

Abstract
Although lymphopenia is a hallmark of severe infection with highly pathogenic H5N1 and the newly emerged H7N9 influenza viruses in humans, the mechanism(s) by which lethal H5N1 viruses cause lymphopenia in mammalian hosts remains poorly understood. Because influenza-specific T cell responses are initiated in the lung draining lymph nodes, and lymphocytes subsequently traffic to the lungs or peripheral circulation, we compared the immune responses in the lung draining lymph nodes following infection with a lethal A/HK/483/97 or non-lethal A/HK/486/97 (H5N1) virus in a mouse model. We found that lethal H5N1, but not non-lethal H5N1 virus infection in mice enhances Fas ligand (FasL) expression on plasmacytoid dendritic cells (pDCs), resulting in apoptosis of influenza-specific CD8+ T cells via a Fas-FasL mediated pathway. We also found that pDCs, but not other DC subsets, preferentially accumulate in the lung draining lymph nodes of lethal H5N1 virus-infected mice and that the induction of FasL expression on pDCs correlates with high levels of IL-12p40 monomer/homodimer in the lung draining lymph nodes. Our data suggest that one of the mechanisms of lymphopenia associated with lethal H5N1 virus infection involves a deleterious role for pDCs.
Introduction
H5N1 influenza A viruses that transmitted from poultry to humans in 1997 claimed the lives of six of the 18 people infected (1, 2). The virus re-emerged in 2003 and continues to cause infection, with a current cumulative total of 630 confirmed human cases, of which 375 have died (www.who.int/influenza/human_animal_interface/H5N1_cumulative_table_archives/en/). Leukopenia or lymphopenia at the time of admission to the hospital was a prominent feature in H5N1 infected patients with a severe or fatal outcome, but was not reported in individuals who had less severe disease. Indeed, lymphopenia is also a hallmark of severe H7N9 influenza virus infection (3). The mouse model has been used extensively to investigate the pathogenesis of H5N1 virus infection (4–6); the viruses are associated with a range of morbidity and mortality (7–9). With some exceptions the virulence in mice infected with human H5N1 isolates corresponds to the severity of disease in humans (5, 7, 10–12). The conventional approach to investigate the molecular basis for virulence is to study a pair of viruses that are associated with different levels of virulence in mice (8, 12–14). One such pair of viruses is A/Hong Kong/483/97 (HK/483) and A/Hong Kong/486/97 (HK/486). The case patient from whom HK/483 was isolated had a low total peripheral leukocyte count at hospital admission and ultimately succumbed to infection. In contrast, the HK/486 case patient did not display leukopenia and recovered (15). The outcome of infection with H5N1 viruses in mice also correlates strongly with a reduction in circulating numbers of leukocytes (8). Transient leukopenia that rebounded 4 to 5 days post infection was observed in mice infected with HK/486 or the control H1N1 virus influenza A/Puerto Rico/8/34 (PR8), while profound lymphopenia was observed following HK/483 infection in mice (8). The authors observed that lymphopenia in lethal HK/483 infection was associated with an increase in apoptosis in the spleen and lungs and they concluded that depletion of lymphocytes contributed to the virulence of HK/483 in mice (8). Indeed, Influenza viruses induce apoptosis in vitro in tissue culture (16, 17) and in peripheral blood monocytes (18, 19). Early lymphopenia has been described in influenza-infected patients, and experimental inoculation of humans with influenza virus caused a decrease in both T- and B- cell numbers during illness (20, 21).
The clearance of influenza virus by influenza-specific CD8+ T cells is primarily mediated by Fas-FasL, perforin, and TRAIL destruction of virus–infected cells (22–24). However, activated T cells are also Fas+ and are therefore susceptible to FasL- mediated killing (25). Previous studies have shown that a reduction in CD8+ T cell responses in lethal H2N2 influenza virus infection in mice is mediated by lymph node (LN) resident dendritic cells (DCs), especially plasmacytoid dendritic cells (pDCs) that express FasL and drive FasL-Fas induced T cell apoptosis (26, 27) in a dose-dependent manner. In addition, Fujikura et al. reported that FasL expression was induced in the lungs, including on CD11c+ cells (i.e. dendritic cells and alveolar macrophages), of mice following infection with a lethal dose of the laboratory strain influenza A/Puerto Rico/8/34 (H1N1) virus and prevention of FasL/Fas interaction by administration of a recombinant decoy receptor for FasL or a functional mutation in the FasL gene resulted in protection from lethal infection (28). In this study, we investigated the role of LN DCs in lymphopenia associated with H5N1 virus infection, comparing the degree of influenza-specific CD8+ T cell apoptosis in mice infected with lethal (HK/483) and non-lethal (HK/486) H5N1 viruses. Lymphopenia can result from impaired development or destruction of lymphocytes. Vogel et al. reported that H5N1 virus infection in mice led to massive lung damage and infection of respiratory DCs, and proposed that the migration of infected DCs into the thymus interferes with T lymphocyte selection processes (29). We show that lethal highly pathogenic avian influenza (HPAI) H5N1 influenza virus infection results in increased destruction of lymphocytes by up-regulation of FasL expression on pDCs and that FasL+ pDCs are responsible for apoptosis of influenza-specific CD8+ T cells. Adoptive transfer of wild-type pDCs into FasL-deficient mice confirmed that pDCs trigger Fas-FasL mediated apoptosis of influenza-specific CD8+ T cells. Thus, the elimination of influenza -specific CD8+ T cells by pDC-induced apoptosis contributes to lymphopenia in lethal HPAI H5N1 infection.
Materials and Methods
Mice
Male and female BALB/c mice (6–8 wk old) were obtained from Taconic (Taconic Farms, Inc; Germantown, NY). Clone-4 (CL-4) TCR transgenic mice (B10.Cg-H2d Tg(TcraCl4,TcrbCl4)1Shrm/ShrmJ) specific for HA533/HA529 epitope of H5N1 influenza viruses and BALB/c gld mice (CPt.C3-Tnfsf6gld/J) were obtained from University of Iowa, Iowa City, IA. All animal experiments were performed at the National Institutes of Health, in compliance with the guidelines of the NIAID/NIH Institutional Animal Care and Use Committee.
Virus
A/Hong Kong/483/97 (HK/483) and A/Hong Kong/486 (HK/486) H5N1 influenza viruses were amplified in the allantoic cavity of 10-day-old embryonated specific pathogen-free chicken eggs and stored at −80°C. The titer of the virus stock was determined in MDCK cells and is expressed as a 50% tissue culture infectious dose (TCID50).
Laboratory facility
All experiments using highly pathogenic H5N1 avian influenza viruses, including work with animals were conducted using enhanced biosafety level 3+ containment procedures.
Virus infection
BALB/c mice were anesthetized using isoflurane and were infected intranasally with either 10 LD50 or 0.1 LD50 of A/Hong Kong/483 or A/Hong Kong/486 in 50 µl of Leibovitz-15 (L-15) media (Life Technologies, Grand Island, NY).
Flow cytometry
Lymph node cells were stained with the following monoclonal Abs: rat anti-mouse CD8α (53-6.7), hamster anti-mouse CD11c (HL3) or rat anti-mouse CD45R (RA-6B2) (BD Bioscience, San Jose, CA). The anti-FasL (CD178) (MFL3) was purchased from eBioscience (San Diego, CA). For FasL staining, the cells were stained as previously described (27). Briefly, the cells were blocked with 1:100 rat serum, 1:100 hamster serum and 1:400 free streptavidin (Molecular Probe, Eugene, OR) on ice for 25 min. Cell were then washed twice and stained with biotin conjugated anti-FasL (MFL3) follow by streptavidin PE (BD Bioscience). All flow cytometry data were acquired by BD FACS Calibur or LSRII. The data were analyzed using Flowjo software (TreeStar, Ashland, OR).
CD8 CL-4 T cell purification and adoptive transfer
Spleens from CL-4 mice were removed and processed into a single cell suspension. CD8 T cells were purified using Dynabead Untouched mouse CD8 cell kit according to manufacturer’s instructions (Invitrogen, Carlsbad, CA). 2×106 of purified CD 90.1+ CL-4 cells were adoptively transferred intravenously into CD90.2+ BALB/c mice. At 24 h post-transfer, the recipient mice were infected I.N. with H5N1 influenza virus. Lung draining lymph nodes from infected CD90.2+ CL-4 transferred recipient mice were harvested on day 3 post-infection.
In vivo pDC transfer
Spleens from naive BALB/c wt or gld mice were harvested, and single cell suspensions were stained with anti-PDCA-1 microbeads and purified according to manufacturer’s instructions (Miltenyi Biotec, Auburn, CA). 2×106 of naïve BALB/c or gld pDCs were adoptively transferred intravenously into recipient wt or gld mice 24 h prior to influenza virus infection. On day 4 post-infection, lung draining LNs were harvested to evaluate influenza-specific T cell responses in the LNs.
In vitro culture of intact LNs
Lung draining LNs were harvested from naïve and infected BALB/c mice, immersed in Iscove’s media with 10% FCS (5LNs/500 µl) and cultured intact for 48 h at 37 °C in 95% O2/5%CO2 (26, 30). Supernatants from intact LN cultures were analyzed for total IL-12p40, IL-12p70 and IL-23 by capture ELISA according to manufacturer’s instructions (eBioscience, San Diego, CA). The levels of IL-1α, IL-1β, IL-6 and IL-10 in culture supernatant of LNs were determined by Bio-Plex multiplex system (Bio-Rad, Hercules, CA) according to manufacturer’s instructions.
Results
HK/483 and HK/486 achieve similar titers in the lungs of infected mice but only HK/483 causes leukopenia
To determine the levels of replication of HK/483 and HK/486, we infected mice with a low (0.1 50% mouse lethal dose (MLD50) that corresponds to 10 50% tissue culture-infective dose (TCID50)) or high (10 MLD50 or 106TCID50) doses of each virus and determined their titers in the lungs 3 days later. Although mortality was observed with high and low doses of HK/483 (Fig.1A), no significant difference was observed in lung virus titers in mice infected with either high or low doses of HK/486 or HK/483 (Fig.1B). We further compared the number of circulating leukocytes in mice infected with high or low doses of HK/483 and HK/486 and found that only infection with HK/483 at either dose caused a profound reduction of total leukocytes in the circulation (leukopenia)(Fig.1C).
Figure 1.

Percent survival (A), lung virus titers 3 days post infection (B) and lymphocyte count in peripheral blood (C) following high dose (106 TCID50) or low dose (10 TCID50) of HK/483 and HK/486 virus infection. The data represent the average for each group (n=5), ns = not significant, Student’s t test.
FasL expression on lymph node pDCs is increased with high dose HK/483 but not HK/486 infection
Because the reduction of influenza-specific CD8+ T cells in lethal H2N2 influenza virus infection was linked to FasL expression on DCs in lung draining LNs but not in sublethal infection (26), we speculated that HK/483 and HK/486 viruses might also differentially induce FasL expression on LN DCs. In this study, we determined the levels of FasL expression of lung draining lymph node DC subsets (CD11cmodCD8α+CD45R+ (pDC), CD11c+CD8α+CD45R− (CD8α+DCs) and CD11c+CD8α−CD45R− (CD8α−DC s); please see Supplementary Fig.1 for gating strategy). Our results demonstrate that FasL expression is substantially increased on pDCs following high dose (Fig.2 upper panel) and low dose (Fig.2 lower panel) HK/483 infection. This increase was not seen when mice were infected with the HK/486 virus. Likewise while we observed an increased level of FasL on the CD8α+ and CD8α− DC subsets following high dose infection the differences in FasL expression on these cells between the HK/483 and HK/486 infections was modest at best (Fig.2, middle and right panels). Of note the increase of FasL expression on the pDC observed following high and low dose HK/483 and on CD8α+ DC after both high dose HK/483 and HK/486 infection (Fig.2) appears to represent an increase in expression versus that observed on these subsets from naïve mice (Fig 2 and Supplementary Fig.2). Because these results suggested an involvement of pDCs in apoptosis of influenza-specific CD8+ T cells, we next determined the magnitude of their recruitment or expansion in LNs during HK/483 and HK/486 infection. We found that the number of pDCs in LNs dramatically increased on day 3 post-infection (p<0.001) (Fig.3A). Although we found a significant increase in the number of CD8α+ DCs but not CD8α− DCs (Fig.3B and 3C), FasL expression on these cells did not increase during high dose HK/483 versus HK/486 infection (Fig. 2). Interestingly, the number of pDCs present in the LNs was greater than other DC subsets during either HK/483 or HK/486 infection. Because pDCs exhibited enhanced recruitment to the LN in HK/483 infection, especially following high dose infection (Fig.3A), the pDCs expressed high levels of FasL in HK/483 but not HK/486 infection, and lymph node resident pDCs have been shown to lack MHC I-influenza viral peptide presentation and therefore the ability to rescue T cells from FasL induced apoptosis (27), we hypothesized that pDCs may be the predominant population responsible for the elimination of influenza-specific CD8+ T cells in lung draining LNs via Fas-FasL mediated apoptosis in lethal HK/483 infection.
Figure 2.

FasL expression on dendritic cell subsets during HK/483 and HK/486 virus infection. Mice were infected with high dose (106 TCID50) or low dose (10TCID50) of HK/483 or HK/486. 3 days post infection, cells from mediastinal and peribronchiolar lymph nodes from each group (n=5 per group) were evaluated for FasL expression. Shown is representative FasL expression on dendritic cell subsets; isotype control (filled grey), HK/486 infection (dotted line) and HK/483 infection (black line) from three independent experiments with 5 animals per group. Number inserts represent the mean fluorescence intensity (MFI) of FasL expression on DC subsets infected with HK/483 (black) or HK/486 (Grey)..
Figure 3.
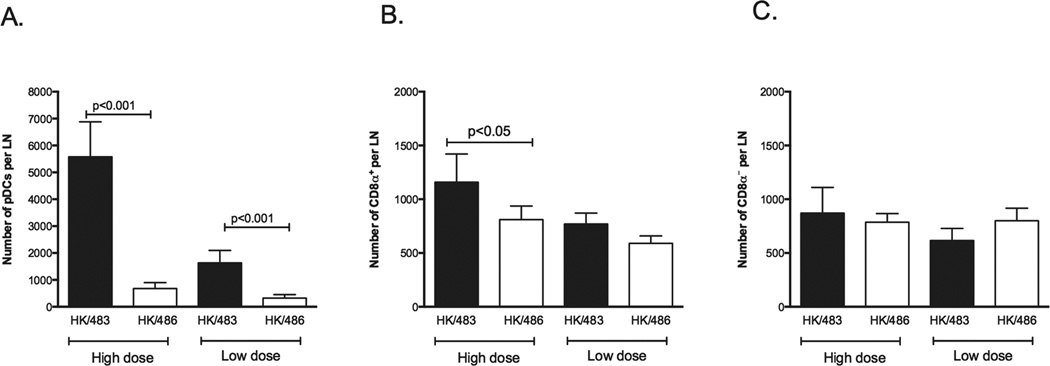
pDCs preferentially accumulate in the lung draining LNs of HK/483 infected mice. Mice were infected with high dose (106TCID50) or low dose (10 TCID50) of HK/483 (solid bar) and HK/486 (open bar). All of the lung draining LNs from each mouse were pooled and the numbers of CD11cmodCD45R+ cells (pDCs), P < 0.001, Student’s t test (A), CD11c+CD45R−CD8+(CD8α+ DCs), P<0.05, Student’s t test (B) and CD11c+CD45R−CD8− (CD8α− DCs) (C) in the lung draining LNs were determined. The data represent the average ± standard deviation (SD) for each group and are from three independent experiments with 5 animals per group.
Different cytokine profiles are observed in lung draining LNs during HK/483 and HK/486 infection
We isolated lung draining LNs from control (uninfected) and low dose or high dose HK/483 or HK/486 infected mice 24 h post-infection, cultured the intact LNs in vitro for 48 h and quantified cytokines that were released into culture supernatants (Fig. 4A). We found that high dose infection with either HK/483 or HK/486 caused substantial release of IL-1α and IL-6 (Fig.4B). Interestingly, higher levels of IL-10, which have been shown to control pulmonary inflammation and limit excessive tissue damage during acute influenza virus infection (31), were observed in LNs from mice infected with low dose HK/486 compared to low dose HK/483 (Fig.4B).
Figure 4.

Secretion of different cytokines in lung draining LNs following high or low dose HK/483 and HK/486 infection. Schematic diagram for experimental setup, the lung draining LNs were pooled from mice (n=5) 24 h post infection and cultured for 48h, the cytokines were measured in the culture supernatants by Bioplex system or ELISA (A) Secretion of IL-1α, IL-1β, IL-6, IL-10, P <0.05, Student’s t test (B) and IL-12 cytokine family in lung draining lymph node culture following high dose (106TCID50) or low dose (10TCID50) of HK/483 or HK/486 virus infection, IL-12p40 monomer/homodimer levels were calculated by subtracting the IL-12p70 and IL-23 values from the total IL-12p40 amounts, P < 0.05, P < 0.001, Student’s t test (C). The data represent the average ± standard deviation (SD) for each group (n=5).
Because IL-12p40 homodimer was shown be the regulator of FasL expression on DCs in LNs of lethal H2N2 influenza infection in mice (26), we compared the production of IL-12p40 in LNs of HK/483 and HK/486 infected mice. Total levels of IL-12p40-containing cytokines (i.e. IL-12p40 monomer/homodimer, IL-12p70 (p40+p35) and IL-23 (p40+p19) were 3-fold higher in LNs isolated from mice infected with high dose HK/483 compared to HK/486 (Fig.4C upper left, p<0.001). While IL-12p70 levels were significantly higher in LNs of mice infected with high dose HK/483 than high dose HK/486 (Fig.4C lower left, p<0.05), IL-23 levels were consistently higher in LNs of mice infected with either high or low dose HK/486 (Fig.4C upper right, p<0.001). Importantly, IL-12p40 monomer/homodimer was the predominant form of IL-12 secreted from the LNs of high dose HK/483 infected mice. These results, along with our previous studies (27), suggest that high levels of IL-12p40 monomer/homodimers in LNs can affect the up-regulation of FasL on pDCs during high dose HK/483 infection.
High dose infection with HK/483 results in increased apoptosis of influenza-specific CD8+ T cells
To study apoptosis of influenza-specific CD8+ T cells in the LNs of mice infected with lethal HK/483 and non-lethal HK/486 viruses, we transferred CD90.1+ CL-4 CD8+ T cells (which recognize the HA512–520 peptide of H5N1 influenza virus) into naïve CD90.2+ BALB/c recipient mice. We then infected the recipient mice with either a 106 TCID50 or 10 TCID50 of HK483 or HK/486 viruses. We identified apoptosis of influenza-specific CD8+T cells in the LNs 3 days post-infection by co-staining for CD90.1 and Annexin V and analyzing cell populations by flow cytometry (Fig.5A). We observed high numbers of CD90.1+ CL-4 CD8α+ T cells in the LNs, associated with low numbers of these cells in the lungs of high dose HK/483 infected mice (Fig. 5B and C). In the LNs, the CD90.1+ CL-4 CD8α+ T cells showed statistically significantly higher levels of apoptosis following high dose HK/483 infection than high dose HK/486 infection (Fig. 5D). In contrast, when the T cells were able to escape apoptosis in the lymph nodes during high dose HK/486 infections they were able to significantly accumulate in the lungs (Fig 5).
Figure 5.

Influenza specific T cells undergo apoptosis in lung draining lymph nodes during infection with HK/483 but not HK/486. Schematic diagram for experimental setup for in vivo apoptosis assay (A). The percentage and number of adoptively transferred clone-4 T cells in lung draining lymph nodes (B) [p=0.004; 0.003 and ns = not significant, Student’s t test] and in the lungs (C) [p <0.05, 0.04 and ns = not significant, Student’s t test] of infected mice. (D) The percentage and number of apoptotic clone-4 T cells in lung draining lymph nodes of infected mice [p = 0.0079, Student’s t test, one mouse in the HK/486 group was an outlier; Grubbs outlier test p<0.01].
pDCs enhance apoptosis of influenza-specific CD8+ T cells
To assess the impact of pDCs on apoptosis of influenza-specific CD8+ T cells in the LNs (Fig. 5D) and to determine whether wild-type pDCs (FasL+) can mediate apoptosis of influenza-specific CD8+ T cells during high dose HK/483 infection, we adoptively transferred pDCs into gld mice that lack functional FasL (32). When wild-type (BALB/c) pDCs (FasL+) were adoptively transferred into gld mice, apoptosis of CL-4 T cells was significantly increased 3 days post-infection with HK/483 (Fig.6) and the levels of influenza-specific CD8+ T cell apoptosis were similar to those in wild-type (BALB/c) mice during high dose HK/483 infection. In contrast, we did not observe significant levels of influenza-specific CD8+ T cell apoptosis in gld mice that received pDCs transferred from gld mice. Together, these data suggest a critical role for pDCs in apoptosis of influenza-specific CD8+ T cells in lethal H5N1 infection.
Figure 6.

pDCs expressing FasL kill influenza specific CD8+ T cells in lethal HK/483 infection. BALB/c or FasL-deficient (gld) recipient mice received 106 clone-4 T cells (I.V.) 48 h prior to infection. 106 purified pDCs from BALB/c or gld mice were transferred I.V. into recipient mice 18 h prior to infection. Four days post infection, the lung draining lymph nodes were harvested, and the numbers of clone-4 T cell undergoing apoptosis was determined by flow cytometry. The data represent the average for each group (n=5), P < 0.001, Student’s t test.
Discussion
The aim of our study was to identify immunopathological mechanism(s) that contributed to lymphopenia, which is a prominent binary disease outcome of infection with two closely related H5N1 viruses in mice. Our results provide compelling evidence that FasL expression on pDCs is an important mechanism underlying the lymphopenia associated with lethal H5N1 infection. Interestingly, lymphopenia is not unique to severe or fatal H5N1 virus infection; it is also associated with severe outcomes in human infections with avian H7N9 influenza viruses (3, 33, 34).
We performed a series of experiments in mice using a pair of well-characterized HPAI H5N1 viruses isolated from patients in Hong Kong in 1997. As has been reported previously, we found that HK/483 but not HK/486 was associated with lethality in mice and that the severity of infection correlated with leukopenia (8). Although the replication of HK/483 and HK/486 were similar in the lungs of mice, lymphopenia was only observed in lethal HK/483 infection, suggesting that the host response induced by HK/483 infection differs from that induced by HK/486 (Fig.1). Molecular characterization of these two viruses showed that they share a multi-basic amino acid motif in hemagglutinin (HA) and are highly pathogenic for chickens, but the viruses differ in their pathogenicity in mice (4, 5, 7, 12). There are 55 coding differences between HK/483 and HK/486 viruses throughout the genome (14).
pDCs play a deleterious role during infection with a lethal dose of H2N2 influenza virus by limiting the CD8 T cell response (27). We observed differential high levels of FasL expression on pDCs but not other DC subsets in lethal HK/483 infection, and also not in non-lethal HK/486 infection (Fig. 2). pDCs are the only DC subset that differentially expressed high levels of FasL expression and they were the predominant DC subset recruited to the LNs during HK/483 infection (Fig.3). These data suggested that pDCs maybe responsible for the elimination of T cells through Fas-FasL interactions in the LNs during HK/483 infection. Unlike the studies with H2N2 influenza virus, the induction of FasL on pDCs did not correlate with the dose of virus because high dose HK/486 infection also failed to induce FasL expression on LN DCs and low dose HK/483 infection induced FasL expression on pDCs albeit to a lesser extent. Therefore, our data suggest that FasL induction by HPAI H5N1 viruses is restricted by the strain rather than the dose of virus. We hypothesize that this may result from genetic differences between HPAI H5N1 and H2N2 viruses.
A cytokine “storm” has been proposed to contribute to the increased severity of lethal H5N1 infection (35). We found that infection with HK/483 induced high levels of pro-inflammatory cytokines (IL-1α, IL-6) whereas increased levels of IL-10, a key cytokine that controls pulmonary inflammation and limits excessive tissue damage during acute influenza virus infection (31) was observed in HK/486 infected mice (Fig.4). The nature of the cytokine environment in the LNs where contact between DCs and T cells occurs influences the nature of T cell responses (36). For instance, substantial production of IL-12p40 in the peribronchial LNs maintains elevated FasL on LN resident DCs and results in increased apoptosis of activated, proliferating CD8+T cells in the LN and decreased generation of effector T cells (26). In this study we found a correlation between the up-regulation of FasL expression and the induction of IL-12p40 production in LNs during lethal HK/483 infection. Our results suggest a role for pDCs in regulating the magnitude of the CD8+ T cell response through IL-12p40 dependent FasL expression.
Although other investigators have previously reported apoptosis of T cells in HK/483 infection (8), they did not study the LNs, where the initial interaction between DCs and T cells occurs. We observed increased apoptosis of influenza-specific T cells in the LNs of HK/483 but not in HK/486 infected mice (Fig. 5D). Although we found a large number of influenza-specific CD8+ T cells in the LNs of HK/483 infected mice (Fig.5B), the majority of the cells were undergoing apoptosis (Fig.5D). Interestingly, we found higher numbers of influenza-specific CD8+ T cells in the lungs of HK/486 than HK/483 infected mice (Fig.5C). The lower numbers of influenza-specific T cells in the lungs of HK/483 infected mice may be a consequence of apoptosis of T cells in the LNs, resulting in fewer numbers of T cells trafficking from LNs to the lungs (Fig.5B and 5C). We demonstrated additional support for the likely role of pDCs in regulating apoptosis of influenza-specific CD8+ T cells by adoptively transferring wild-type pDCs into FasL-deficient mice followed by lethal HK/483 infection. Strikingly, passive transfer of wild-type pDCs into FasL-deficient recipient mice enhanced influenza-specific T cell apoptosis in lung draining LNs. The mechanism(s) by which FasL+ pDCs dampen T cell numbers warrants further study.
In summary, we demonstrate that elevated FasL on pDCs leads to apoptosis of influenza-specific CD8+ T cells in lung draining LNs of mice infected with a lethal H5N1 virus. This finding is relevant for understanding the immunopathological mechanisms that underlie severe outcomes of influenza virus infection.
Supplementary Material
Acknowledgements
This research was supported by the Intramural Research Program of the NIAID, NIH and the Department of Pathology at the University of Iowa. We would like to thank Ms. Betty M. Young, University of Iowa for arranging animal transportation. We also thank Dr. Lesley W. Shupert, Laboratory of Virology, Aaron Carmody, Flow Cytometry Unit, Rachel Lacasse, Veterinary Branch and staff of RML, NIAID, Montana for excellent technical support.
References
- 1.de Jong JC, Claas EC, Osterhaus AD, Webster RG, Lim WL. A pandemic warning? Nature. 1997;389:554. doi: 10.1038/39218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Subbarao K, Klimov A, Katz J, Regnery H, Lim W, Hall H, Perdue M, Swayne D, Bender C, Huang J, Hemphill M, Rowe T, Shaw M, Xu X, Fukuda K, Cox N. Characterization of an avian influenza A (H5N1) virus isolated from a child with a fatal respiratory illness. Science. 1998;279:393–396. doi: 10.1126/science.279.5349.393. [DOI] [PubMed] [Google Scholar]
- 3.Chen Y, Liang W, Yang S, Wu N, Gao H, Sheng J, Yao H, Wo J, Fang Q, Cui D, Li Y, Yao X, Zhang Y, Wu H, Zheng S, Diao H, Xia S, Zhang Y, Chan KH, Tsoi HW, Teng JL, Song W, Wang P, Lau SY, Zheng M, Chan JF, To KK, Chen H, Li L, Yuen KY. Human infections with the emerging avian influenza A H7N9 virus from wet market poultry: clinical analysis and characterisation of viral genome. Lancet. 2013;381:1916–1925. doi: 10.1016/S0140-6736(13)60903-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dybing JK, Schultz-Cherry S, Swayne DE, Suarez DL, Perdue ML. Distinct pathogenesis of hong kong-origin H5N1 viruses in mice compared to that of other highly pathogenic H5 avian influenza viruses. J. Virol. 2000;74:1443–1450. doi: 10.1128/jvi.74.3.1443-1450.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gao P, Watanabe S, Ito T, Goto H, Wells K, McGregor M, Cooley AJ, Kawaoka Y. Biological heterogeneity, including systemic replication in mice, of H5N1 influenza A virus isolates from humans in Hong Kong. J. Virol. 1999;73:3184–3189. doi: 10.1128/jvi.73.4.3184-3189.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gubareva LV, McCullers JA, Bethell RC, Webster RG. Characterization of influenza A/HongKong/156/97 (H5N1) virus in a mouse model and protective effect of zanamivir on H5N1 infection in mice. The Journal of infectious diseases. 1998;178:1592–1596. doi: 10.1086/314515. [DOI] [PubMed] [Google Scholar]
- 7.Katz JM, Lu X, Tumpey TM, Smith CB, Shaw MW, Subbarao K. Molecular correlates of influenza A H5N1 virus pathogenesis in mice. J. Virol. 2000;74:10807–10810. doi: 10.1128/jvi.74.22.10807-10810.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tumpey TM, Lu X, Morken T, Zaki SR, Katz JM. Depletion of lymphocytes and diminished cytokine production in mice infected with a highly virulent influenza A (H5N1) virus isolated from humans. J. Virol. 2000;74:6105–6116. doi: 10.1128/jvi.74.13.6105-6116.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maines TR, Lu XH, Erb SM, Edwards L, Guarner J, Greer PW, Nguyen DC, Szretter KJ, Chen LM, Thawatsupha P, Chittaganpitch M, Waicharoen S, Nguyen DT, Nguyen T, Nguyen HH, Kim JH, Hoang LT, Kang C, Phuong LS, Lim W, Zaki S, Donis RO, Cox NJ, Katz JM, Tumpey TM. Avian influenza (H5N1) viruses isolated from humans in Asia in 2004 exhibit increased virulence in mammals. J. Virol. 2005;79:11788–11800. doi: 10.1128/JVI.79.18.11788-11800.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Claas EC, Osterhaus AD, van Beek R, De Jong JC, Rimmelzwaan GF, Senne DA, Krauss S, Shortridge KF, Webster RG. Human influenza A H5N1 virus related to a highly pathogenic avian influenza virus. Lancet. 1998;351:472–477. doi: 10.1016/S0140-6736(97)11212-0. [DOI] [PubMed] [Google Scholar]
- 11.Suarez DL, Perdue ML, Cox N, Rowe T, Bender C, Huang J, Swayne DE. Comparisons of highly virulent H5N1 influenza A viruses isolated from humans and chickens from Hong Kong. J. Virol. 1998;72:6678–6688. doi: 10.1128/jvi.72.8.6678-6688.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu X, Tumpey TM, Morken T, Zaki SR, Cox NJ, Katz JM. A mouse model for the evaluation of pathogenesis and immunity to influenza A (H5N1) viruses isolated from humans. J. Virol. 1999;73:5903–5911. doi: 10.1128/jvi.73.7.5903-5911.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen H, Bright RA, Subbarao K, Smith C, Cox NJ, Katz JM, Matsuoka Y. Polygenic virulence factors involved in pathogenesis of 1997 Hong Kong H5N1 influenza viruses in mice. Virus research. 2007;128:159–163. doi: 10.1016/j.virusres.2007.04.017. [DOI] [PubMed] [Google Scholar]
- 14.Fornek JL, Gillim-Ross L, Santos C, Carter V, Ward JM, Cheng LI, Proll S, Katze MG, Subbarao K. A single-amino-acid substitution in a polymerase protein of an H5N1 influenza virus is associated with systemic infection and impaired T-cell activation in mice. J. Virol. 2009;83:11102–11115. doi: 10.1128/JVI.00994-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yuen KY, Chan PK, Peiris M, Tsang DN, Que TL, Shortridge KF, Cheung PT, To WK, Ho ET, Sung R, Cheng AF. Clinical features and rapid viral diagnosis of human disease associated with avian influenza A H5N1 virus. Lancet. 1998;351:467–471. doi: 10.1016/s0140-6736(98)01182-9. [DOI] [PubMed] [Google Scholar]
- 16.Hinshaw VS, Olsen CW, Dybdahl-Sissoko N, Evans D. Apoptosis: a mechanism of cell killing by influenza A and B viruses. J. Virol. 1994;68:3667–3673. doi: 10.1128/jvi.68.6.3667-3673.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takizawa T, Matsukawa S, Higuchi Y, Nakamura S, Nakanishi Y, Fukuda R. Induction of programmed cell death (apoptosis) by influenza virus infection in tissue culture cells. The Journal of general virology. 1993;74(Pt 11):2347–2355. doi: 10.1099/0022-1317-74-11-2347. [DOI] [PubMed] [Google Scholar]
- 18.Fesq H, Bacher M, Nain M, Gemsa D. Programmed cell death (apoptosis) in human monocytes infected by influenza A virus. Immunobiology. 1994;190:175–182. doi: 10.1016/S0171-2985(11)80292-5. [DOI] [PubMed] [Google Scholar]
- 19.Hofmann P, Sprenger H, Kaufmann A, Bender A, Hasse C, Nain M, Gemsa D. Susceptibility of mononuclear phagocytes to influenza A virus infection and possible role in the antiviral response. Journal of leukocyte biology. 1997;61:408–414. doi: 10.1002/jlb.61.4.408. [DOI] [PubMed] [Google Scholar]
- 20.Criswell BS, Couch RB, Greenberg SB, Kimzey SL. The lymphocyte response to influenza in humans. The American review of respiratory disease. 1979;120:700–704. doi: 10.1164/arrd.1979.120.3.700. [DOI] [PubMed] [Google Scholar]
- 21.Dolin R, Richman DD, Murphy BR, Fauci AS. Cell-mediated immune responses in humans after induced infection with influenza A virus. The Journal of infectious diseases. 1977;135:714–719. doi: 10.1093/infdis/135.5.714. [DOI] [PubMed] [Google Scholar]
- 22.Topham DJ, Tripp RA, Doherty PC. CD8+ T cells clear influenza virus by perforin or Fas-dependent processes. J. Immunol. 1997;159:5197–5200. [PubMed] [Google Scholar]
- 23.Lowin B, Hahne M, Mattmann C, Tschopp J. Cytolytic T-cell cytotoxicity is mediated through perforin and Fas lytic pathways. Nature. 1994;370:650–652. doi: 10.1038/370650a0. [DOI] [PubMed] [Google Scholar]
- 24.Brincks EL, Katewa A, Kucaba TA, Griffith TS, Legge KL. CD8 T cells utilize TRAIL to control influenza virus infection. J. Immunol. 2008;181:4918–4925. doi: 10.4049/jimmunol.181.7.4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wolfe T, Asseman C, Hughes A, Matsue H, Takashima A, von Herrath MG. Reduction of antiviral CD8 lymphocytes in vivo with dendritic cells expressing Fas ligand-increased survival of viral (lymphocytic choriomeningitis virus) central nervous system infection. J. Immunol. 2002;169:4867–4872. doi: 10.4049/jimmunol.169.9.4867. [DOI] [PubMed] [Google Scholar]
- 26.Legge KL, Braciale TJ. Lymph node dendritic cells control CD8+ T cell responses through regulated FasL expression. Immunity. 2005;23:649–659. doi: 10.1016/j.immuni.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 27.Langlois RA, Legge KL. Plasmacytoid dendritic cells enhance mortality during lethal influenza infections by eliminating virus-specific CD8 T cells. J Immunol. 2010;184:4440–4446. doi: 10.4049/jimmunol.0902984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fujikura D, Chiba S, Muramatsu D, Kazumata M, Nakayama Y, Kawai T, Akira S, Kida H, Miyazaki T. Type-I interferon is critical for FasL expression on lung cells to determine the severity of influenza. PLoS One. 2013;8:e55321. doi: 10.1371/journal.pone.0055321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vogel AB, Haasbach E, Reiling SJ, Droebner K, Klingel K, Planz O. Highly pathogenic influenza virus infection of the thymus interferes with T lymphocyte development. J. Immunol. 2010;185:4824–4834. doi: 10.4049/jimmunol.0903631. [DOI] [PubMed] [Google Scholar]
- 30.Logan AC, Chow KP, George A, Weinstein PD, Cebra JJ. Use of Peyer's patch and lymph node fragment cultures to compare local immune responses to Morganella morganii. Infection and immunity. 1991;59:1024–1031. doi: 10.1128/iai.59.3.1024-1031.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun J, Madan R, Karp CL, Braciale TJ. Effector T cells control lung inflammation during acute influenza virus infection by producing IL-10. Nature medicine. 2009;15:277–284. doi: 10.1038/nm.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hahne M, Peitsch MC, Irmler M, Schroter M, Lowin B, Rousseau M, Bron C, Renno T, French L, Tschopp J. Characterization of the non-functional Fas ligand of gld mice. International immunology. 1995;7:1381–1386. doi: 10.1093/intimm/7.9.1381. [DOI] [PubMed] [Google Scholar]
- 33.Liem NT, Tung CV, Hien ND, Hien TT, Chau NQ, Long HT, Hien NT, Mai le Q, Taylor WR, Wertheim H, Farrar J, Khang DD, Horby P. Clinical features of human influenza A (H5N1) infection in Vietnam: 2004–2006. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2009;48:1639–1646. doi: 10.1086/599031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao HN, Lu HZ, Cao B, Du B, Shang H, Gan JH, Lu SH, Yang YD, Fang Q, Shen YZ, Xi XM, Gu Q, Zhou XM, Qu HP, Yan Z, Li FM, Zhao W, Gao ZC, Wang GF, Ruan LX, Wang WH, Ye J, Cao HF, Li XW, Zhang WH, Fang XC, He J, Liang WF, Xie J, Zeng M, Wu XZ, Li J, Xia Q, Jin ZC, Chen Q, Tang C, Zhang ZY, Hou BM, Feng ZX, Sheng JF, Zhong NS, Li LJ. Clinical findings in 111 cases of influenza A (H7N9) virus infection. The New England journal of medicine. 2013;368:2277–2285. doi: 10.1056/NEJMoa1305584. [DOI] [PubMed] [Google Scholar]
- 35.de Jong MD, Simmons CP, Thanh TT, Hien VM, Smith GJ, Chau TN, Hoang DM, Chau NV, Khanh TH, Dong VC, Qui PT, Cam BV, Ha do Q, Guan Y, Peiris JS, Chinh NT, Hien TT, Farrar J. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nature medicine. 2006;12:1203–1207. doi: 10.1038/nm1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guermonprez P, Valladeau J, Zitvogel L, Thery C, Amigorena S. Antigen presentation and T cell stimulation by dendritic cells. Annual review of immunology. 2002;20:621–667. doi: 10.1146/annurev.immunol.20.100301.064828. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.