

Abstract
Primary adrenal mesenchymal tumors are extremely rare. These tumors are hard to diagnose, and similar to certain adrenal tumors, as they do not produce hormones, and they can only manifest themselves when the tumor reaches an advanced size. These tumors are generally detected incidentally. This study reports a rare case of primary leiomyosarcoma of the right adrenal gland with vena cava invasion, in a 70-year-old woman who presented with right flank pain. Computerized tomography showed an adrenal mass with a diameter of 78 mm, which exerted pressure on the vena cava inferior. The invasive part was excised by using adrenalectomy and cavatomy. Tumor invasion was determined on the wall of the vena cava. Histopathological examination on 10× magnification showed 8-10 mitotic events. Immunohistochemical staining showed that the cells were SMA (+), desmin (+), cytokeratin (-), and Bcl-2 (-). The Ki67 proliferation index was 70%. Widespread metastasis developed six months after the adrenalectomy.
Key words: leiomyosarcoma, adrenal leiomyosarcoma, sarcoma, adrenalectomy
Introduction
Leiomyosarcomas are aggressive, malignant tumors with high metastatic potential that generally originate from the smooth muscles of soft tissues and uterus. Adrenal leiomyosarcomas especially originate from the smooth muscles of the adrenalin-rich vena.1 Adrenal leiomyosarcomas reach a large diameter, because they originate from mesenchymal components, and unlike other tumors, they are devoid of natural barriers. Sarcomas typically carry a pseudocapsule. According to the literature, there are only 13 cases of primary adrenal leiomyosarcoma, and the current case represents the 14th case in the literature.2 It has a wide range of differential diagnosis including primary adrenal carcinoma, malignant peripheral nerve sheath tumor, angiosarcoma, malignant melanoma, gastrointestinal stromal tumor, rhabdomyosarcoma, fibrosarcoma, carcinosarcoma, malignant fibrous histiocytoma, angiosarcoma and malignant hemangioperistoma, and metastatic carcinoma. A complete workup, including radiological, biochemical, histomorphological and immunohistochemical investigation, is essential for the final diagnosis of primary adrenal leiomyosarcoma. Diagnosis is especially made with immunohistochemical examination.
While the current case is the 14th case in the literature, it represents the only primary adrenal leiomyosarcoma case with a vena cava invasion.
Case Report
A 70-year-old patient was admitted with pain on the right side persisting for six months. Biochemical analyses showed that AST and ALT levels were normal, fasting glucose level was 123 mg/dL, TSH level was 1.89 IU/mL, creatinine level was 0.75 mg/dL, urea was 26 mg/dL, HCT was 29.7, hemoglobin was 9.89 g/dL, white blood cells count was 14,500, and platelet count was 191,000.
The physical examination showed pain in the left and right flanks upon palpation, and the patient had a history of hypertension and hypothyroidy. Ultrasonography showed a hyperechogenic solid mass with a 78×67×75 mm dimension on the right adrenal gland. The computed tomography (CT) scan showed an invasive solid mass in the right adrenal gland, with a 78×76×70 mm dimension, which exerted pressure on the vena cava inferior. The mass was hormone-inactive. The patient underwent transperitoneal right adrenalectomy plus cavatomy and excision for the invased vena cava. PET-CT showed no distant metastasis. The patient was enrolled to a follow-up program. The CT showed widespread metastasis at the sixth month follow-up, and the patient was transferred to the Medical Oncology Department for chemotherapy.
Following the histopathological examination, the case was reported as a primary adrenal leiomyosarcoma. Macroscopic examination showed a pinkish-yellow colored, uniformly-bound tumor with a maximum diameter of 8 cm in the right adrenal, which had cystic features and bleeding at a focal point. Necrosis was not observed. The tumor was excised with negative surgical margins. Serosal involvement and invasion was observed in the vena cava inferior. Microscopic examination showed that tumor consisted of cells with spindle-shaped nuclei and pink cytoplasm that crossed each other at varying angles and pleomorphism at certain foci were remarkable. Mitosis was 8-10/10HPF (Figure 1A) (Hematoxylin and Eosin, 200×). Immunohistochemical staining showed that tumor cells were SMA (+), desmin (+), cytokeratin (-), and Bcl-2 (-) (Figure 1B,C). The Ki-67 proliferation index was 70%.
Figure 1.
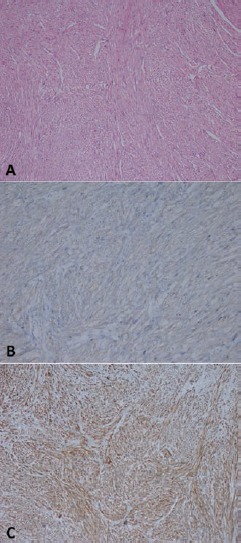
A) Hematoxilyn-eosin staining demostrating high-grade sarcomatoid cells (200×). B) Immunohistochemistry showing difuse actin expression in smooth muscle fiber cytoplasm (Actin, 200×). C) Immunohistochemistry showing desmin (100×).
Discussion
Primary adrenal leiomyosarcomas are extremely rare tumors, and are believed to especially originate from the smooth muscle cells of the adrenal vena branches.3 According to the literature, only 13 primary adrenal leiomyosarcoma cases have been reported so far. The current case represents the 14th case in the literature.2
To the best of our knowledge, our case is the only case with a vena cava invasion. Regarding the 13 cases in the literature, the mean age distribution ranged between 30-75 years, and the mean tumor diameter ranged between 11-25 cm. The incidence of the tumor is equal in men and women. In this regard, the current case, with a diameter of 8 cm, is lower compared to the mean tumor size. In only one case report, the tumor was 3 cm in size.4,5 It is rather difficult to distinguish adrenal leiomyosarcoma from adrenocortical cancer showing sarcomatoid differentiation, as they share similar clinical, radiological, and pathological features. When a lumbar or abdominal mass reaches a clinically palpable size, pain or nonspecific dyspeptic complaints appear in the lumbar or abdominal region. Tumors can reach enormous sizes. Sarcomas should be considered in the case of fast-growing adrenal masses. Sarcomas have pseudocapsules, but this capsule is an unreliable surgical barrier, as in most cases it is infiltrated by the tumor. High-grade sarcomas have generally high metastatic potential, and the most frequent site for metastasis is the lung. Liver and lymph nodes are the second most frequent metastatic sites. Low-grade sarcomas frequently lead to local recurrence, and surgical removal of the local recurrence prolongs the survival.
Liposarcoma, rhabdomyosarcoma, fibrosarcoma, carcinosarcoma, malignant fibrous histiocytoma, angiosarcoma and malignant hemangioperistoma, malignant melanoma, adrenal invasion by a retroperitoneal leiomyosarcoma, and metastatic cancers should be considered in the differential diagnosis of adrenal leiomyosarcomas.
Histopathologically, the presence of five or more mitotic cells in 50× magnification field is considered as malignancy in smooth muscle cell tumors. The presence of necrosis within the tumor is a major finding that supports the malignancy.6 While necrosis was not observed in the histopathological examination, cells with spindle-shaped nuclei and pink cytoplasm that crossed each other at varying angles and pleomorphism at certain foci were detected. Mitosis was 8-10/10HPF. The current case had typical pathological features of leiomyosarcoma, except necrosis. Despite the low number of mitosis, early recurrence and metastases emerged due to local invasion. Rather than determining the tumor type, immunohistochemical examination is indispensable for identifying tumor features such as metastasis or recurrence. Conventional leiomyosarcomas invariably show reactivity for smooth muscle markers such as smooth muscle actin and/or muscle specific actin in 90 to 95% of cases, and desmin in 70-90% of cases.3,4,7,8 Adrenal leiomyosarcomas have a poor prognosis with respect to their metastatic potential or frequent and early local recurrence. The median life expectancy is 20 months.8 In the current case, widespread metastasis was detected in the sixth month, and the patient was enrolled in a doxorubicin-based adjuvant chemotherapy program. In the quantitative meta-analysis of 1568 patients in 14 studies, doxorubicin-based adjuvant chemotherapy achieved 6% disease-free survival.9
Conclusions
Adrenal primary leiomyosarcomas are rare tumors with a rather aggressive behavior, and carry high metastatic potential. The diagnosis and prognosis are determined by the tumor’s radiological, histomorphological and immunohistochemical features. The prognosis is poor in the case of lymphovascular invasion, high levels of mitosis, necrosis, and tumors with large diameters. For these patients, the major treatment is complete surgical resection. Due to the high metastatic potential, the patients should be followed up closely. Surgical excision significantly increases survival in cases of low-grade adrenal leiomyosarcoma, showing low level of mitosis and lacking necrosis. Adrenal leiomyosarcoma should be treated with a multidisciplinary approach involving sufficient surgical excision, radiotherapy, and adjuvant chemotherapy.
References
- 1.Rosai J, Adrenal gland and other paraganglia. : Rosai and Ackerman’s surgical pathology. 9th ed.Edinburgh: Mosby; 2004. 1115–1162 [Google Scholar]
- 2.Deshmukh SD, Babanagare SV, Anand M, et al. Primary adrenal leiomyosarcoma: a case report with immunohistochemical study and review of literature. J Cancer Res Ther 2013;9:114–6 [DOI] [PubMed] [Google Scholar]
- 3.Lack EE, Graham CW, Azumi N, et al. Primary leiomyosarcoma of adrenal gland. Case report with immunohistochemical and ultrastructural study. Am J Surg Pathol 1991;15:899–905 [DOI] [PubMed] [Google Scholar]
- 4.Lujan MG, Hoang MP. Pleomorphic leiomyosarcoma of the adrenal gland. Arch Pathol Lab Med 2003;127:32–5 [DOI] [PubMed] [Google Scholar]
- 5.Lee CW, Tsang YM, Liu KL. Primary adrenal leiomyosarcoma. Abdom Imaging 2006;31:123–4 [DOI] [PubMed] [Google Scholar]
- 6.Rosai J, Peritoneum, retroperitoneum, and related structures. Rosai and Ackerman’s Surgical Pathology. 9th ed Edinburgh: Mosby; 2004. 2373–2415 [Google Scholar]
- 7.Shmookler BM, Lauer DH. Retroperitoneal leiomyosarcoma: a clinicopathologic analysis of 36 cases. Am J Surg Pathol 1983;7:269–80 [PubMed] [Google Scholar]
- 8.Zetler PJ, Filipenko JD, Bilbey JH, Schmidt N. Primary adrenal Leiomyosarcoma in a man with acquired immunodeficiency syndrome (AIDS). Further evidence for an increase in smooth muscle tumors related to Epstein-Barr infection in AIDS. Arch Pathol Lab Med 1995;119:1164–7 [PubMed] [Google Scholar]
- 9.Sarcoma Meta-analysis Collaboration (SMAC). Adjuvant chemotherapy for localised resectable soft tissue sarcoma in adults. Cochrane Database Syst Rev 2000. :CD001419. [DOI] [PubMed] [Google Scholar]