 66, 4274–4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady L. J., Maddocks S. E., Larson M. R., Forsgren N., Persson K., Deivanayagam C. C., Jenkinson H. F. (2010). The changing faces of Streptococcus antigen I/II polypeptide family adhesins. Mol Microbiol 77, 276–286. 10.1111/j.1365-2958.2010.07212.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broxmeyer L. (2002). Parkinson’s: another look. Med Hypotheses 59, 373–377. 10.1016/S0306-9877(02)00188-3 [DOI] [PubMed] [Google Scholar]
- Burne R. A., Ahn S. J., Wen Z. T., Zeng L., Lemos J. A., Abranches J., Nascimento M. (2009). Opportunities for disrupting cariogenic biofilms. Adv Dent Res 21, 17–20. 10.1177/0895937409335593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butko P., Buford J. P., Goodwin J. S., Stroud P. A., McCormick C. L., Cannon G. C. (2001). Spectroscopic evidence for amyloid-like interfacial self-assembly of hydrophobin Sc3. Biochem Biophys Res Commun 280, 212–215. 10.1006/bbrc.2000.4098 [DOI] [PubMed] [Google Scholar]
- Cegelski L., Pinkner J. S., Hammer N. D., Cusumano C. K., Hung C. S., Chorell E., Aberg V., Walker J. N., Seed P. C. & other authors (2009). Small-molecule inhibitors target Escherichia coli amyloid biogenesis and biofilm formation. Nat Chem Biol 5, 913–919. 10.1038/nchembio.242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman M. R., Robinson L. S., Pinkner J. S., Roth R., Heuser J., Hammar M., Normark S., Hultgren S. J. (2002). Role of Escherichia coli curli operons in directing amyloid fiber formation. Science 295, 851–855. 10.1126/science.1067484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernoff Y. O. (2004). Amyloidogenic domains, prions and structural inheritance: rudiments of early life or recent acquisition? Curr Opin Chem Biol 8, 665–671. 10.1016/j.cbpa.2004.09.002 [DOI] [PubMed] [Google Scholar]
- Claessen D., Rink R., de Jong W., Siebring J., de Vreugd P., Boersma F. G., Dijkhuizen L., Wosten H. A. (2003). A novel class of secreted hydrophobic proteins is involved in aerial hyphae formation in Streptomyces coelicolor by forming amyloid-like fibrils. Genes Dev 17, 1714–1726. 10.1101/gad.264303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley P. J., Brady L. J., Michalek S. M., Bleiweis A. S. (1999). Virulence of a spaP mutant of Streptococcus mutans in a gnotobiotic rat model. Infect Immun 67, 1201–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DaSilva K. A., Shaw J. E., McLaurin J. (2010). Amyloid-beta fibrillogenesis: structural insight and therapeutic intervention. Exp Neurol 223, 311–321. 10.1016/j.expneurol.2009.08.032 [DOI] [PubMed] [Google Scholar]
- de Jong W., Wösten H. A., Dijkhuizen L., Claessen D. (2009). Attachment of Streptomyces coelicolor is mediated by amyloidal fimbriae that are anchored to the cell surface via cellulose. Mol Microbiol 73, 1128–1140. 10.1111/j.1365-2958.2009.06838.x [DOI] [PubMed] [Google Scholar]
- de Vocht M. L., Reviakine I., Wösten H. A., Brisson A., Wessels J. G., Robillard G. T. (2000). Structural and functional role of the disulfide bridges in the hydrophobin SC3. J Biol Chem 275, 28428–28432. 10.1074/jbc.M000691200 [DOI] [PubMed] [Google Scholar]
- Díaz-Corrales F. J., Colasante C., Contreras Q., Puig M., Serrano J. A., Hernández L., Beaman B. L. (2004). Nocardia otitidiscaviarum (GAM-5) induces parkinsonian-like alterations in mouse. Braz J Med Biol Res 37, 539–548. 10.1590/S0100-879X2004000400011 [DOI] [PubMed] [Google Scholar]
- Dueholm M. S., Petersen S. V., Sønderkær M., Larsen P., Christiansen G., Hein K. L., Enghild J. J., Nielsen J. L., Nielsen K. L. & other authors (2010). Functional amyloid in Pseudomonas. Mol Microbiol 77, 1009–1020. [DOI] [PubMed] [Google Scholar]
- Ehrnhoefer D. E., Bieschke J., Boeddrich A., Herbst M., Masino L., Lurz R., Engemann S., Pastore A., Wanker E. E. (2008). EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat Struct Mol Biol 15, 558–566. 10.1038/nsmb.1437 [DOI] [PubMed] [Google Scholar]
- Ellefsen B., Holm-Pedersen P., Morse D. E., Schroll M., Andersen B. B., Waldemar G. (2008). Caries prevalence in older persons with and without dementia. J Am Geriatr Soc 56, 59–67. 10.1111/j.1532-5415.2007.01495.x [DOI] [PubMed] [Google Scholar]
- Epstein E. A., Chapman M. R. (2008). Polymerizing the fibre between bacteria and host cells: the biogenesis of functional amyloid fibres. Cell Microbiol 10, 1413–1420. 10.1111/j.1462-5822.2008.01148.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein E. A., Reizian M. A., Chapman M. R. (2009). Spatial clustering of the curlin secretion lipoprotein requires curli fiber assembly. J Bacteriol 191, 608–615. 10.1128/JB.01244-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler D. M., Koulov A. V., Balch W. E., Kelly J. W. (2007). Functional amyloid – from bacteria to humans. Trends Biochem Sci 32, 217–224. 10.1016/j.tibs.2007.03.003 [DOI] [PubMed] [Google Scholar]
- Garcia M. C., Lee J. T., Ramsook C. B., Alsteens D., Dufrêne Y. F., Lipke P. N. (2011). A role for amyloid in cell aggregation and biofilm formation. PLoS ONE 6, e17632. 10.1371/journal.pone.0017632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebbink M. F., Claessen D., Bouma B., Dijkhuizen L., Wösten H. A. (2005). Amyloids – a functional coat for microorganisms. Nat Rev Microbiol 3, 333–341. 10.1038/nrmicro1127 [DOI] [PubMed] [Google Scholar]
- Grelle G., Otto A., Lorenz M., Frank R. F., Wanker E. E., Bieschke J. (2011). Black tea theaflavins inhibit formation of toxic amyloid-β and α-synuclein fibrils. Biochemistry 50, 10624–10636. 10.1021/bi2012383 [DOI] [PubMed] [Google Scholar]
- Hammer N. D., Schmidt J. C., Chapman M. R. (2007). The curli nucleator protein, CsgB, contains an amyloidogenic domain that directs CsgA polymerization. Proc Natl Acad Sci U S A 104, 12494–12499. 10.1073/pnas.0703310104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper J. D., Lansbury P. T., Jr (1997). Models of amyloid seeding in Alzheimer’s disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu Rev Biochem 66, 385–407. 10.1146/annurev.biochem.66.1.385 [DOI] [PubMed] [Google Scholar]
- Heiser V., Scherzinger E., Boeddrich A., Nordhoff E., Lurz R., Schugardt N., Lehrach H., Wanker E. E. (2000). Inhibition of huntingtin fibrillogenesis by specific antibodies and small molecules: implications for Huntington’s disease therapy. Proc Natl Acad Sci U S A 97, 6739–6744. 10.1073/pnas.110138997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hett E. C., Hung D. T. (2009). Targeting multiple biofilm pathways. Chem Biol 16, 1216–1218. 10.1016/j.chembiol.2009.12.003 [DOI] [PubMed] [Google Scholar]
- Howie A. J., Brewer D. B., Howell D., Jones A. P. (2008). Physical basis of colors seen in Congo red-stained amyloid in polarized light. Lab Invest 88, 232–242. 10.1038/labinvest.3700714 [DOI] [PubMed] [Google Scholar]
- Jenkinson H. F., Demuth D. R. (1997). Structure, function and immunogenicity of streptococcal antigen I/II polypeptides. Mol Microbiol 23, 183–190. 10.1046/j.1365-2958.1997.2021577.x [DOI] [PubMed] [Google Scholar]
- Jin L. W., Claborn K. A., Kurimoto M., Geday M. A., Maezawa I., Sohraby F., Estrada M., Kaminksy W., Kahr B. (2003). Imaging linear birefringence and dichroism in cerebral amyloid pathologies. Proc Natl Acad Sci U S A 100, 15294–15298. 10.1073/pnas.2534647100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk W. E., Jacob R. F., Mason R. P. (1999). Quantifying amyloid beta-peptide (Abeta) aggregation using the Congo red-Abeta (CR-abeta) spectrophotometric assay. Anal Biochem 266, 66–76. 10.1006/abio.1998.2933 [DOI] [PubMed] [Google Scholar]
- Koga T., Asakawa H., Okahashi N., Hamada S. (1986). Sucrose-dependent cell adherence and cariogenicity of serotype c Streptococcus mutans. J Gen Microbiol 132, 2873–2883. [DOI] [PubMed] [Google Scholar]
- Kolodkin-Gal I., Romero D., Cao S., Clardy J., Kolter R., Losick R. (2010). d-Amino acids trigger biofilm disassembly. Science 328, 627–629. 10.1126/science.1188628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuner P., Bohrmann B., Tjernberg L. O., Näslund J., Huber G., Celenk S., Grüninger-Leitch F., Richards J. G., Jakob-Roetne R. & other authors (2000). Controlling polymerization of beta-amyloid and prion-derived peptides with synthetic small molecule ligands. J Biol Chem 275, 1673–1678. 10.1074/jbc.275.3.1673 [DOI] [PubMed] [Google Scholar]
- Kwan A. H., Winefield R. D., Sunde M., Matthews J. M., Haverkamp R. G., Templeton M. D., Mackay J. P. (2006). Structural basis for rodlet assembly in fungal hydrophobins. Proc Natl Acad Sci U S A 103, 3621–3626. 10.1073/pnas.0505704103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam H., Oh D. C., Cava F., Takacs C. N., Clardy J., de Pedro M. A., Waldor M. K. (2009). d-Amino acids govern stationary phase cell wall remodeling in bacteria. Science 325, 1552–1555. 10.1126/science.1178123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamont R. J., El-Sabaeny A., Park Y., Cook G. S., Costerton J. W., Demuth D. R. (2002). Role of the Streptococcus gordonii SspB protein in the development of Porphyromonas gingivalis biofilms on streptococcal substrates. Microbiology 148, 1627–1636. [DOI] [PubMed] [Google Scholar]
- Larsen P., Nielsen J. L., Dueholm M. S., Wetzel R., Otzen D., Nielsen P. H. (2007). Amyloid adhesins are abundant in natural biofilms. Environ Microbiol 9, 3077–3090. 10.1111/j.1462-2920.2007.01418.x [DOI] [PubMed] [Google Scholar]
- Larsen P., Nielsen J. L., Otzen D., Nielsen P. H. (2008). Amyloid-like adhesins produced by floc-forming and filamentous bacteria in activated sludge. Appl Environ Microbiol 74, 1517–1526. 10.1128/AEM.02274-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson M. R., Rajashankar K. R., Patel M. H., Robinette R. A., Crowley P. J., Michalek S., Brady L. J., Deivanayagam C. (2010). Elongated fibrillar structure of a streptococcal adhesin assembled by the high-affinity association of alpha- and PPII-helices. Proc Natl Acad Sci U S A 107, 5983–5988. 10.1073/pnas.0912293107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson M. R., Rajashankar K. R., Crowley P. J., Kelly C., Mitchell T. J., Brady L. J., Deivanayagam C. (2011). Crystal structure of the C-terminal region of Streptococcus mutans antigen I/II and characterization of salivary agglutinin adherence domains. J Biol Chem 286, 21657–21666. 10.1074/jbc.M111.231100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. F., Progulske-Fox A., Erdos G. W., Piacentini D. A., Ayakawa G. Y., Crowley P. J., Bleiweis A. S. (1989). Construction and characterization of isogenic mutants of Streptococcus mutans deficient in major surface protein antigen P1 (I/II). Infect Immun 57, 3306–3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez del Amo J. M., Fink U., Dasari M., Grelle G., Wanker E. E., Bieschke J., Reif B. (2012). Structural properties of EGCG-induced, nontoxic Alzheimer’s disease Aβ oligomers. J Mol Biol 421, 517–524. 10.1016/j.jmb.2012.01.013 [DOI] [PubMed] [Google Scholar]
- Lundmark K., Westermark G. T., Nyström S., Murphy C. L., Solomon A., Westermark P. (2002). Transmissibility of systemic amyloidosis by a prion-like mechanism. Proc Natl Acad Sci U S A 99, 6979–6984. 10.1073/pnas.092205999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundmark K., Westermark G. T., Olsén A., Westermark P. (2005). Protein fibrils in nature can enhance amyloid protein A amyloidosis in mice: cross-seeding as a disease mechanism. Proc Natl Acad Sci U S A 102, 6098–6102. 10.1073/pnas.0501814102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald A. B. (2006). Plaques of Alzheimer’s disease originate from cysts of Borrelia burgdorferi, the Lyme disease spirochete. Med Hypotheses 67, 592–600. 10.1016/j.mehy.2006.02.035 [DOI] [PubMed] [Google Scholar]
- Mackay J. P., Matthews J. M., Winefield R. D., Mackay L. G., Haverkamp R. G., Templeton M. D. (2001). The hydrophobin EAS is largely unstructured in solution and functions by forming amyloid-like structures. Structure 9, 83–91. 10.1016/S0969-2126(00)00559-1 [DOI] [PubMed] [Google Scholar]
- Macrina F. L., Tobian J. A., Jones K. R., Evans R. P., Clewell D. B. (1982). A cloning vector able to replicate in Escherichia coli and Streptococcus sanguis. Gene 19, 345–353. 10.1016/0378-1119(82)90025-7 [DOI] [PubMed] [Google Scholar]
- Maury C. P. (2009a). The emerging concept of functional amyloid. J Intern Med 265, 329–334. 10.1111/j.1365-2796.2008.02068.x [DOI] [PubMed] [Google Scholar]
- Maury C. P. (2009b). Self-propagating β-sheet polypeptide structures as prebiotic informational molecular entities: the amyloid world. Orig Life Evol Biosph 39, 141–150. 10.1007/s11084-009-9165-6 [DOI] [PubMed] [Google Scholar]
- McArthur W. P., Rhodin N. R., Seifert T. B., Oli M. W., Robinette R. A., Demuth D. R., Brady L. J. (2007). Characterization of epitopes recognized by anti-Streptococcus mutans P1 monoclonal antibodies. FEMS Immunol Med Microbiol 50, 342–353. 10.1111/j.1574-695X.2007.00260.x [DOI] [PubMed] [Google Scholar]
- Miklossy J., Kis A., Radenovic A., Miller L., Forro L., Martins R., Reiss K., Darbinian N., Darekar P., Mihaly L. (2006). Beta-amyloid deposition and Alzheimer’s type changes induced by Borrelia spirochetes. Neurobiol Aging 27, 228–236. 10.1016/j.neurobiolaging.2005.01.018 [DOI] [PubMed] [Google Scholar]
- Necula M., Kayed R., Milton S., Glabe C. G. (2007). Small molecule inhibitors of aggregation indicate that amyloid beta oligomerization and fibrillization pathways are independent and distinct. J Biol Chem 282, 10311–10324. 10.1074/jbc.M608207200 [DOI] [PubMed] [Google Scholar]
- Nilsson M. R. (2004). Techniques to study amyloid fibril formation in vitro. Methods 34, 151–160. 10.1016/j.ymeth.2004.03.012 [DOI] [PubMed] [Google Scholar]
- Nobbs A. H., Vajna R. M., Johnson J. R., Zhang Y., Erlandsen S. L., Oli M. W., Kreth J., Brady L. J., Herzberg M. C. (2007). Consequences of a sortase A mutation in Streptococcus gordonii. Microbiology 153, 4088–4097. 10.1099/mic.0.2007/007252-0 [DOI] [PubMed] [Google Scholar]
- Nobbs A. H., Lamont R. J., Jenkinson H. F. (2009). Streptococcus adherence and colonization. Microbiol Mol Biol Rev 73, 407–450. 10.1128/MMBR.00014-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nylander A., Forsgren N., Persson K. (2011). Structure of the C-terminal domain of the surface antigen SpaP from the caries pathogen Streptococcus mutans. Acta Crystallogr Sect F Struct Biol Cryst Commun 67, 23–26. 10.1107/S174430911004443X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otoo H. N., Lee K. G., Qiu W., Lipke P. N. (2008). Candida albicans Als adhesins have conserved amyloid-forming sequences. Eukaryot Cell 7, 776–782. 10.1128/EC.00309-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otzen D., Nielsen P. H. (2008). We find them here, we find them there: functional bacterial amyloid. Cell Mol Life Sci 65, 910–927. 10.1007/s00018-007-7404-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer S. R., Crowley P. J., Oli M. W., Ruelf M. A., Michalek S. M., Brady L. J. (2012). YidC1 and YidC2 are functionally distinct proteins involved in protein secretion, biofilm formation and cariogenicity of Streptococcus mutans. Microbiology 158, 1702–1712. 10.1099/mic.0.059139-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porat Y., Abramowitz A., Gazit E. (2006). Inhibition of amyloid fibril formation by polyphenols: structural similarity and aromatic interactions as a common inhibition mechanism. Chem Biol Drug Des 67, 27–37. 10.1111/j.1747-0285.2005.00318.x [DOI] [PubMed] [Google Scholar]
- Ramsook C. B., Tan C., Garcia M. C., Fung R., Soybelman G., Henry R., Litewka A., O’Meally S., Otoo H. N. & other authors (2010). Yeast cell adhesion molecules have functional amyloid-forming sequences. Eukaryot Cell 9, 393–404. 10.1128/EC.00068-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauceo J. M., Gaur N. K., Lee K. G., Edwards J. E., Klotz S. A., Lipke P. N. (2004). Global cell surface conformational shift mediated by a Candida albicans adhesin. Infect Immun 72, 4948–4955. 10.1128/IAI.72.9.4948-4955.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinette R. A., Oli M. W., McArthur W. P., Brady L. J. (2011). A therapeutic anti-Streptococcus mutans monoclonal antibody used in human passive protection trials influences the adaptive immune response. Vaccine 29, 6292–6300. 10.1016/j.vaccine.2011.06.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero D., Aguilar C., Losick R., Kolter R. (2010). Amyloid fibers provide structural integrity to Bacillus subtilis biofilms. Proc Natl Acad Sci U S A 107, 2230–2234. 10.1073/pnas.0910560107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero D., Vlamakis H., Losick R., Kolter R. (2011). An accessory protein required for anchoring and assembly of amyloid fibres in B. subtilis biofilms. Mol Microbiol 80, 1155–1168. 10.1111/j.1365-2958.2011.07653.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Römling U., Bian Z., Hammar M., Sierralta W. D., Normark S. (1998). Curli fibers are highly conserved between Salmonella typhimurium and Escherichia coli with respect to operon structure and regulation. J Bacteriol 180, 722–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldaña Z., Xicohtencatl-Cortes J., Avelino F., Phillips A. D., Kaper J. B., Puente J. L., Girón J. A. (2009). Synergistic role of curli and cellulose in cell adherence and biofilm formation of attaching and effacing Escherichia coli and identification of Fis as a negative regulator of curli. Environ Microbiol 11, 992–1006. 10.1111/j.1462-2920.2008.01824.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T., Hu J. P., Ohki K., Yamaura M., Washio J., Matsuyama J., Takahashi N. (2003). Identification of mutans streptococci by restriction fragment length polymorphism analysis of polymerase chain reaction-amplified 16S ribosomal RNA genes. Oral Microbiol Immunol 18, 323–326. 10.1034/j.1399-302X.2003.00095.x [DOI] [PubMed] [Google Scholar]
- Sawyer E. B., Claessen D., Haas M., Hurgobin B., Gras S. L. (2011). The assembly of individual chaplin peptides from Streptomyces coelicolor into functional amyloid fibrils. PLoS ONE 6, e18839. 10.1371/journal.pone.0018839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeken M. J., van der Hoeven J. S., Franken H. C. (1986). Comparative recovery of Streptococcus mutans on five isolation media, including a new simple selective medium. J Dent Res 65, 906–908. 10.1177/00220345860650060901 [DOI] [PubMed] [Google Scholar]
- Schwartz K., Syed A. K., Stephenson R. E., Rickard A. H., Boles B. R. (2012). Functional amyloids composed of phenol soluble modulins stabilize Staphylococcus aureus biofilms. PLoS Pathog 8, e1002744. 10.1371/journal.ppat.1002744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp A., Crabb S. J., Johnson P. W., Hague A., Cutress R., Townsend P. A., Ganesan A., Packham G. (2009). Thioflavin S (NSC71948) interferes with Bcl-2-associated athanogene (BAG-1)-mediated protein–protein interactions. J Pharmacol Exp Ther 331, 680–689. 10.1124/jpet.109.153601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shewmaker F., McGlinchey R. P., Thurber K. R., McPhie P., Dyda F., Tycko R., Wickner R. B. (2009). The functional curli amyloid is not based on in-register parallel β-sheet structure. J Biol Chem 284, 25065–25076. 10.1074/jbc.M109.007054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J. F., Knowles T. P., Dobson C. M., Macphee C. E., Welland M. E. (2006). Characterization of the nanoscale properties of individual amyloid fibrils. Proc Natl Acad Sci U S A 103, 15806–15811. 10.1073/pnas.0604035103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N., Nyvad B. (2008). Caries ecology revisited: microbial dynamics and the caries process. Caries Res 42, 409–418. 10.1159/000159604 [DOI] [PubMed] [Google Scholar]
- Ton-That H., Marraffini L. A., Schneewind O. (2004). Protein sorting to the cell wall envelope of Gram-positive bacteria. Biochim Biophys Acta 1694, 269–278. 10.1016/j.bbamcr.2004.04.014 [DOI] [PubMed] [Google Scholar]
- Troffer-Charlier N., Ogier J., Moras D., Cavarelli J. (2002). Crystal structure of the V-region of Streptococcus mutans antigen I/II at 2.4 Å resolution suggests a sugar preformed binding site. J Mol Biol 318, 179–188. 10.1016/S0022-2836(02)00025-6 [DOI] [PubMed] [Google Scholar]
- Wang X., Chapman M. R. (2008). Sequence determinants of bacterial amyloid formation. J Mol Biol 380, 570–580. 10.1016/j.jmb.2008.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Blanco L. P., Smith D. R., Chapman M. R. (2012). Bacterial amyloids. Methods Mol Biol 849, 303–320. 10.1007/978-1-61779-551-0_21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zogaj X., Bokranz W., Nimtz M., Römling U. (2003). Production of cellulose and curli fimbriae by members of the family Enterobacteriaceae isolated from the human gastrointestinal tract. Infect Immun 71, 4151–4158. 10.1128/IAI.71.7.4151-4158.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]