

Abstract
Annexin II (AXII), a calcium-dependent phospholipid-binding protein, has been recently found to be an osteoclast (OCL) stimulatory factor that is also secreted by OCLs. In vitro studies showed that AXII induced OCL formation and bone resorption. However, the mechanism of action by which AXII acts as a soluble extracellular protein to induce OCL formation is unknown. In this paper, we demonstrate that AXII gene expression is upregulated by 1,25-dihydroxyvitamin D3 [1,25-(OH)2D3] and that addition of AXII significantly increased OCL-like multinucleated cell formation. Time-course studies suggested that AXII acted on the proliferative stage of OCL precursors and that AXII increased thymidine incorporation in OCL precursors. Moreover, AXII enhanced the growth of CFU-GM, the earliest identifiable OCL precursor, when bone marrow cultures were treated with low concentrations of GM-CSF. This capacity of AXII to induce OCL precursor proliferation was due to induction of GM-CSF expression, because the addition of neutralizing antibodies to GM-CSF blocked the stimulatory effect of AXII on OCL formation. RT-PCR analysis using RNA from highly purified subpopulations of marrow cells demonstrated that T cells, especially CD4+ T cells, produced GM-CSF in response to AXII. Furthermore, FACS® analysis of T-cell subpopulations treated with fluorescein-labeled AXII suggested that the CD4+, but not CD8+, subpopulation of T cells express an AXII receptor. Taken together, these data suggest that AXII stimulates OCL formation by activating T cells through a putative receptor to secrete GM-CSF. GM-CSF then expands the OCL precursor pool to enhance OCL formation.
J. Clin. Invest. 103:1605–1613 (1999).
Introduction
The osteoclast (OCL), the primary cell responsible for bone resorption, is under control of several factors that regulate its formation and/or activation. These factors can be systemic, such as 1,25-(OH)2D3, calcitonin, and parathyroid hormone (PTH); or can be produced locally, such as IL-6 and RANK ligand (1), by cells in the bone microenvironment such as osteoblasts, stromal cells, and immune cells (1). Recently, we and others have demonstrated that the OCL secretes factors that enhance or inhibit OCL formation and/or activation. IL-6 was one of the first cytokines identified among the autocrine/paracrine factors produced by OCLs that stimulated OCL formation (2). TGF-β is also produced by OCLs and can inhibit OCL activity (3). We have also identified and cloned a novel factor secreted by OCLs (OIP-1) that blocks OCL formation and bone resorption (4). Recently, Takahashi et al. (5), using an expression cloning approach, identified annexin II (AXII) as an OCL-secreted product that stimulates human and murine OCL formation and can also enhance the stimulatory effects of suboptimal concentrations of 1,25-(OH)2D3 on OCL formation in marrow cultures and on bone resorption in long bones of fetal rats. Others have shown that rabbit OCL activity is inhibited by neutralizing antibodies to AXII (6). However, the mechanisms responsible for the stimulatory effects of AXII on OCL activity are unclear.
Until recently, AXII was considered an intracellular or a membrane protein that played a role in cell-cell adhesion and plasminogen activation and acted as a cell surface receptor (7). Nesbitt and Horton (6) proposed that intracellular AXII in the OCL was involved in the clearance of matrix protein degraded by OCLs by acting as a binding protein. However, this mechanism does not explain the actions of secreted AXII on OCL formation.
OCLs are multinucleated cells that share a common ancestor with monocytes/macrophages and CFU-GM, the granulocyte-macrophage colony-forming cell. CFU-GM proliferate and differentiate under the influence of osteoclastogenic factors and fuse to form multinucleated OCLs that finally are activated to resorb bone (1). Osteotrophic factors enhance OCL formation by expanding the OCL precursor pool through cell proliferation (e.g., GM-CSF or IL-6), fusion of precursors [e.g., PTH and 1,25-(OH)2D3], or both (e.g., IL-1) (8). These effects could be direct, such as GM-CSF, or through induction of a second secreted factor, as with PTH, whose stimulatory effects are through the induction of RANK ligand (9). In this report, we demonstrate that AXII increases OCL formation by enhancing proliferation of CFU-GM. This is accomplished through induction of GM-CSF expression by CD4+ T cells. The effects of AXII appear to be receptor mediated through a putative cell surface receptor.
Methods
Material.
Recombinant human GM-CSF (hGM-CSF) was purchased from Immunex (Seattle, Washington, USA). The 1,25-(OH)2D3 was a generous gift from Milan Uskokovic (Hoffman La Roche Inc., Nutley, New Jersey, USA). The 23c6 mAb that identifies human OCLs was provided by Michael Horton (St. Bartholomew’s Hospital, London, United Kingdom). Highly purified bovine lung AXII was purchased from BIODESIGN International (Kennebunk, Maine, USA). Neutralizing mAb to GM-CSF was purchased from R&D Systems Inc. (Minneapolis, Minnesota, USA).
Human bone marrow cultures.
After obtaining informed consent, 2 mL of bone marrow was aspirated from the posterior superior iliac crest of healthy normal volunteers into a 20-mL syringe containing 1 mL of α-MEM and 1,000 U/mL of preservative-free heparin. The bone marrow was then processed as described previously (10). Briefly, the mononuclear cell fraction was obtained by density gradient centrifugation over Ficoll-Hypaque, and the mononuclear cells were incubated in α-MEM/20% FCS at 5 × 106 cells/mL overnight at 37°C in 100-mm tissue culture plates (Fisher Scientific, Houston, Texas, USA) to deplete adherent cells. The bone marrow nonadherent mononuclear cells were collected and used for cultures of immature mononuclear OCL precursors (CFU-GM) and formation of OCL-like multinucleated cells, as described previously (11–14). These studies were approved by the Institutional Review Board of the University of Texas Health Science Center at San Antonio.
In brief, CFU-GM colonies were formed by culturing 105 nonadherent mononuclear marrow cells per milliliter in methylcellulose in 35-mm tissue culture plates (Fisher Scientific). AXII (0–100 ng/mL) was added to the cultures in the presence or absence of 50–100 pg/mL hGM-CSF. The number of colonies (40 or more cells) were scored after 14 days of culture.
OCL-like multinucleated cells were formed by culturing 105 nonadherent bone marrow mononuclear cells in 0.1 mL of α-MEM/20% horse serum for 3 weeks in 96-well plates (Fisher Scientific). Half the media was changed weekly. Cells were incubated with either 100 ng/mL AXII, 10–8 M 1,25-(OH)2D3, or media alone for 1, 2, or 3 weeks. After 3 weeks, the cultures were fixed and then stained with the 23c6 anti–vitronectin receptor mAb, which identifies multinucleated cells that express calcitonin receptors and form resorption lacunae on calcified matrices, as described previously (14). Cells that contained 3 or more nuclei and reacted with the 23c6 antibody were scored as OCL-like multinucleated cells. To determine the average number of nuclei per multinucleated cell, the number of nuclei in 100 random multinucleated cells per culture were scored in 3 independent experiments.
Autoradiography.
Human marrow nonadherent mononuclear cells (5 × 105 per well) were cultured in the presence or absence of 100 ng/mL AXII in 4-well LabTek chamber slides (Nalge Nunc International, Naperville, Illinois, USA). [3H]thymidine (0.5 mCi/mL) was added to the cells for 48 hours and then replaced with fresh media that did not contain [3H]thymidine; cells were incubated for 3 weeks as described previously (15). The cells were then fixed in glutaraldehyde for 15 minutes, stained for tartrate-resistant acid phosphatase (TRAP) using a commercially available kit (Sigma Chemical Co., St. Louis, Missouri, USA), and then processed for autoradiography as described by Takahashi et al. (16). Results are expressed as the number of labeled nuclei in multinucleated cells containing 1 or more labeled nuclei and as the number of labeled nuclei in OCL-like multinucleated cells (assessed from the first 100 multinucleated cells).
Expression of AXII mRNA in OCL-like cells.
The EcoRI fragment of the AXII cDNA was excised from the pcDNA1 and subcloned into pBluescript II KS+ (Stratagene, La Jolla, California, USA). We prepared 35S-labeled RNA probes with T7 and T3 RNA polymerase. After ethanol precipitation, the probes were degraded into small fragments (∼150 bp) in 0.1 M sodium carbonate buffer (pH 10.2) for 30 minutes. Highly purified OCL-like giant cells from human giant cell tumors were prepared as described previously (2) and were fixed in 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.3) in 8-well chamber slides. The slides were rinsed in 4× SSC and then prehybridized in 2× SSC containing 50% formamide at 50°C for 3 hours. Antisense or sense RNA probes were added with the hybridization buffer (50% formamide, 2× SSC, 1 μg/mL tRNA, and 10% dextran sulfate) at a concentration of 104 dpm/μL and hybridized for 24 hours at 42°C. The slides were then washed, dipped in autoradiographic emulsion NR-M2 (Konica, Tokyo, Japan), exposed for 7 days, and developed.
Effect of anti–GM-CSF on the stimulating effect of AXII on OCL formation.
To examine the possible role of GM-CSF as mediator for the effect of AXII, different concentrations of a neutralizing polyclonal anti–GM-CSF (R&D Systems Inc.) or control rabbit IgG were added to the bone marrow culture in the presence of 100 ng/mL of AXII or 1,25-(OH)2D3 (10–8 M). One microgram of the anti–GM-CSF can neutralize 1 ng of GM-CSF. After 3 weeks of culture, OCLs were scored as described above.
Effect of AXII on GM-CSF expression.
To determine if AXII can regulate GM-CSF expression, GM-CSF concentrations were determined at the protein and mRNA levels in human marrow cultures treated with or without 200 ng/mL of AXII for 48 hours. GM-CSF protein concentration was determined using an ELISA kit following the manufacturer’s protocol (Genzyme Pharmaceuticals, Cambridge, Massachusetts, USA). For GM-CSF mRNA levels, semiquantitative RT-PCR was performed as follows: RNA was reverse transcribed using Maloney virus reverse transcriptase, and 5 μL from each reaction was amplified using 5′-AGCCCTGGGAGCATGTGAAT-3′ (85–102 bp) as the sense primer and 5′-GTTGCACAGGAAGTTTCCGG-3′ (344–325 bp) as the antisense primer (GenBank accession no. M10663). The parameters for PCR were as follows: 94°C for 1 minute, 60°C for 1 minute, and 60°C for 7 minutes as the final step. Different numbers of cycles were used to quantify the amount of GM-CSF mRNA expressed. To monitor the amount of RNA in each reaction, β-actin mRNA expression was used. In each reaction, 0.5 μL of [32P]dCTP (3,000 Ci/mmol) was added. Nondenaturing PAGE (5%) was used for separation of PCR products, and the bands were quantified by autoradiography.
Effect of 1,25-(OH)2D3 on AXII expression during OCL differentiation.
AXII mRNA expression was evaluated in early human OCL precursors, more differentiated OCL precursors, and mature OCL-like cells, as described previously (14). RNA was extracted from highly purified populations of each cell type, and RT-PCR was performed using AXII-specific primers (GenBank accession no. M14043): 5′-GCTTCCTTCGAATTCAATGTCTACTGTTCAC-3′ (corresponding to +38–64 bp of the sequence) for sense and 5′-GCCCCTTCATGGAAGCTTTTAGCTCAGAAG-3′ (corresponding to +381–410 bp of the sequence) for antisense. To monitor the amount of RNA, β-actin mRNA expression was used. PCR products were separated in 2% agarose gels and quantitated as described previously (17).
Fluorescence labeling of AXII.
AXII was conjugated to NHS- fluorescein using the NHS-Fluorescein Labeling Kit (Pierce Chemical Co., Rockford, Illinois, USA) according to the manufacturer’s instructions. Briefly, 100 μg of AXII (BIODESIGN International) was dissolved in 100 μL of 50 mM bicarbonate buffer (pH 8.5). NHS-fluorescein (0.1 mg in 200 μL of DMSO) was admixed with AXII, and coupling was continued for 2 hours at 4°C in the dark. Untreated NHS-fluorescein was removed using molecular size filtration by adding 0.1 M phosphate buffer (pH 7.0) to the reaction mixture, followed by concentration of the fluorescent AXII conjugate using a Centricon-30 microconcentrator (Millipore Corp., Danvers, Massachusetts, USA).
Binding of AXII-fluorescein to PSV10 stromal cells.
PSV10 cells, a human marrow stromal cell line that supports OCL formation (18), were cultured in T75 flasks in α-MEM culture medium containing 10% FCS (both from GIBCO BRL, Grand Island, New York, USA), and the cells were detached from plastic by scraping, washed 2 times with PBS, and resuspended to 106 cells/mL in PBS. Aliquots of 105 cells per tube were incubated with increasing amounts of AXII-fluorescein (0–4 μg/mL). Ten microliters of 10× CaCl2-containing annexin-binding buffer (CLONTECH Laboratories Inc., Palo Alto, California, USA) was admixed to each tube, and binding was continued for 30 minutes at 4°C. Unbound AXII was removed by centrifugation, and the amount of bound AXII-fluorescein was determined by flow cytometry (FACStar Plus; Becton Dickinson Immunocytometry Systems, San Jose, California, USA). Analysis of fluorescence intensity and peak mean channel (PMC) was determined using CellQuest software (Becton Dickinson Immunocytometry Systems). Fifty thousand cells were analyzed. To test the requirement of Ca2+ for binding AXII, the binding buffer (containing Ca2+) was omitted. To test the specificity of AXII binding, unlabeled AXII (2.5 μg) was added first to the cells, and the incubation was continued for 10 minutes at 4°C, after which 0.5 μg of AXII-fluorescein was added for 30 minutes at 4°C.
Sorting of T cells and binding of AXII-fluorescein to flow-sorted T cells.
Bone marrow mononuclear cells were prepared as described above and washed 3 times with α-MEM culture medium containing 20% FCS. Cells were resuspended at 2 × 106 cells/mL in the same media and were allowed to adhere to plastic in T75 flasks for 4–16 hours to remove monocytes. The monocyte-depleted fraction was collected and stained with either anti-CD3– PE (for purification of T cells), anti-CD4– PE, or anti-CD8– PE (for purification of T-cell subsets). All antibodies were from Becton Dickinson Immunocytometry Systems and were used at 10 μL/106 cells in 0.1 mL PBS + 1% BSA. Tubes were incubated for 30 minutes at 4°C. Unbound antibody was removed by centrifugation. Cells were sorted using the FACStar Plus cell sorter, and an aliquot of each fraction was reanalyzed to determine its purity. Purified T cells or T-cell subset aliquots of 100,000 cells per tube were incubated with increasing amounts of AXII-fluorescein (0–4 μg/mL). Ten microliters of 10× binding buffer (containing CaCl2) was admixed to each tube, and binding continued for 30 minutes at 4°C. Dependency of AXII binding on Ca2+, the specificity of binding, and analysis of the results were determined as described above for PSV10 cells.
Statistical analysis.
Results, expressed as mean ± SEM, were calculated. Differences between treatment groups were analyzed by a two-way ANOVA for repeated measures. Differences were considered significant for P < 0.05.
Results
AXII mRNA expression during OCL precursor differentiation.
We previously cloned AXII from a cDNA library prepared from highly purified OCL-like cells (5) and wanted to determine when AXII was expressed during OCL precursor differentiation. As shown in Figure 1a, AXII transcripts were induced by 1,25-(OH)2D3 in early OCL precursors (CFU-GM) after 24 hours, and AXII expression was sustained during the later stages of OCL precursor differentiation. We confirmed that AXII was also expressed in OCL-like multinucleated cells formed in human marrow cultures, as shown by RT-PCR analysis of AXII mRNA expression by highly purified multinucleated cells and by freshly isolated OCL-like giant cells from human osteoclastomas by in situ hybridization (Figure 1b).
Figure 1.

(a) Effects of 1,25-(OH)2D3 on AXII mRNA expression during OCL differentiation. Total RNA has been isolated from mature OCL-like multinucleated cells and OCL precursors treated with 1,25-(OH)2D3 (10–8 M) at the times indicated. Semiquantitative RT-PCR was performed using specific primers for AXII as described in Methods. β-actin was used as internal control for amount of RNA isolated and the integrity of the RNA. (b) In situ hybridization of AXII RNA in multinucleated OCLs from human giant cell tumors of bone. Giant cells were prepared, fixed, and incubated with sense and antisense probes for AXII in 4-well LabTek chamber slides, and autoradiography was performed as described in Methods. The dark granules on the cells are magnetic beads used in the immunopurification of the OCL-like cells.
Effects of AXII on OCL formation.
We have previously shown that AXII increases OCL-like cell formation in both human and murine marrow cultures (5). Therefore, to characterize the time course for the effects of AXII on OCL formation, human bone marrow cultures were treated with AXII for varying time periods. As shown in Figure 2, AXII increased OCL-like multinucleated cell formation in human bone marrow cultures. However, the stimulating effects of AXII occurred only if AXII was present during the first week of culture. No increase in OCL formation was observed if AXII was added only during the second or third week of culture (Figure 2). These results suggested that AXII affected the proliferative stage of OCL precursors.
Figure 2.

Effect of varying treatment time with AXII on formation of 23c6Ab+ multinucleated cells in culture of normal human bone marrow. Human bone marrow mononuclear cells (105 cells per well) were cultured in 96-well plates in α-MEM and 20% horse serum, with AXII (100 ng/mL) for 1, 2, or 3 weeks, followed by treatment with media alone. In selected experiments, AXII was added to cultures for only the second and third weeks, or the third week only, of culture. Values are mean ± SEM of triplicate determinations in 5 independent experiments and are expressed as a percentage of the number of multinucleated cells formed by cells cultured in media alone for 3 weeks. *P < 0.02, significantly different from control cultures (3 weeks media alone).
To confirm that AXII was stimulating proliferation of precursors for OCL-like multinucleated cells, [3H]thymidine incorporation studies were performed. These data showed that treatment of cultures with AXII (100 ng/mL) significantly increased the number of multinucleated cells per culture (Figure 3a) (Table 1), as well as the percentage of multinucleated cells containing labeled nuclei (Figure 3b) and total number of labeled nuclei in multinucleated cells (Table 1). Most multinucleated cells contained 1 labeled nucleus per cell, whether treated with AXII, 1,25-(OH)2D3, or untreated controls. Although AXII increased the total number of multinucleated cells, it did not increase the average number of nuclei per multinucleated cell (Table 1), suggesting that it was not affecting the rate of cell fusion in these cultures. In contrast, 1,25-(OH)2D3, which enhances cell fusion but not OCL precursor proliferation (16), did increase the average number of nuclei per multinucleated cell. AXII did not enhance the growth of other cell types. For example, 48-hour AXII treatment of subconfluent stromal cells (2 × 105 cells) derived from giant cell tumors of bone did not result in increased cell growth compared with untreated cells (control: 106 cells; AXII treatment: 1.1 × 106 cells; mean of duplicate determinations).
Figure 3.

[3H]thymidine incorporation of OCL-like multinucleated cells formed in response to AXII. Normal human bone marrow nonadherent mononuclear cells (5 × 105 cells per culture) were cultured in 4-well LabTek chamber slides in the presence or absence of AXII. After 24 hours of culture, [3H]thymidine (0.5 mCi/well) was added for the next 48 hours. Cells were fixed and stained for TRAP and then processed for autoradiography. The number of TRAP+ multinucleated cells (a) and percentage of multinucleated cells containing labeled nuclei (b) were scored. Results are expressed as the mean ± SEM for 3 or 4 cultures from a typical experiment. Similar results were seen in 3 independent experiments. *P < 0.05, significantly different from control cultures (media alone).
Table 1.
Effect of AXII on formation of 23c6Ab+ MNC and lack of effect on number of nuclei per MNC in culture of normal human bone marrow

We then determined if the increase in OCL precursor proliferation by AXII was due to enhanced growth of CFU-GM, the earliest identifiable OCL precursor. AXII was added to marrow cultures treated with GM-CSF, and the number of CFU-GM colonies formed was determined. AXII significantly increased CFU-GM colony formation in methyl cellulose cultures treated with low doses of GM-CSF in a dose-dependent manner (Figure 4). Addition of AXII to methyl cellulose cultures treated with maximum stimulatory doses of GM-CSF (1 ng/mL) did not further enhance CFU-GM colony formation (data not shown). These data suggest that AXII’s effects on CFU-GM growth may result from induction of GM-CSF.
Figure 4.

Effect of AXII on CFU-GM colony formation. Human bone marrow mononuclear cells (105 cells/mL) were cultured in 35-mm plates in α-MEM with 30% FCS in methylcellulose. Varying concentrations of AXII (0–100 ng/mL) were added to the cultures in the presence of 100 pg/mL hGM-CSF. CFU-GM–derived colonies that are composed of 40 or more cells were counted. Results are expressed as the mean ± SEM for 4 cultures from a typical experiment. Similar results were seen in 3 independent experiments. *P < 0.05, significantly different from cultures treated with 100 pg/mL GM-CSF alone.
Effects of GM-CSF neutralization on the stimulating effects of AXII on OCL formation.
To assess further if GM-CSF was mediating the stimulatory effects of AXII on OCL formation, human bone marrow cultures were treated with AXII (100 ng/mL) in the presence or absence of different concentrations of a neutralizing antibody against GM-CSF. As shown in Figure 5a, the stimulatory effects of AXII on OCL formation were blocked by addition of a neutralizing antibody to GM-CSF. This effect was dose dependent and significant at 0.1–1 μg/mL of antibody. In contrast, anti–GM-CSF did not affect OCL formation stimulated by 1,25-(OH)2D3 (Figure 5b). These results demonstrated that the anti–GM-CSF was not toxic to the cultures, and suggested that GM-CSF mediated the effects of AXII on OCL formation.
Figure 5.

Effects of anti–GM-CSF on AXII-stimulated OCL formation. Human bone marrow (105 cells/mL) was cultured with AXII (0–100 ng/mL) (a) or 1,25-(OH)2D3 (10–8 M) (b) in the presence or absence of increasing concentrations of neutralizing antibodies to GM-CSF (0.001–5.0 μg/mL) in 96-well plates. Cells were fed weekly by changing half the media. After 3 weeks, multinucleated OCL-like cells were scored as described. Results represent the mean ± SEM of quadruplicate determinations from a typical experiment. The results were similar in 2 experiments.
Effects of AXII on GM-CSF expression.
Because the effects of AXII were blocked by a neutralizing antibody to GM-CSF, we then determined if AXII upregulated GM-CSF gene expression or simply induced release of GM-CSF. For this purpose, GM-CSF levels were determined in the conditioned media of human bone marrow cultures. As determined by ELISA, GM-CSF levels were increased in AXII-treated cultures (250 ± 30 pg/mL/2.5 × 105 cells; mean ± SEM) compared with control cultures (<10 pg/mL/2.5 × 105 cells; mean ± SEM). Semiquantitative RT-PCR demonstrated that AXII also upregulated GM-CSF mRNA expression. As shown in Figure 6, 48-hour exposure of nonadherent bone marrow cultures to AXII increased GM-CSF mRNA expression more than 3-fold above control values after normalization for β-actin (determined by densitometry of data presented in Figure 6 during the log phase of the PCR reaction).
Figure 6.

Effects of AXII on GM-CSF mRNA expression in human bone marrow cultures. Total RNA was extracted from human bone marrow cultures treated with or without 100 ng/mL of AXII for 48 hours. GM-CSF mRNA expression was measured by RT-PCR using specific primers as described. β-actin was used as control for the amount and integrity of the RNA isolated. In parallel experiments, conditioned media were collected and used to quantify GM-CSF protein levels by ELISA, following the manufacturer’s protocol.
AXII expression by cells in the bone marrow microenvironment.
To determine if other cells expressed AXII in the marrow microenvironment, Northern blot analysis was performed with different types of hematopoietic and nonhema-topoietic cells. As shown in Figure 7, AXII was highly expressed in stromal and osteoblast cell lines. However, it was expressed at lower levels — or not expressed at all — in cells exhibiting monocyte-macrophage characteristics, such as HL-60 cells (none detected) or U937 cells.
Figure 7.
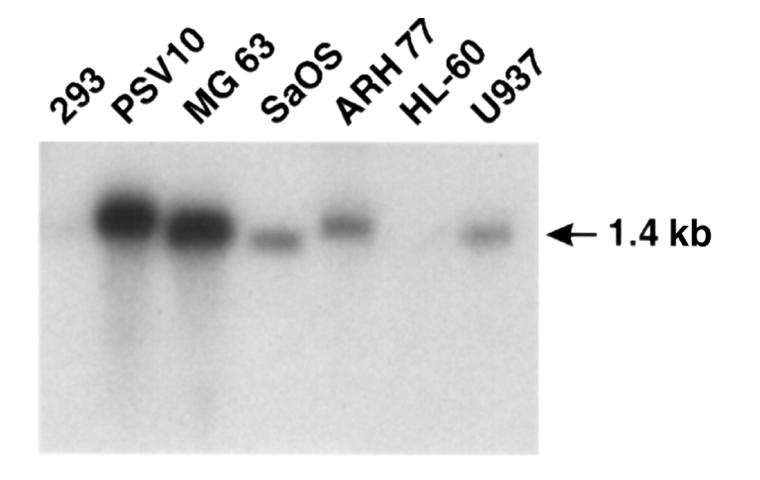
Northern blot analyses for expression of AXII mRNA. Northern blot analysis of AXII mRNA was performed using multiple cell lines, and the blot was probed with a 32P-labeled AXII cDNA probe or a β-actin probe, as described in Methods.
Effect of AXII on GM-CSF expression in stromal cells and T cells.
To identify the cellular target in bone marrow for the induction of GM-CSF by AXII, Saka and PSV10 cells, which are human marrow stromal cell lines derived from normal volunteers and Paget’s disease patients, respectively, were treated with 100 ng/mL of AXII for 48 hours. In parallel experiments, highly purified (∼90%) T cells were also used as targets. Semiquantitative RT-PCR analysis showed that AXII induced GM-CSF mRNA expression in T cells (Figure 8) but did not increase the already high expression levels of GM-CSF by stromal cells (data not shown).
Figure 8.

Effects of AXII on GM-CSF gene expression in T cells. GM-CSF expression was assessed by RT-PCR analysis using total RNA isolated from T cells incubated with or without 0–1 μg/mL of AXII. Different cycles of PCR were used as described. β-actin was used as control for the integrity and amount of RNA in each sample. Densitometry analysis was performed to compare the expression levels of GM-CSF.
Binding of AXII to T cells and marrow stromal cells.
Because AXII enhanced GM-CSF expression in T cells, FACS analysis was used to determine if T cells and stromal cells expressed a putative AXII receptor. The PMC for the fluorescence intensity was used to estimate the number of putative AXII receptor molecules on the surface of cells. CD3+ T cells were purified by flow sorting using a FACStar Plus cell sorter. A T-cell fraction with >97% T cells was obtained (Figure 9a). The T cell–negative fraction was also sorted, with a purity of >99% (Figure 9b). Titration of AXII-fluorescein binding to the flow-sorted CD3+ and CD3– cells is depicted in Figure 9c. The results indicate a linear binding of AXII to CD3+ cells between 0.1 and 0.5 μg/mL, with a saturation between 0.5 and 1 μg/mL. Omitting calcium from the binding assay resulted in a ∼40% decrease in the binding of AXII-fluorescein to T cells. Furthermore, a 5-fold excess of unlabeled AXII effectively competed (>50% decrease) for the binding of AXII-fluorescein to T cells. In contrast to the CD3+ cell fraction, the CD3– cell fraction did not bind AXII-fluorescein (Figure 9c).
Figure 9.

Binding of AXII to CD3+ cells. Sorting was performed as described in Methods. The purity of each sorted fraction, CD3+ (a) and CD3– (b), was determined by analysis of the histogram. Binding of AXII and analyses were performed as described in Methods. Two independent experiments were performed and very similar results were obtained. Binding values were based on 100% conjugation of fluorescein to AXII. Ten thousand cells were analyzed. (c) Binding of AXII to CD3+ cells and CD3– cells.
To characterize further the T-cell subset that bound AXII, CD4+ and CD8+ T cells were sorted to a purity of 90% and 93%, respectively (data not shown), and we tested the binding of each subpopulation of T cells to AXII. The results are summarized in Figure 10. CD4+ T cells bound AXII-fluorescein between 0.1 and 0.5 μg/mL, with a maximum at 18 PMC, and was competed (40%) by unlabeled AXII. In contrast, CD8+ cells did not bind AXII-fluorescein (Figure 10).
Figure 10.

Analysis of AXII binding to flow-sorted CD4+ and CD8+ T-cell subsets. Sorting of bone marrow–derived T-cell subsets was performed as described in Methods. The purity of each sorted fraction (CD4+, CD8+) was determined by analysis of histograms, as was done for the CD3+ fraction. AXII-fluorescein binding and AXII competition assays were performed as described above for CD3+ (a) or CD3– (b) bone marrow cells. Ten thousand cells were analyzed. Results are reported as the percent change in fluorescence when the cells were exposed to AXII fluorescein. Basal fluorescence was assigned as 100% to permit comparisons of the 2 T-cell subpopulations. Three independent experiments were performed and very similar results were obtained.
Marrow stromal cells also expressed a putative AXII receptor. Figure 11 depicts a titration curve for AXII-fluorescein binding to PSV10 cells between 0 and 1 μg/mL. Linear binding of AXII was observed in the range of 0.1–0.25 μg/mL, assuming a 100% efficiency of conjugation of AXII to fluorescein. Under our experimental conditions, complete saturation was observed at ∼0.7 μg/mL. Binding of AXII-fluorescein to PSV10 cells was reduced to 35% in the absence of calcium and was effectively competed (>50%) by a 5-fold excess of unlabeled AXII (Figure 11). The fluorescence intensity was about 1 log higher in the PSV10 cells than in T cells, suggesting a smaller number of AXII receptors per cell on T cells compared with the number of receptors per cell on PSV10 cells. However, correcting for the size difference between the 2 cell types (7 μm and 20 μm in diameter), the density per surface area was approximately equal.
Figure 11.

AXII binding to PSV10 cells. Cells (105/0.1 mL of PBS) were used for each analysis, and 10,000 cells were collected and analyzed as described in Figure 9. Four experiments were performed and 1 typical experiment is shown. Binding values are based on the assumption of 100% conjugation of fluorescein to AXII.
Discussion
The OCL secretes factors that can regulate its own formation and/or activation (1). In addition to IL-6, AXII, a calcium-dependent phospholipid-binding protein, was identified as a secreted stimulator of OCL formation and activation (5). Our data demonstrate that AXII expression is regulated by 1,25-(OH)2D3. We found that AXII expression was induced by 1,25-(OH)2D3 during OCL differentiation. AXII expression could be detected as early as 24 hours after treatment with 1,25-(OH)2D3.
Other cells in the bone microenvironment express AXII, including stromal cells and osteoblasts. These data suggest that AXII may have both autocrine and paracrine effects on OCL formation and activation. Such a possibility is supported by studies by Nesbitt and Horton (6), who demonstrated that AXII plays a role in the clearance of degraded bone matrix by OCLs. These authors suggested that AXII binds the degraded matrix protein and carries it across the cell in order for the degraded matrix to be released on the opposite side of the bone-resorbing surface.
In this report, we examined the mechanism(s) by which AXII, as a soluble extracellular protein, increases OCL formation. During OCL formation, OCL precursors undergo several stages of differentiation until they become multinucleated bone-resorbing OCLs. During early stages of OCL precursor differentiation, the cells proliferate in response to factors such as IL-6 and GM-CSF, thereby increasing the OCL precursor pool for subsequent OCL formation (1). This proliferative phase is followed by a terminal differentiation and fusion phase leading to formation of multinucleated cells (14). Factors such as RANK ligand act predominantly at this later stage of OCL precursor differentiation (1).
Our data suggest that AXII acts at the proliferative stage of OCL precursor differentiation. This conclusion is based on the following results. First, the stimulating effect of AXII on OCL formation occurred during the early stages of the bone marrow culture (week 1). In contrast, no significant effect of AXII was observed when it was added during the later stages of the cultures. Second, AXII was able to increase the number of labeled nuclei per OCL-like multinucleated cell, further suggesting that it was enhancing OCL precursor proliferation. Third, AXII enhanced the effects of GM-CSF on the growth of CFU-GM, the earliest identifiable OCL precursor (14). Taken together, these data suggested that AXII enhanced the proliferation of early OCL precursors. Our results do not support the possibility that the stimulating effect of AXII was partly due to increasing the cell fusion rate of more mature OCL precursors, because no effect was seen when AXII was added to highly purified OCL late precursors, which are postmitotic cells (14) (data not shown). Consistent with these results was the fact that there was no increase in the average number of nuclei per multinucleated cell, an indirect measure of the fusion rate of OCL precursors (19).
We then examined how AXII increased the proliferation of OCL precursors. As noted above, AXII enhanced the effects of GM-CSF on CFU-GM colony formation. AXII by itself had no effect on CFU-GM growth, suggesting that it was not by itself a colony-stimulating factor, but could induce GM-CSF production in marrow cultures. Consistent with this possibility are our findings that neutralizing antibodies to GM-CSF blocked the stimulatory effects of AXII on OCL formation in a dose-dependent manner. Furthermore, AXII induced GM-CSF production in bone marrow cultures, as determined by ELISA. RT-PCR analysis showed that this increase in GM-CSF was due, in part, to the increase in levels of GM-CSF mRNA.
We then determined which of the cell types present in these marrow cultures may be producing GM-CSF in response to AXII. GM-CSF is secreted by activated T cells (20), activated macrophages (21), and endothelial cells (22), and it is also expressed by osteoblastic-like marrow stromal cells (20–23). Therefore, because T cells and stromal cells are present in the microenvironment of OCL precursors, we determined if AXII was able to induce GM-CSF expression in a human stromal cell line and highly purified T cells. As shown in Figure 6, AXII increased GM-CSF expression after 48 hours in bone marrow cultures, as assessed by RT-PCR; this increase in GM-CSF expression was not observed when human stromal cells such as PSV10 or Saka cells, which produced GM-CSF mRNA constitutively, were treated with AXII. Furthermore, flow cytometric studies demonstrated that CD4+ T cells appear to be the T-cell subpopulation responding to AXII. These data support the finding of others for the role of T cells in OCL formation through the production of cytokines such as IL-17, IL-6, and IL-4 (24).
Preliminary studies were then undertaken to determine if T cells or stromal cells expressed AXII receptors. Recently, Hajjar et al. (25) demonstrated that AXII binds to the endothelial cell membranes in a saturable, equilibrium-based manner. Similarly, Liu et al. (26) have reported calcium-dependent and calcium-independent binding of AXII to A549 cells. Our FACS analysis studies using FITC-conjugated AXII demonstrated that AXII binds to the CD4+ T-cell membrane in a saturable manner. Moreover, this binding is partially inhibited by an excess of nonlabeled AXII in a dose-dependent manner and is calcium dependent. These data suggest that CD4+ T cells may express a receptor for AXII. It is unlikely that the binding of AXII to T cells simply represents nonspecific binding of AXII to phospholipid, a property of AXII (27), because AXII does not bind to CD8+ T cells under the same conditions and could be competed in a dose-dependent manner by excess unlabeled AXII.
In summary, AXII is an autocrine/paracrine factor produced by OCLs, as well as marrow stromal cells and osteoblasts, that increases OCL formation. AXII expression is regulated by 1,25-(OH)2D3 and increases with OCL precursor differentiation. This effect of AXII on OCL formation is due to an increase in the proliferation of early OCL precursors through the induction of GM-CSF by CD4+ T cells, although we cannot rule out a role for stromal cells in this process. This increase in GM-CSF results from an increase in the steady-state levels of GM-CSF mRNA in CD4+ T cells. The interactions of AXII with T cells and stromal cells appear to be mediated through a putative AXII receptor. Thus, the effects of AXII are similar to those of IL-6 and TGF-α, factors that stimulate the proliferation of early OCL precursors (14, 16) and represent a potentially important mechanism for regulating OCL activity.
Acknowledgments
The authors wish to thank Judy Anderson for technical assistance with human bone marrow preparation, and Bibi Cates for excellent preparation of this manuscript. Supported by research funds from the Veterans Administration; grant AG-13625 from the National Institute on Aging; grant CA-40035 from the National Cancer Institute; and grants AR-44603 and AR-41336 from the National Institute of Arthritis and Musculoskeletal and Skin Diseases.
References
- 1.Reddy SV, Roodman GD. Control of osteoclast differentiation. Crit Rev Eukaryot Gene Expr. 1998;8:1–17. doi: 10.1615/critreveukargeneexpr.v8.i1.10. [DOI] [PubMed] [Google Scholar]
- 2.Ohsaki Y, et al. Evidence for an autocrine/paracrine role for interleukin-6 in bone resorption by giant cells from giant cell tumors of bone. Endocrinology. 1992;131:2229–2234. doi: 10.1210/endo.131.5.1425421. [DOI] [PubMed] [Google Scholar]
- 3.Oursler MJ. Osteoclast synthesis, secretion and activation of latent transforming growth factor beta. J Bone Miner Res. 1994;9:443–452. doi: 10.1002/jbmr.5650090402. [DOI] [PubMed] [Google Scholar]
- 4.Choi SJ, et al. Cloning and identification of human Sca as a novel inhibitor of osteoclast formation and bone resorption. J Clin Invest. 1998;12:1360–1368. doi: 10.1172/JCI2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takahashi S, et al. Cloning and identification of AXII as an autocrine/paracrine factor that increases osteoclast formation and bone resorption. J Biol Chem. 1994;269:28696–28701. [PubMed] [Google Scholar]
- 6.Nesbitt S, Horton MA. Osteoclast annexins bind collagen and play a role in bone resorption. J Bone Miner Res. 1995;10(Suppl. 1):S221. (Abstr.) [Google Scholar]
- 7.Siever DA, Erickson HP. Extracellular annexin II. Int J Biochem Cell Biol. 1997;29:1219–1223. doi: 10.1016/s1357-2725(97)00057-5. [DOI] [PubMed] [Google Scholar]
- 8.Uy HL, et al. Use of an in vivo model to determine the effects of interleukin-1 on cells at different stages in the osteoclast lineage. J Bone Miner Res. 1995;10:295–301. doi: 10.1002/jbmr.5650100217. [DOI] [PubMed] [Google Scholar]
- 9.Yasuda H, et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/ osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci USA. 1998;95:3597–3602. doi: 10.1073/pnas.95.7.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.MacDonald B, et al. Formation of multinucleated cells that respond to osteotropic hormones in long term human bone marrow cultures. Endocrinology. 1987;120:2326–2333. doi: 10.1210/endo-120-6-2326. [DOI] [PubMed] [Google Scholar]
- 11.Kukita T, McManus L, Miller M, Civin C, Roodman GD. Osteoclast-like cells formed in long-term human bone marrow cultures express a similar surface phenotype as authentic osteoclasts. Lab Invest. 1989;60:532–538. [PubMed] [Google Scholar]
- 12.Takahashi N, et al. Osteoclast-like cells form in long-term human bone marrow but not in peripheral blood cultures. J Clin Invest. 1989;83:543–550. doi: 10.1172/JCI113916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pfeilschifter J, Chenu C, Bird A, Mundy GR, Roodman GD. Interleukin-1 and tumor necrosis factor stimulate the formation of human osteoclast-like cells in vitro. J Bone Miner Res. 1989;4:113–118. doi: 10.1002/jbmr.5650040116. [DOI] [PubMed] [Google Scholar]
- 14.Kurihara N, Chenu C, Miller M, Civin C, Roodman GD. Identification of committed mononuclear precursors for osteoclast-like cells formed in long-term human marrow cultures. Endocrinology. 1990;126:2733–2741. doi: 10.1210/endo-126-5-2733. [DOI] [PubMed] [Google Scholar]
- 15.Roodman GD, Hutton J, Bollum F. DNA polymerase, thymidine kinase and DNA synthesis in erythropoietic mouse spleen cells separated on bovine serum albumin gradients. Biochim Biophys Acta. 1976;425:478–491. doi: 10.1016/0005-2787(76)90012-5. [DOI] [PubMed] [Google Scholar]
- 16.Takahashi N, et al. Recombinant human transforming growth factor-alpha stimulates the formation of osteoclast-like cells in long-term human marrow cultures. J Clin Invest. 1986;78:894–898. doi: 10.1172/JCI112677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ausubel, F.M., et al. 1996. Current protocols in molecular biology, volume 2. John Wiley & Sons. New York, NY. 15.3.1–15.3.5.
- 18.Takahashi S, Singer FR, Roodman GD. Pagetic marrow stromal cell lines enhance CFU-GM and osteoclast-like cell formation. J Bone Miner Res. 1993;8:S285. (Abstr.) [Google Scholar]
- 19.Hentunen TA, et al. Development of an osteoclast cell line by targeting of bcl-XL and simian virus 40 large T antigen to the osteoclast lineage in transgenic mice. J Clin Invest. 1998;102:88–97. doi: 10.1172/JCI2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tobler A, Gasson J, Reichel H, Norman AW, Koeffler HP. Granulocyte-macrophage colony-stimulating factor. Sensitive and receptor-mediated regulation by 1,25-dihydroxyvitamin D3 in normal human peripheral blood lymphocytes. J Clin Invest. 1986;79:1700–1705. doi: 10.1172/JCI113009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thorens B, Mermod JJ, Vassalli P. Phagocytosis and inflammatory stimuli induce GM-CSF mRNA in macrophages through post-transcriptional regulation. Cell. 1987;48:671–679. doi: 10.1016/0092-8674(87)90245-5. [DOI] [PubMed] [Google Scholar]
- 22.Munker R, Gasson JC, Ogawa M, Koeffler HP. Recombinant human tumor necrosis factor induces production of granulocyte-macrophage colony-stimulating factor mRNA and protein from lung fibroblasts and vascular endothelial cells in vitro. Nature. 1986;323:79–82. doi: 10.1038/323079a0. [DOI] [PubMed] [Google Scholar]
- 23.Weir EC, Insogna KL, Horowitz MC. Osteoblast-like cells secrete granulocyte-macrophage colony-stimulating factor in response to parathyroid hormone and lipopolysaccharide. Endocrinology. 1989;124:899–904. doi: 10.1210/endo-124-2-899. [DOI] [PubMed] [Google Scholar]
- 24.Tamai M, Sagawa K, Kawabata R, Inoue A, Itoh K. Production of IL-6 by T cells from the femoral head of patients with rapidly destructive coxopathy (RDC) Clin Exp Immunol. 1996;103:506–513. doi: 10.1111/j.1365-2249.1996.tb08309.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hajjar KA, Guevara CA, Lev E, Dowling K, Chako J. Interaction of the fibrinolytic receptor, Annexin II, with the endothelial cell surface. J Biol Chem. 1996;271:21652–21659. doi: 10.1074/jbc.271.35.21652. [DOI] [PubMed] [Google Scholar]
- 26.Liu L, Tao JQ, Zimmerman UJ. Annexin II binds to the membrane of A549 cells in a calcium-dependent and calcium-independent manner. Cell Signal. 1997;9:299–304. doi: 10.1016/s0898-6568(97)89891-9. [DOI] [PubMed] [Google Scholar]
- 27.Raynal P, Pollard HB. Annexins: the problem of assessing the biological role for a gene family of multifunctional calcium- and phospholipid-binding proteins. Biochim Biophys Acta. 1994;1197:63–93. doi: 10.1016/0304-4157(94)90019-1. [DOI] [PubMed] [Google Scholar]