

Abstract
Skeletal muscle is able to restore contractile functionality after injury thanks to its ability to regenerate. Following muscle necrosis, debris is removed by macrophages, and muscle satellite cells (MuSCs), the muscle stem cells, are activated and subsequently proliferate, migrate, and form muscle fibers restoring muscle functionality. In most muscle dystrophies (MDs), MuSCs fail to properly proliferate, differentiate, or replenish the stem cell compartment, leading to fibrotic deposition. However, besides MuSCs, interstitial nonmyogenic cells and inflammatory cells also play a key role in orchestrating muscle repair. A complete understanding of the complexity of these mechanisms should allow the design of interventions to attenuate MDs pathology without disrupting regenerative processes. In this review we will focus on the contribution of immune cells in the onset and progression of MDs, with particular emphasis on Duchenne muscular dystrophy (DMD). We will briefly summarize the current knowledge and recent advances made in our understanding of the involvement of different innate immune cells in MDs and will move on to critically evaluate the possible role of cell populations within the acquired immune response. Revisiting previous observations in the light of recent evidence will likely change our current view of the onset and progression of the disease.
1. Background
Only a small number of immune cells reside within intact skeletal muscle, but they are recruited during injury and play important roles in the regeneration process, critically contributing to its resolution. Upon injury, immune cells rapidly infiltrate the muscle to remove necrotic tissue and secrete soluble factors that serve initially to activate muscle satellite cells (MuSCs) [1–3]. As such, immune cells constitute a transient local environment for MuSCs. Satellite cells and immune cells attract one another through chemokines (chemoattraction). Satellite cells have been demonstrated to secrete a panel of proinflammatory cytokines, such as IL-1, IL-6, and TNF-α to facilitate immune cell infiltration and function [4, 5]. In turn, immune cells secrete a wealth of diffusible factors, such as growth factors, IL-6, globular adiponectin, extracellular matrix (ECM) components, and ECM remodeling MMPs. These diffusible factors generate ECM chemoattractive fragments, which help satellite cells escape from the basal lamina of myofibers, and promote satellite cell proliferation [6]. In addition, cell-to-cell contact between immune and satellite cells protects satellite cells from apoptosis [7]. All these events must be timely regulated and alterations in quality, amount, and time lead to impaired regeneration, increased muscle wasting, and deposition of fibrotic tissue, as it occurs in muscle aging or in muscular dystrophies, such as Duchenne muscular dystrophy (DMD) [8, 9]. DMD is a lethal X-linked genetic disorder caused by deficiency of dystrophin, a critical component of the dystrophin glycoprotein complex (DGC), acting as a link between the cytoskeleton and extracellular matrix in skeletal and cardiac muscles [10]. A consequence of the DGC inefficiency is muscle fragility, contraction-induced damage, necrosis, and inflammation. Although satellite cells compensate for muscle fiber loss in the early stages of disease, eventually these progenitors become exhausted [11]. Moreover, aberrant intracellular signalling cascades that regulate both inflammatory and immune processes contribute substantially to the degenerative process. As a result, fibrous and fatty connective tissue overtakes the functional myofibers. These changes culminate in progressive muscle wasting, with the majority of patients being wheelchair-bound in their early teens, succumbing to cardiac/respiratory failure in their twenties [12]. Among the animal models of DMD, the mdx mouse model is the best characterized. It lacks dystrophin expression, and, though with a milder phenotype, exhibits extensive limb muscle degeneration and inflammation, as well as myocardial lesions [13–15]. Available data sets, although limited and not comprehensive, suggest that early immune cell infiltration in DMD patients and mdx mice represent an important, but underappreciated, aspect of dystrophic muscle pathology. In fact, although lack of dystrophin makes myofibers susceptible to fragility and degeneration when contracting, this mechanical defect hypothesis for dystrophic muscle death has been unable to explain many aspects of the pathophysiology of DMD and emerging clues attribute an active role to the immune response in these events [16]. From a therapeutic point of view, a clear understanding of the cell populations and of the mechanisms involved in the inflammatory response in DMD may allow the design of valuable anti-inflammatory therapeutic strategies to ameliorate muscular dystrophy. Although blunting inflammation would not restore the primary defect, the emerging clue is that multiple strategies, addressing different aspects of the pathology, which may eventually converge, may be successful.
2. Innate Immune Response in Muscular Dystrophies
As a consequence of the dystrophin-null myofibers fragility, DMD muscle is characterized by continuous cycles of myofibers necrosis and repair. Myofibers undergoing degeneration/necrosis, independently of the injury insult, release Th1 inflammatory stimuli, which recruit neutrophils and monocytes/macrophages required to clear cell debris, followed by a Th2 immune response which promotes muscle healing [17]. Much of the current information about the role of immune cell populations of the innate response in muscle repair comes from studies on healthy regenerating muscle [1, 18–20]. Although many similarities were found between acute and chronic muscle injury, the kinetics, quality, and outcome diversify in many aspects [21, 22].
2.1. Neutrophils
In acute injury, neutrophils, identified as Ly6C+/F4/80- cells, are the first cells to invade injured muscle, followed by macrophages. In acutely injured muscle in mice, they begin to appear at elevated numbers within 2 h of muscle damage, typically peaking in concentration between 6 and 24 h after injury and then rapidly decline in numbers [23]. Their function mostly involves phagocytic activity to remove debris but also release of TNFα, as a Th1 stimuli, and production of myeloperoxidase (MPO), inducing muscle membrane damage and increasing macrophage proinflammatory activity [24–26]. As in acute injury, neutrophils, together with macrophages, invade mdx dystrophic muscle as early as 2 weeks of age [24]. Indeed, initial muscle injury and membrane lysis are caused by superoxide production mediated by these early infiltrating neutrophils. Previous studies demonstrated that anti-GR1 antibody-mediated depletion of neutrophils, starting at age 19 days, significantly reduces muscle necrosis at age ~21 days and subsequent regeneration at age ~28 days [25]. However, GR-1 is highly expressed in neutrophils, but it is also expressed in macrophages [27, 28], so whether the observed phenotype was specifically related to neutrophils depletion is not as clear. In any case, although neutrophils activity appears to overlap with that of M1 macrophages (see below), since they are one of the earliest infiltrating cell types, they might promote initial muscle lesion, playing an important role in the early onset of the pathology.
2.2. Macrophages
A comprehensive survey of the current knowledge on macrophage involvement in MDs and muscle regeneration is beyond the scope of this review. The reader is referred to many systematic reviews already available on this topic [16, 21, 29]. Briefly, two subpopulations of macrophages have been identified in regenerating muscle tissue that may influence muscle degeneration and regeneration depending on the proportion of these cells present [18, 30]. As a simplistic point of view, the M1 population are proinflammatory, characterized by the expression of iNOS and secretion of proinflammatory cytokines (e.g., TNFα, IL-1β, and IL-6), and promote muscle cell lysis; by contrast, the M2 population is characterized by the expression of arginase-1, CD163, and CD206 mannose receptor (usually in noninflammatory, repair conditions) and/or anti-inflammatory cytokines (e.g., IL-10) [19, 21, 31, 32]. They are believed to enhance muscle regeneration, by inducing satellite cell proliferation [19]. However, beyond this broad definition, mainly determined from in vitro polarization experiments, macrophages exhibit a wide variety of intermediate phenotype, including M2b and M2c, M1 and M2 being the extremes of a continuum in activation states. M2b macrophages are known to release large amounts of IL-10, which promotes the proliferation of nonmyeloid cells, although, like M1 macrophages, they can also release proinflammatory cytokines, such as IL-1β and TNFα. IL-10 can also induce M2c macrophages, which have anti-inflammatory functions [33]. However, it has been recently highlighted that several markers should be examined at the cellular level in order to properly assess the macrophage inflammatory state in vivo, since the population remains heterogeneous [34]. While in acute muscle injury the sequential waves of M1, followed by M2 macrophage invasion, leads to resolution of inflammation and efficient muscle repair, in muscular dystrophies, repetitive cycles of myofiber degeneration lead to muscle invasion by M1 macrophages together with M2a macrophages, which may reduce the cytotoxic activity of M1 macrophages [35]. Therefore, the inflammatory milieu in dystrophic muscle is similar, but not the same as found in acute injury. Subsequent invasion of dystrophic muscle by M2c macrophages is associated with progression to the regenerative phase in pathophysiology. However, the number of M2 macrophages declines upon resolution of the damage in acute-injured muscle, while in mdx muscle their number increases with age, and promotes fibrosis [36]. Thus, increased and persistent presence of macrophages modifies the intensity, duration, and interactions of the different released factors, leading to increased myofibers necrosis, ECM accumulation, and replacement of muscle with fibrotic and fat tissue. Early studies showed that depletion of macrophages from mdx mice before the onset of histopathology caused great reduction in muscle pathology in 4-week-old mice [37], reinforcing the notion that macrophages induce muscle lesions, along with clearing debris. However, given that myofiber degeneration occurs in the absence of dystrophin, depletion of macrophages might not be as beneficial in long term, as it would prevent removal of debris. Indeed, whether or not phagocytosis is required for proper muscle repair is still debatable. Depletion of phagocytes was shown to slow cellular debris removal following freeze-injury of muscle but to have no effect on early postinjury regeneration, in terms of satellite cells activation [38]. By contrast, other studies have concluded that reducing the numbers of phagocytic leukocytes (neutrophils and monocytes/macrophages) in mice prior to toxin-induced muscle injury slows the removal of cellular debris and slows muscle regeneration, since reduced frequency of central-nucleated muscle fibers was observed [18, 39]. However, considering that, as mentioned before, phagocytic leukocytes also promote muscle damage, it could be argued that reduced number of regenerating fibers was dependent on reduced muscle damage because of the depletion of leukocytes. In this context, a recent study by Mounier et al. showed that inhibiting phagocytic activity in macrophages prevents macrophage skewing from a pro- to anti-inflammatory phenotype at the time of resolution of inflammation, in regenerating healthy muscle, suggesting that phagocytosis is required not only to clear debris, but also to promote resolution of the damage [34].
Although these studies are related to healthy regenerating muscle, deciphering the mechanisms regulating macrophages activation and plasticity should greatly facilitate the design of novel therapeutic strategies to modify the dystrophic muscle environment, reduce muscle damage, and increase muscle repair.
2.3. Mast Cells
Very little is known about mast cell involvement in muscular dystrophies. Nevertheless, mast cell proliferation and degranulation are often observed in areas of grouped fiber necrosis [40]. Mast cell degranulation involves the release of histamine and cytokines (including TNFα), which contribute to a pro-inflammatory environment that promotes muscle necrosis. In addition, proteases released during mast cell degranulation induce membrane lysis of nearby cells promoting local areas of ischemia [41]. Indeed, treatment of mdx mice prior to disease onset (age 19 days) with sodium cromoglycate (cromolyn), a blocker of mast cell degranulation, results in ~59% decrease in cumulative TA muscle damage by age 28 days [42]. Further studies are needed to gain insight into the mechanisms that regulate the microenvironment and ischemic conditions that result in fiber necrosis in muscular dystrophies. In a mouse model of arthritis, an inflammatory disease not related to muscle, mast cell-mediated increase of vascular permeability has been shown to be involved in the recruitment of inflammatory cells into the site of inflammation [43]. Interestingly, it has been recently shown that mast cells deficiency in a mouse model of polymyositis results in reduced susceptibility to C-protein-induced myositis, as compared with WT [44]. The reduced susceptibility to C-protein-induced myositis was associated with a reduced number of macrophages and CD8+ T cells, mainly due to reduced vascular permeability. The observed reduced susceptibility was restored by reconstitution of mast cells, highlighting the crucial role of these cells in promoting massive inflammatory cells infiltration [44]. Polymyositis (PM) belongs to the class of idiopathic inflammatory myopathies, IIM, which also includes adult and juvenile dermatomyositis (DM), myositis, and inclusion body myositis (IBM), a heterogeneous group of acquired disorders characterized by chronic inflammation of striated muscle leading to predominantly proximal muscle weakness. Although the precise pathogenesis is unknown, many evidences support their autoimmune basis and they likely result from immune-mediated processes initiated by environmental factors in genetically susceptible individuals [45, 46].
Increase in vascular permeability is certainly one of the events contributing to the immune cell infiltration in dystrophic muscle, as well. These aspects have been poorly addressed in muscular dystrophy, as only recently the disease was recognized as an “inflammatory” disease; investigating the disease from this new perspective may give unexpected insights into the mechanisms regulating its onset and progression.
2.4. Eosinophils
Eosinophils are generally associated with immune responses to infection by parasites or with allergic reactions [47]. However, they can play a significant role in the cellular immune response, in which their involvement appears to be tightly coupled both temporally and functionally to T lymphocyte activity; indeed, they are activated by the Th2 cytokine IL-5 [48, 49]. Although eosinophilia occurs only rarely and inexplicably in muscle disease, eosinophil invasion was found in both DMD and mdx dystrophies [50, 51]. Previous studies observed that eosinophils increased within mdx dystrophic muscle at about 4 weeks of age, together with cytotoxic T cells invasion, and, although their number decreases during the regenerative phase, their concentration remains higher at 30–32 weeks of age, as compared to healthy muscle of age-matched wild type mice, depending on the muscle examined [51]. Interestingly, transplantation of splenocytes derived from mdx mice into irradiated WT animal induced an increased number of eosinophils in muscle, as compared to irradiated control mice, suggesting that eosinophils invasion is dependent on lymphocytes activity [51]. Indeed, prednisone treatment (the major drug used in DMD patients) reduces eosinophil infiltration [52]. Moreover, despite the original expectation that eosinophilia in dystrophic muscle was a nonspecific consequence of the Th2 inflammatory environment, eosinophils were shown to modulate injury and regeneration. In particular, genetic ablation of major basic protein-1 (MBP-1), a cytolytic protein expressed by eosinophils, caused an increase in the numbers of cytotoxic T cells in dystrophic muscle [50]. That finding is significant to mdx dystrophy because a Th1-driven cellular immune response in which cytotoxic T cells promote apoptosis of mdx muscle fibers is an early feature of the disease that is attenuated as muscle regeneration begins. Thus, eosinophils may mediate the regeneration of dystrophic muscle by promoting the transition from Th1 to Th2 inflammatory environment. An interesting insight on the possible role of eosinophils in promoting muscle regeneration was recently suggested by Heredia and colleagues [17]. They showed that eosinophils invade acute injured muscle very early, even before neutrophils invasion. This observation is in contrast to other studies which showed, using the same injury model, that eosinophils are exceeding scarce in muscle following toxin injection and they appear to increase after other inflammatory cell populations [34]. However, using the eosinophil-deficient ΔdblGATA1 mouse model, which presents a 90% reduction in eosinophils, Heredia et al. observed persistent and unresolved muscle damage after acute injury. The authors identified IL-4 as the key cytokine produced by eosinophils, whose lack is known to prevent muscle regeneration [53]. Surprisingly, they demonstrate that the primary target of this cytokine produced by eosinophils at the very early stage of muscle damage is not macrophage, as expected, but fibroadipogenic precursor (FAP) cells. In fact, while lack of IL-4 receptor in macrophages did not result in regenerative defect, the lack of this receptor in FAPs is deleterious and leads to fat deposition by promoting their differentiation into adipocytes. FAPs are recently identified multipotent mesenchymal cells residing in skeletal muscle interstitium, who represent a critical component of the cellular niche required for effective satellite cell-mediated regeneration [54–57]. Thus, eosinophils may form a transitional niche for proliferating FAPs via secretion of IL-4; in the presence of IL-4, FAPs proliferate as fibroblasts to support myogenesis [17]. So far, whether these observations are relevant to muscular dystrophy is a matter of speculation; it can be hypothesized that, in dystrophic muscle, the eosinophils promote the transition from a Th1 to a Th2 inflammatory environment, which would in turn promote FAPs proliferation as fibroblasts to support myogenesis. On the other hand, persistence of eosinophils within the dystrophic muscle would sustain fibroblast proliferation, which promotes fibrotic tissue deposition, accelerating the clinical decline of the disease [36]. Thus, further studies are needed to gain insight into the role of eosinophils in the pathophysiology of muscular dystrophy and to verify whether manipulating eosinophils activity would be therapeutically beneficial.
3. Lymphocytes and the Adaptive Immune Response in Muscle Repair
3.1. T-Cell Response in DMD
It is widely believed that lymphocytes do not play a relevant role in healthy regenerating muscle, due to the inability of skeletal muscle to activate a T-cell response. Neither MHC class I nor class II molecules has been detected on muscle fibers from healthy muscle tissues [58, 59]. By contrast, appearance of MHC class I and/or II was observed in muscle tissue of patients with idiopathic inflammatory myopathies (IIM), where an autoimmune pathogenesis is now recognized, but also in regenerating fibers of patients with DMD [58]. These findings, together with the observation that an inflammatory milieu may upregulate HLA-DR and costimulatory molecules (ICOS-L and CD40), led to the idea that myoblasts can become facultative APCs in both MHCI and II-dependent immune reactions, forming functional immunological synapses with T cells [59, 60]. Indeed, T cells are found in degenerating muscle after acute injury, but their recruitment is more robust and persistent in chronic diseases, such as myositis or muscular dystrophies, where also B cells can be detected [61, 62].
The recruitment of T cells into injured muscle implies an adaptive immune response, which normally depends on antigen exposure. While this mechanism appears now conceivable in inflammatory myositis, it is less clear in regenerating muscle after acute injury or in chronic diseases such as muscular dystrophies. Many observations suggest that muscle-specific autoantigens drive T-cell expansion in IIM [46], but no such observations are available in regenerating muscle or in muscular dystrophies yet. Furthermore, whether or not T-cell infiltrate critically contributes to muscle damage in muscular dystrophies is still debatable. Persistence of T cells in dystrophic muscle may actually modulate inflammatory milieu and immune cells activity but may also directly interfere with muscle cell function through lymphocyte-released cytokines and chemokines [63, 64]. Indeed, it was recently shown that conditioned medium from activated T-cells, previously cultured in anti-CD3 coated dishes in the presence of IL2 [65], induced proliferation of C2C12 muscle cell, a mouse derived muscle cell line widely used as a model of myogenesis in vitro, and prevents myogenic differentiation, as compared to conditioned medium derived from resting T cells [66].
Very early studies correlated the reduction in T cells observed in prednisone-treated DMD patients, with reduction in muscle necrosis and fibrosis [67, 68]. Further studies identified T cells in muscles of several DMD patients, characterized by a specific T-cell receptor (TCR) rearrangement [69]. The overrepresentation of a T-cell population expressing a restricted set of TCR variable genes might indicate a selective T-cell response directed to a muscle-specific antigen. Their persistence in DMD muscle could derive from either clonal expansion or conserved antigen recognition, or from the emergence of a regulatory population. Interestingly, specific TCR arrangement, different from the one found in DMD, was initially found in the T cells invading muscle in PM patients [70]. Since then, many studies aimed to characterize T-cell populations and their role in MDs and muscle regeneration, though the results were not as exhaustive and were sometimes contradictory.
3.2. T-Cell Response in mdx Muscle
Elevated concentrations of activated cytotoxic CD8+ and helper CD4+ T cells (Th) are present in affected muscles of mdx mice aged 4–8 weeks but rapidly decrease in concentration by 14 weeks of age [63]. However, no increase in these cells has been observed in axillary or inguinal lymph nodes of mdx mice, suggesting that their activation is occurring in muscle tissue, and not systemically [63]. CD8+ T cells are the first to invade dystrophic muscle, peaking at 4 weeks of age; their activation is generally driven by a Th1 cellular immune response to kill their target cells through perforin-mediated processes [51]. Around 2 weeks later, CD4+ T cells also invade dystrophic muscle; T helper CD4+ T cells can generally differentiate into Th effector inflammatory cells, mainly Th1 and Th2, or into regulatory T cells (Treg), both of which participate in immune responses. Th1 cells are known to support macrophage M1 polarization by producing IL-1, IL-2, TNF-α, and INF-γ, while Th2 produce IL-4, IL-13, and IL-6 sustaining the M2 macrophage polarization [71]. On the other hand, Treg cells are required for the resolution of the immune response. These cells are characterized by the expression of Foxp3 transcription factor and produce anti-inflammatory cytokines such as IL-10 [72]. They were originally described as regulators of T-cell activity but were later recognized to also regulate B cells and several other innate immune response players [73].
One of the first studies addressing the possible role of lymphocytes in mdx showed that antibody-mediated depletion of CD8+ or CD4+ cells in mdx mice, beginning at 6 days of age and continuing until the age of 4 weeks, resulted in a 75% and 61% reduction in muscle histopathology, respectively [63]. This positive outcome suggested an important role for these cells in the development of muscle lesions. In another study, scid/mdx mice, which are deficient in functional T and B lymphocytes, were shown to develop much less diaphragm fibrosis at 1 year of age and a decrease in activated TGFβ in skeletal muscle, compared with mdx mice. Improvement in muscle regeneration was also observed in these mice, but not in muscle functionality [74]. Accordingly, in nu/nu/mdx mice, the lack of functional T cells alone was associated with less diaphragm fibrosis at 3 months of age [75]. Altogether, these results support the pathogenic role of T cells in mdx muscle and reveal this lymphocyte subclass as an important source of TGFβ1. By contrast, more recently, an additional dystrophic immunodeficient mouse model was generated, the Rag2(−)Il2rb(−)Dmd(−) mouse, which lacks T, B, and NK cells, and also carries a mutant Dmd allele that prevents the production of any dystrophin isoform [76]. Although not systemically analyzed, the authors reported that there was no difference in the pathological features of the disease in these mice, compared to mdx mice. Indeed, previous studies have shown that thymectomy at 1 month of age, which completely depletes circulating T cells, failed to prevent diaphragm fibrosis at 6 months of age [75]. Although this result suggested that the fibrotic process is self-sustaining from a very early stage, it can be argued that depleting circulating T cells at 1 month of age was already late, as early muscle invading T cells may eventually expand within the muscle and contribute to fibrous tissue deposition. However, the possibility of muscle-specific T cells clonal expansion has been poorly investigated, so far.
T cells represent approximately 3% of all infiltrating cells in mdx muscle, with over half present as double-negative T cells (lacking both CD4 and CD8 expression), 8%–10% of which were recently identified as being NKT-like cells, which express both T and NK markers [77]. Interestingly, a predominant T-cell population expressing Vβ8.1/8.2 was identified within the muscle infiltrate in mdx mice at 1 month of age. These Vβ8.1/8.2+ cells are not generally overrepresented in circulating cells, and, therefore, it is likely that these cells are either selectively home to or expand within mdx muscle. These cells express high level of osteopontin (OPN), which modulates cellular immune profiles in mdx muscles; in fact, its ablation in mdx mice results in significant reduction in neutrophils, CD3+/Vβ8.1/8.2+ cells, and NKT-like cells, but not macrophages. Interestingly, an increase in CD3+/CD4+/FoxP3+ Treg cells was observed, highlighting the importance of modulating T cells subclasses in muscular dystrophy. In this context, OPN appears to be an immune modulator that specifically impacts the concentration of NKT-like cells, neutrophils, and Tregs in dystrophic muscle [77]. As a result, lack of OPN in mdx mice reduces intramuscular TGFβ and fibrosis in both muscle and heart at 6 months of age, and importantly, it improves muscle strength. Vβ8.1/8.2+ cells are not the only source of OPN in dystrophic muscle as other immune cells, as well as muscle tissue, which could also produce it. Thus, besides the identification of OPN as a potential therapeutic target to ameliorate inflammation and progression of muscular dystrophy, these data clearly demonstrated a crucial role of T-cell subclasses in modulating the inflammatory milieu in dystrophic muscle. In line with this view, treatment with the immunosuppressant drug Rapamycin ameliorates the mdx phenotype with a reduction of both CD4+ and CD8+ cells, but not of regulatory Foxp3+ cells [64]. In this context, we recently showed that lack of protein kinase C θ (PKCθ) in mdx mice prevents muscle degeneration and inflammation, thus improving muscle regeneration and, importantly, muscle performance [78]. PKCθ is a member of the PKC family, predominantly expressed in muscle, where we and others have shown that it is an upstream regulator of several intracellular pathways leading to muscle homeostasis [78–83]. Furthermore, PKC θ, which is a key regulator of T-cell activation and proliferation, is being proposed as an attractive target to prevent immune response and alloreactivity [84, 85]. Indeed, it appears to be required for the development of a robust inflammatory response in vivo [86, 87]. Interestingly, PKCθ −/− mice fail to develop experimental allergic encephalomyelitis, display drastically reduced lung inflammation after induction of allergic asthma, and have a significantly diminished response in experimental colitis, and a type II collagen induced arthritis model [86, 88, 89]. Of note, PKCθ −/− mice can still mount a normal protective immune response to clear viral infections, and, importantly, maintain Treg function, since PKCθ is known to inhibit Treg differentiation [90, 91]. Indeed, we showed by bone marrow transplantation experiments that the improved phenotype observed in the double mutant mdx/PKCθ −/−mice is primarily due to lack of PKCθ in the hematopoietic cell compartment [78]. Thus, the possibility that lack of PKCθ in T cells may alter the T-cell subclasses within dystrophic muscle, and, in turn, the inflammatory milieu, including macrophage subclasses and function, is currently under investigation.
3.3. T-Cell Response in Dysferlinopathies
The involvement of lymphocytes activity appears to be a general feature in muscular dystrophies. In fact, it has been recently demonstrated that lack of functional T/B cells improves regeneration in a mouse model of dysferlinopathies [92]. These diseases belong to the so-called limb girdle muscular dystrophies (LGMDs), characterized by predominant weakness and wasting of muscles of the pelvic and shoulder girdle. Among them, LGMD-2B and Miyoshi myopathy (MM) develop due to defects in the dysferlin gene, coding for a membrane protein mainly required for vesicle traffic and membrane repair [93]. Inflammatory cells were detected in both MM and LGMD patients, scattered or organized into clusters, around necrotic fibers. Inflammatory infiltrates around vessels mainly consisted of macrophages, whereas CD4+ and CD8+ cells were found in endomysial infiltrates. Abnormal MHC-I expression was observed in degenerating/regenerating fibers usually close to inflammatory cluster cells but was absent in normal fibers [58, 59]. Indeed, the dysferlin-deficient AJ mice, one of the mouse models of dysferlinopathies, lacking functional T/B cells (AJ/SCID mice) develop a milder pathologic phenotype with increased muscle regeneration and reduced percentage of proinflammatory M1 macrophages. Importantly, these mice showed an increase in muscle force. The rescue of the phenotype was primarily due to a reduced IL-6 production by macrophages [74]. Indeed, liposome-clodronate macrophage depletion impairs IL-6 production also in control AJ mice, supporting the notion that the beneficial effect of T/B cells absence is due to a switch in macrophage phenotype and function.
3.4. The Emerging Role of Treg Cells in the Immune Response to Muscle Injury
As mentioned above, although it has long been recognized that lymphocytes invade acutely injured muscle, very little is known about their role in these conditions, mainly due to the very small number of these cells within the muscle, making their characterization difficult.
Recently, several mouse models have been generated to further understand the mechanisms underlying T cells recruitment, expansion, and persistence in acutely injured muscle.
Interestingly, it was recently shown that acute myofiber damage itself transiently activates muscle-antigen-specific CD8+ T cells in draining lymph nodes, supporting the idea of a transient autoreactive immune response upon muscle injury [94]. These activated CD8+ T cells transiently invade acute injured muscle, but their negative impact on muscle repair is promptly controlled by immunosuppressive cues [94]. The authors proposed that inadequate control of this CD8+ T-cell response might favor the emergence of sustained autoimmune myositis. In line with this view, Young et al. showed that aberrant muscle antigen exposure is sufficient to induce myositis in a Treg deficient milieu [95]. They generated a FoxP3/Syt VII double mutant, in which lack of synaptotagmin VII (Syt VII, a member of synaptotagmin family of membrane-trafficking proteins) impairs membrane resealing thus exposing endogenous muscle antigens [96]. Interestingly, while Syt VII mutant mice develop a mild, self-limiting inflammatory response involving muscle, FoxP3/Syt VII double mutants develop a significant inflammatory response in muscle, histologically resembling polymyositis in humans. Taken together, these studies suggest that, although muscle injury may evoke an autoreactive immune response to endogenous muscle antigens, this reaction is blunted within muscle through local immune-suppressive Treg dependent mechanism. In support of this, a muscle specific Treg population capable of potentiating muscle repair was recently identified [72]. Following cardiotoxin (CTX) muscle injury, a Treg cell population began to accumulate at day 4 after injury, just as the myeloid cell infiltrate switched from a proinflammatory to a proregenerative phenotype. Their frequency peaked at 2 weeks after injury, accounting for about 50% of the CD4+ T cells. However, while the number of CD4 T cells dropped to levels characteristic of uninjured muscle by 28 days after CTX injection, the number of Treg cells remained elevated. Interestingly, the increase in their number was not due to further recruitment, rather to clonal expansion within the muscle. Although these muscle Treg cells expressed the canonical gene signature characteristic of Treg cells, microarray gene expression analysis showed that the muscle Treg cells were closely related to Treg cells derived from visceral adipose tissue (VAT), but different from Treg cells in lymphoid organs. The authors provide a large data set of genes differentially expressed in VAT, muscle, colonic lamina propria, and prediabetic NOD pancreas derived Treg cells, supporting the notion that the Treg compartment is heterogeneous, has multiple functions, and exerts effects beyond the boundaries of the immune system [97]. Importantly, using the Foxp3-DTR mouse, in which Diphtheria Toxin is expressed under the control of the promoter of the Treg-specific Foxp3 gene, they showed that ablation of Treg cells by Diphtheria Toxin injection, after muscle injury, prevented muscle regeneration [72]. These authors identified a key molecule released by muscle Treg, responsible for the observed beneficial effect, named Amphiregulin (Areg), an EGF family growth factor. Administration of Areg in mice lacking Tregs restored efficient muscle regeneration, suggesting that this growth factor is part of fundamental cues during muscle repair. In this context, previous studies have suggested that Areg might act directly on muscle cells [98, 99]. Interestingly, a Treg population was enriched in dystrophic muscle derived from dysferlin-deficient and mdx mice. In particular, in mdx muscle, during both the acute and the chronic phase of the disease, this enriched Treg appears to be similar to muscle Treg. Although their number is very low, loss- and gain-of-function experiments showed that reducing Treg cells exacerbates the disease, while enriching them ameliorates it, as measured by serum creatine kinase level, an enzyme released into the blood upon muscle damage and a standard indicator of muscle damage in dystrophic mouse models, as well as by gene expression analysis. Taken together, these findings raise the possibility that a muscle antigen might be involved in recruiting Treg cells to the site of injury and/or retaining them therein. Since endogenous muscle antigens are released in physiological conditions upon injury as well as during pathological conditions by mechanical stress, such as in muscular dystrophies, it is conceivable that Treg cells, which are recruited and/or clonally expanded in muscle following injury, function to prevent a potential systemic autoimmune response by suppressing and regulating the inflammatory response. Taken together, these studies are very important and identify Treg cells and their products as potential new players in the orchestrated series of events underlying muscle repair in both acute and chronic context.
4. Concluding Remarks
Tissue damage induces a series of complex events including inflammation, which culminate in deposition of ECM. If this process is faulty, excessive and persistent ECM deposition takes place, and normal tissue is substituted by collagen scar, resulting in tissue dysfunction. In muscle, dysregulated repair with persistent fibrosis rather than efficient regeneration plays a prominent role in the clinical decline and reduced life expectancy associated with severe muscular dystrophies. A deep knowledge of the complex events following muscle damage will hopefully allow to design strategies aimed at promoting efficient regeneration.
Although inflammation is now considered a pathological feature of muscle repair, the role and regulation of this process have not been sufficiently examined. Moreover, the relative role of the innate or adaptive immunity in muscle regeneration and dystrophies is still unclear. Although blunting inflammation would not be a “cure” for these diseases, the emerging picture is that multiple strategies, addressing different aspects of the repair process, which may eventually converge, may be successful. In this context, although macrophages are emerging as indispensable for damage control and tissue remodelling following muscle injury, and as principal mediators of pathological skeletal remodelling in diseases such as IIMs and dystrophies, the involvement of other immune cells in promoting or preventing muscle damage resolution is also emerging.
In the last two decades, much research has pointed out the active interactions between muscle cells and the immune system and has clarified some of the mechanisms involved in IIMs. According to most of these studies, muscle cells are now emerging as possible facultative APCs, able, within an inflammatory milieu, to drive the activation and proliferation of CD4+ T cells, previously primed against exogenous or endogenous peptides [100]. Whether similar mechanisms may take place in muscle regeneration and muscular dystrophies is not clear yet. It is well known that T cells invade muscle upon both acute injury and chronic diseases. T cells can be recruited by muscle or other infiltrating cell populations, through cytokine release, but their persistence and activity might be sustained by an antigen-specific response. The possibility that an adaptive response to endogenous muscle antigens released upon muscle fibers degeneration, which may represent a common feature in acute injury as well as in IIM or MDs, would attribute to all of these conditions an auto-reactive component. In acute injury, the efficient repair process and the ability of MuSCs to proliferate and differentiate may be sustained by local Treg dependent immune regulation. In fact, autoreactive T cells are normally negatively selected in the thymus; if they escape to the periphery, they are normally eliminated by induction of apoptosis or rendered nonfunctional by the induction of anergy [101]. By contrast, in IIM, muscle fibers are direct targets of adaptive response, and the persistence and clonal expansion of autoreactive T cells prevent muscle healing. Although speculative at this stage, it may be hypothesised that in MDs, the continuous cycles of muscle fiber degeneration, due to muscle fiber fragility, allow for a prolonged and sustained endogenous muscle-specific antigen exposure, leading to the persistence and clonal expansion of potential muscle-antigen reactive T cells, eventually leading to breakdown in peripheral tolerance to self-antigens. Thus, if cytotoxic T cells escape the mechanisms of peripheral tolerance, a cellular immune response will accompany the innate immune response to tissue damage. The presence of alloreactive cytotoxic T cells in mdx muscle [102], the ability to transfer pathology from mdx mice to healthy mice by adoptive transfer of immune cells primed with muscle homogenates [63], and the presence of a well-conserved peptide in the hypervariable domain of the T-cell receptor of cytotoxic T cells from DMD patients [69] all support the possibility that a breakdown of peripheral tolerance occurs in muscular dystrophy. Furthermore, the observation that muscle specific clonal expansion of Treg cell population occurs during muscle regeneration and in MDs also suggests that a specific adaptive immune response is operating during muscle repair.
The discovery of the possible autoantigens that may evoke an adaptive immune response in MDs will facilitate therapeutic intervention. Similarly, the dissection of the T-cell response, which contributes to the complexity of the inflammatory milieu (outlined in Figure 1), will also allow the design of novel therapeutic strategies that would modulate the immune response to create a more favorable environment for MuSCs to differentiate and efficiently regenerate muscle lesions.
Figure 1.
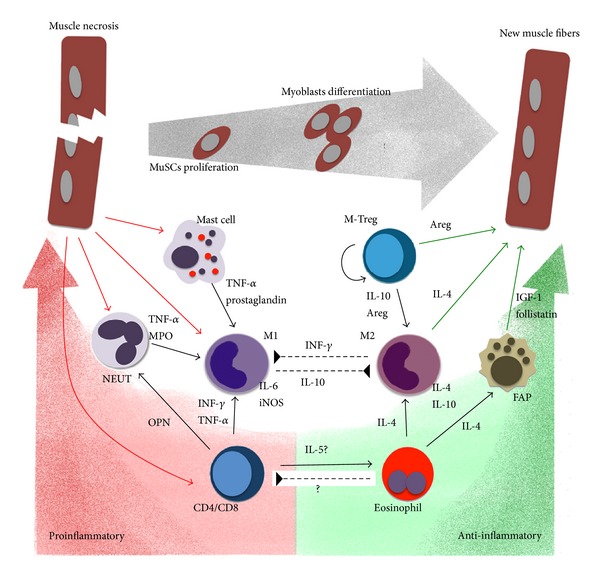
The complexity of the inflammatory milieu in muscle repair. During the initial phases of muscle damage, degenerating fibers release chemokines and cytokines recruiting mast cells, neutrophils (NEUT), and CD8/CD4 T cells which sustain proinflammatory M1 phenotype of recruited monocytes, thus promoting muscle fibers necrosis and debris clearing. These events are primarily mediated by IL-6, NO, TNFα, and IL-1β release. On the other hand, eosinophils are also recruited, which induce FAPs to sustain MuSCs proliferation via IL-4 release. Eosinophils-induced FAPs-produced follistatin promotes MuSCs differentiation, together with the clonally expanding mTreg which produce Areg. Areg also sustains MuSC proliferation and maintains/induces M2 anti-inflammatory/prohealing phenotype, supporting resolution of inflammation. Unbalance in the kinetics, quality, and activity of any of these actors would prevent resolution of inflammation, making muscle environment unfavourable for muscle regeneration, as it occurs in muscular dystrophies.
Acknowledgments
The authors thank Dr. B. Lozanoska-Ochser at DAHFMO, Unit of Histology, for helpful discussion and critical reading of the paper. During the preparation of this review, support was received by the French Muscular Dystrophy Association (AFM) Grant no. 15820 and the Telethon Italy Grant no. GGP13233 to MB. LM was recipient of a Dutch Parent Project NL Fellowship, and, presently, of an AFM Fellowship grant.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Saclier M, Cuvellier S, Magnan M, Mounier R, Chazaud B. Monocyte/macrophage interactions with myogenic precursor cells during skeletal muscle regeneration. FEBS Journal. 2013;280(17):4118–4130. doi: 10.1111/febs.12166. [DOI] [PubMed] [Google Scholar]
- 2.Mauro A. Satellite cell of skeletal muscle fibers. The Journal of Biophysical and Biochemical Cytology. 1961;9:493–495. doi: 10.1083/jcb.9.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seale P, Ishibashi J, Scimè A, Rudnicki MA. Pax7 is necessary and sufficient for the myogenic specification of CD45+:Sca1+ stem cells from injured muscle. PLoS Biology. 2004;2(5):p. E130. doi: 10.1371/journal.pbio.0020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palacios D, Mozzetta C, Consalvi S, et al. TNF/p38α/polycomb signaling to Pax7 locus in satellite cells links inflammation to the epigenetic control of muscle regeneration. Cell Stem Cell. 2010;7(4):455–469. doi: 10.1016/j.stem.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Serrano AL, Baeza-Raja B, Perdiguero E, Jardí M, Muñoz-Cánoves P. Interleukin-6 is an essential regulator of satellite cell-mediated skeletal muscle hypertrophy. Cell Metabolism. 2008;7(1):33–44. doi: 10.1016/j.cmet.2007.11.011. [DOI] [PubMed] [Google Scholar]
- 6.Kuang S, Gillespie MA, Rudnicki MA. Niche regulation of muscle satellite cell self-renewal and differentiation. Cell Stem Cell. 2008;2(1):22–31. doi: 10.1016/j.stem.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 7.Ten Broek RW, Grefte S, Von Den Hoff JW. Regulatory factors and cell populations involved in skeletal muscle regeneration. Journal of Cellular Physiology. 2010;224(1):7–16. doi: 10.1002/jcp.22127. [DOI] [PubMed] [Google Scholar]
- 8.Alexakis C, Partridge T, Bou-Gharios G. Implication of the satellite cell in dystrophic muscle fibrosis: a self-perpetuating mechanism of collagen overproduction. The American Journal of Physiology—Cell Physiology. 2007;293(2):C661–C669. doi: 10.1152/ajpcell.00061.2007. [DOI] [PubMed] [Google Scholar]
- 9.Brack AS, Conboy MJ, Roy S, et al. Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science. 2007;317(5839):807–810. doi: 10.1126/science.1144090. [DOI] [PubMed] [Google Scholar]
- 10.Lapidos KA, Kakkar R, McNally EM. The dystrophin glycoprotein complex: signaling strength and integrity for the sarcolemma. Circulation Research. 2004;94(8):1023–1031. doi: 10.1161/01.RES.0000126574.61061.25. [DOI] [PubMed] [Google Scholar]
- 11.Sacco A, Mourkioti F, Tran R, et al. Short telomeres and stem cell exhaustion model duchenne muscular dystrophy in mdx/mTR mice. Cell. 2010;143(7):1059–1071. doi: 10.1016/j.cell.2010.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bach JR, Martinez D. Duchenne muscular dystrophy: continuous noninvasive ventilatory support prolongs survival. Respiratory Care. 2011;56(6):744–750. doi: 10.4187/respcare.00831. [DOI] [PubMed] [Google Scholar]
- 13.Crisp A, Yin H, Goyenvalle A, et al. Diaphragm rescue alone prevents heart dysfunction in dystrophic mice in vitro. Human Molecular Genetics. 2011;20(3):413–421. doi: 10.1093/hmg/ddq477. [DOI] [PubMed] [Google Scholar]
- 14.Mosqueira M, Baby SM, Lahiri S, Khurana TS. Ventilatory chemosensory drive is blunted in the mdx mouse model of Duchenne Muscular Dystrophy (DMD) PLoS ONE. 2013;8(7) doi: 10.1371/journal.pone.0069567.e69567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mourkioti F, Kustan J, Kraft P, et al. Role of telomere dysfunction in cardiac failure in Duchenne muscular dystrophy. Nature Cell Biology. 2013;15(8):895–904. doi: 10.1038/ncb2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Evans NP, Misyak SA, Robertson JL, Bassaganya-Riera J, Grange RW. Dysregulated intracellular signaling and inflammatory gene expression during initial disease onset in duchenne muscular dystrophy. The American Journal of Physical Medicine and Rehabilitation. 2009;88(6):502–522. doi: 10.1097/PHM.0b013e3181a5a24f. [DOI] [PubMed] [Google Scholar]
- 17.Heredia JE, Mukundan L, Chen FM, et al. Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell. 2013;153(2):376–388. doi: 10.1016/j.cell.2013.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arnold L, Henry A, Poron F, et al. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. Journal of Experimental Medicine. 2007;204(5):1057–1069. doi: 10.1084/jem.20070075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deng B, Wehling-Henricks M, Villalta SA, Wang Y, Tidball JG. IL-10 triggers changes in macrophage phenotype that promote muscle growth and regeneration. Journal of Immunology. 2012;189(7):3669–3680. doi: 10.4049/jimmunol.1103180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang C, Li Y, Wu Y, Wang L, Wang X, Du J. Interleukin-6/signal transducer and activator of transcription 3 (STAT3) pathway is essential for macrophage infiltration and myoblast proliferation during muscle regeneration. Journal of Biological Chemistry. 2013;288(3):1489–1499. doi: 10.1074/jbc.M112.419788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kharraz Y, Guerra J, Mann CJ, Serrano AL, Muñoz-Cánoves P. Macrophage plasticity and the role of inflammation in skeletal muscle repair. Mediators of Inflammation. 2013;2013:9 pages. doi: 10.1155/2013/491497.491497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tidball JG, Villalta SA. Regulatory interactions between muscle and the immune system during muscle regeneration. The American Journal of Physiology—Regulatory Integrative and Comparative Physiology. 2010;298(5):R1173–R1187. doi: 10.1152/ajpregu.00735.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shireman PK, Contreras-Shannon V, Ochoa O, Karia BP, Michalek JE, McManus LM. MCP-1 deficiency causes altered inflammation with impaired skeletal muscle regeneration. Journal of Leukocyte Biology. 2007;81(3):775–785. doi: 10.1189/jlb.0506356. [DOI] [PubMed] [Google Scholar]
- 24.Nguyen HX, Lusis AJ, Tidball JG. Null mutation of myeloperoxidase in mice prevents mechanical activation of neutrophil lysis of muscle cell membranes in vitro and in vivo. Journal of Physiology. 2005;565(2):403–413. doi: 10.1113/jphysiol.2005.085506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hodgetts S, Radley H, Davies M, Grounds MD. Reduced necrosis of dystrophic muscle by depletion of host neutrophils, or blocking TNFα function with Etanercept in mdx mice. Neuromuscular Disorders. 2006;16(9-10):591–602. doi: 10.1016/j.nmd.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 26.Nguyen HX, Tidball JG. Null mutation of gp91phox reduces muscle membrane lysis during muscle inflammation in mice. Journal of Physiology. 2003;553(3):833–841. doi: 10.1113/jphysiol.2003.051912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Daley JM, Thomay AA, Connolly MD, Reichner JS, Albina JE. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. Journal of Leukocyte Biology. 2008;83(1):64–70. doi: 10.1189/jlb.0407247. [DOI] [PubMed] [Google Scholar]
- 28.Fleming TJ, Fleming ML, Malek TR. Selective expression of Ly-6G on myeloid lineage cells in mouse bone marrow: RB6-8C5 mAb to granulocyte-differentiation antigen (Gr-1) detects members of the Ly-6 family. Journal of Immunology. 1993;151(5):2399–2408. [PubMed] [Google Scholar]
- 29.Pillon NJ, Bilan PJ, Fink LN, Klip A. Cross-talk between skeletal muscle and immune cells: muscle-derived mediators and metabolic implications. The American Journal of Physiology—Endocrinology and Metabolism. 2013;304(5):E453–E465. doi: 10.1152/ajpendo.00553.2012. [DOI] [PubMed] [Google Scholar]
- 30.Côté CH, Bouchard P, van Rooijen N, Marsolais D, Duchesne E. Monocyte depletion increases local proliferation of macrophage subsets after skeletal muscle injury. BMC Musculoskeletal Disorders. 2013;14, article 359 doi: 10.1186/1471-2474-14-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perdiguero E, Sousa-Victor P, Ruiz-Bonilla V, et al. p38/MKP-1-regulated AKT coordinates macrophage transitions and resolution of inflammation during tissue repair. Journal of Cell Biology. 2011;195(2):307–322. doi: 10.1083/jcb.201104053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496(7446):445–455. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nature Reviews Immunology. 2005;5(12):953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 34.Mounier R, Théret M, Arnold L, et al. AMPKα1 regulates macrophage skewing at the time of resolution of inflammation during skeletal muscle regeneration. Cell Metabolism. 2013;18(2):251–264. doi: 10.1016/j.cmet.2013.06.017. [DOI] [PubMed] [Google Scholar]
- 35.Villalta SA, Nguyen HX, Deng B, Gotoh T, Tidbal JG. Shifts in macrophage phenotypes and macrophage competition for arginine metabolism affect the severity of muscle pathology in muscular dystrophy. Human Molecular Genetics. 2009;18(3):482–496. doi: 10.1093/hmg/ddn376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vidal B, Ardite E, Suelves M, et al. Amelioration of Duchenne muscular dystrophy in mdx mice by elimination of matrix-associated fibrin-driven inflammation coupled to the αmβ2 leukocyte integrin receptor. Human Molecular Genetics. 2012;21(9):1989–2004. doi: 10.1093/hmg/dds012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wehling M, Spencer MJ, Tidball JG. A nitric oxide synthase transgene ameliorates muscular dystrophy in mdx mice. Journal of Cell Biology. 2001;155(1):123–131. doi: 10.1083/jcb.200105110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Summan M, Warren GL, Mercer RR, et al. Macrophages and skeletal muscle regeneration: a clodronate-containing liposome depletion study. The American Journal of Physiology—Regulatory Integrative and Comparative Physiology. 2006;290(6):R1488–R1495. doi: 10.1152/ajpregu.00465.2005. [DOI] [PubMed] [Google Scholar]
- 39.Teixeira CFP, Zamunér SR, Zuliani JP, et al. Neutrophils do not contribute to local tissue damage, but play a key role in skeletal muscle regeneration, in mice injected with Bothrops asper snake venom. Muscle and Nerve. 2003;28(4):449–459. doi: 10.1002/mus.10453. [DOI] [PubMed] [Google Scholar]
- 40.Gorospe JRM, Nishikawa BK, Hoffman EP. Recruitment of mast cells to muscle after mild damage. Journal of the Neurological Sciences. 1996;135(1):10–17. doi: 10.1016/0022-510x(95)00255-z. [DOI] [PubMed] [Google Scholar]
- 41.Abonia JP, Friend DS, Austen WG, Jr., et al. Mast cell protease 5 mediates ischemia-reperfusion injury of mouse skeletal muscle. Journal of Immunology. 2005;174(11):7285–7291. doi: 10.4049/jimmunol.174.11.7285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Radley HG, Grounds MD. Cromolyn administration (to block mast cell degranulation) reduces necrosis of dystrophic muscle in mdx mice. Neurobiology of Disease. 2006;23(2):387–397. doi: 10.1016/j.nbd.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 43.Binstadt BA, Patel PR, Alencar H, et al. Particularities of the vasculature can promote the organ specificity of autoimmune attack. Nature Immunology. 2006;7(3):284–292. doi: 10.1038/ni1306. [DOI] [PubMed] [Google Scholar]
- 44.Yokota M, Suzuki K, Tokoyoda K, et al. Roles of mast cells in the pathogenesis of inflammatory myopathy. Arthritis Research & Therapy. 2014;16(2):p. R72. doi: 10.1186/ar4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dimachkie MM. Idiopathic inflammatory myopathies. Journal of Neuroimmunology. 2011;231(1-2):32–42. doi: 10.1016/j.jneuroim.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 46.Casciola-Rosen L, Nagaraju K, Plotz P, et al. Enhanced autoantigen expression in regenerating muscle cells in idiopathic inflammatory myopathy. Journal of Experimental Medicine. 2005;201(4):591–601. doi: 10.1084/jem.20041367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Silberstein DS, David JR. Tumor necrosis factor enhances eosinophil toxicity to Schistosoma mansoni larvae. Proceedings of the National Academy of Sciences of the United States of America. 1986;83(4):1055–1059. doi: 10.1073/pnas.83.4.1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ben Baruch-Morgenstern N, Shik D, Moshkovits I, et al. Paired immunoglobulin-like receptor A is an intrinsic, self-limiting suppressor of IL-5-induced eosinophil development. Nature Immunology. 2014;15(1):36–44. doi: 10.1038/ni.2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yamaguchi Y, Hayashi Y, Sugama Y, et al. Highly purified murine interleukin 5 (IL-5) stimulates eosinophil function and prolongs in vitro survival. IL-5 as an eosinophil chemotactic factor. Journal of Experimental Medicine. 1988;167(5):1737–1742. doi: 10.1084/jem.167.5.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wehling-henricks M, Sokolow S, Lee JJ, Myung KH, Villalta SA, Tidball JG. Major basic protein-1 promotes fibrosis of dystrophic muscle and attenuates the cellular immune response in muscular dystrophy. Human Molecular Genetics. 2008;17(15):2280–2292. doi: 10.1093/hmg/ddn129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cai B, Spencer MJ, Nakamura G, Tseng-Ong L, Tidball JG. Eosinophilia of dystrophin-deficient muscle is promoted by perforin-mediated cytotoxicity by T cell effectors. The American Journal of Pathology. 2000;156(5):1789–1796. doi: 10.1016/S0002-9440(10)65050-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wehling-Henricks M, Lee JJ, Tidball JG. Prednisolone decreases cellular adhesion molecules required for inflammatory cell infiltration in dystrophin-deficient skeletal muscle. Neuromuscular Disorders. 2004;14(8-9):483–490. doi: 10.1016/j.nmd.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 53.Horsley V, Jansen KM, Mills ST, Pavlath GK. IL-4 acts as a myoblast recruitment factor during mammalian muscle growth. Cell. 2003;113(4):483–494. doi: 10.1016/s0092-8674(03)00319-2. [DOI] [PubMed] [Google Scholar]
- 54.Uezumi A, Fukada S-I, Yamamoto N, Takeda S, Tsuchida K. Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nature Cell Biology. 2010;12(2):143–152. doi: 10.1038/ncb2014. [DOI] [PubMed] [Google Scholar]
- 55.Joe AWB, Yi L, Natarajan A, et al. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nature Cell Biology. 2010;12(2):153–163. doi: 10.1038/ncb2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mozzetta C, Consalvi S, Saccone V, et al. Fibroadipogenic progenitors mediate the ability of HDAC inhibitors to promote regeneration in dystrophic muscles of young, but not old Mdx mice. EMBO Molecular Medicine. 2013;5(4):626–639. doi: 10.1002/emmm.201202096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cordani N, Pisa V, Pozzi L, Sciorati C, Clementi E. Nitric oxide controls fat deposition in dystrophic skeletal muscle by regulating fibro-adipogenic precursor differentiation. Stem Cells. 2014;32(4):874–885. doi: 10.1002/stem.1587. [DOI] [PubMed] [Google Scholar]
- 58.Choi J-H, Park Y-E, Kim S-I, et al. Differential Immunohistological features of inflammatory myopathies and dysferlinopathy. Journal of Korean Medical Science. 2009;24(6):1015–1023. doi: 10.3346/jkms.2009.24.6.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Marino M, Scuderi F, Mazzarelli P, Mannella F, Provenzano C, Bartoccioni E. Constitutive and cytokine-induced expression of MHC and intercellular adhesion molecule-1 (ICAM-1) on human myoblasts. Journal of Neuroimmunology. 2001;116(1):94–101. doi: 10.1016/s0165-5728(01)00287-9. [DOI] [PubMed] [Google Scholar]
- 60.Schmidt J, Rakocevic G, Raju R, Dalakas MC. Upregulated inducible co-stimulator (ICOS) and ICOS-ligand in inclusion body myositis muscle: significance for CD8+ T cell cytotoxicity. Brain. 2004;127(5):1182–1190. doi: 10.1093/brain/awh148. [DOI] [PubMed] [Google Scholar]
- 61.Lerario A, Cogiamanian F, Marchesi C, et al. Effects of rituximab in two patients with dysferlin-deficient muscular dystrophy. BMC Musculoskeletal Disorders. 2010;11, article 157 doi: 10.1186/1471-2474-11-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Salajegheh M, Pinkus JL, Amato AA, et al. Permissive environment for B-cell maturation in myositis muscle in the absence of B-cell follicles. Muscle and Nerve. 2010;42(4):576–583. doi: 10.1002/mus.21739. [DOI] [PubMed] [Google Scholar]
- 63.Spencer MJ, Montecino-Rodriguez E, Dorshkind K, Tidball JG. Helper (CD4+) and cytotoxic (CD8+) T cells promote the pathology of dystrophin-deficient muscle. Clinical Immunology. 2001;98(2):235–243. doi: 10.1006/clim.2000.4966. [DOI] [PubMed] [Google Scholar]
- 64.Eghtesad S, Jhunjhunwala S, Little SR, Clemens PR. Rapamycin ameliorates dystrophic phenotype in mdx mouse skeletal muscle. Molecular Medicine. 2011;17(9-10):917–924. doi: 10.2119/molmed.2010.00256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tsoukas CD, Landgraf B, Bentin J, et al. Activation of resting T lymphocytes by anti-CD3 (T3) antibodies in the absence of monocytes. Journal of Immunology. 1985;135(3):1719–1723. [PubMed] [Google Scholar]
- 66.Al-Shanti N, Durcan P, Al-Dabbagh S, Dimchev GA, Stewart CE. Activated lymphocytes secretome inhibits differentiation and induces proliferation of C2C12 myoblasts. Cellular Physiology and Biochemistry. 2014;33(1):117–128. doi: 10.1159/000356655. [DOI] [PubMed] [Google Scholar]
- 67.Kissel JT, Lynn DJ, Rammohan KW, et al. Mononuclear cell analysis of muscle biopsies in prednisone- and azathioprine-treated Duchenne muscular dystrophy. Neurology. 1993;43(3):532–536. doi: 10.1212/wnl.43.3_part_1.532. [DOI] [PubMed] [Google Scholar]
- 68.Mendell JR, Moxley RT, Griggs RC, et al. Randomized, double-blind six-month trial of prednisone in Duchenne’s muscular dystrophy. The New England Journal of Medicine. 1989;320(24):1592–1597. doi: 10.1056/NEJM198906153202405. [DOI] [PubMed] [Google Scholar]
- 69.Gussoni E, Pavlath GK, Miller RG, et al. Specific T cell receptor gene rearrangements at the site of muscle degeneration in Duchenne muscular dystrophy. Journal of Immunology. 1994;153(10):4798–4805. [PubMed] [Google Scholar]
- 70.Mantegazza R, Andreetta F, Bernasconi P, et al. Analysis of T cell receptor repertoire of muscle-infiltrating T lymphocytes in polymyositis. Restricted Vα/β rearrangements may indicate antigen-driven selection. Journal of Clinical Investigation. 1993;91(6):2880–2886. doi: 10.1172/JCI116533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gordon S. Alternative activation of macrophages. Nature Reviews Immunology. 2003;3(1):23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 72.Burzyn D, Kuswanto W, Kolodin D, et al. A special population of regulatory T cells potentiates muscle repair. Cell. 2013;155(6):1282–1295. doi: 10.1016/j.cell.2013.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang P, Zheng SG. Regulatory T cells and B cells: implication on autoimmune diseases. International Journal of Clinical and Experimental Pathology. 2013;6(12):2668–2674. [PMC free article] [PubMed] [Google Scholar]
- 74.Farini A, Meregalli M, Belicchi M, et al. T and B lymphocyte depletion has a marked effect on the fibrosis of dystrophic skeletal muscles in the scid/mdx mouse. Journal of Pathology. 2007;213(2):229–238. doi: 10.1002/path.2213. [DOI] [PubMed] [Google Scholar]
- 75.Morrison J, Palmer DB, Cobbold S, Partridge T, Bou-Gharios G. Effects of T-lymphocyte depletion on muscle fibrosis in the mdx mouse. The American Journal of Pathology. 2005;166(6):1701–1710. doi: 10.1016/S0002-9440(10)62480-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vallese D, Negroni E, Duguez S, et al. The Rag2−Il2rb−Dmd− mouse: a novel dystrophic and immunodeficient model to assess innovating therapeutic strategies for muscular dystrophies. Molecular Therapy. 2013;21(10):1950–1957. doi: 10.1038/mt.2013.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vetrone SA, Montecino-Rodriguez E, Kudryashova E, et al. Osteopontin promotes fibrosis in dystrophic mouse muscle by modulating immune cell subsets and intramuscular TGF-β . Journal of Clinical Investigation. 2009;119(6):1583–1594. doi: 10.1172/JCI37662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Madaro L, Pelle A, Nicoletti C, et al. PKC theta ablation improves healing in a mouse model of muscular dystrophy. PLoS ONE. 2012;7(2) doi: 10.1371/journal.pone.0031515.e31515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gao Z, Wang Z, Zhang X, et al. Inactivation of PKCθ leads to increased susceptibility to obesity and dietary insulin resistance in mice. The American Journal of Physiology—Endocrinology and Metabolism. 2007;292(1):E84–E91. doi: 10.1152/ajpendo.00178.2006. [DOI] [PubMed] [Google Scholar]
- 80.Madaro L, Marrocco V, Fiore P, et al. PKCθ signaling is required for myoblast fusion by regulating the expression of caveolin-3 and β1D integrin upstream focal adhesion kinase. Molecular Biology of the Cell. 2011;22(8):1409–1419. doi: 10.1091/mbc.E10-10-0821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Messina G, Biressi S, Monteverde S, et al. Nfix regulates fetal-specific transcription in developing skeletal muscle. Cell. 2010;140(4):554–566. doi: 10.1016/j.cell.2010.01.027. [DOI] [PubMed] [Google Scholar]
- 82.D’Andrea M, Pisaniello A, Serra C, et al. Protein kinase C theta co-operates with calcineurin in the activation of slow muscle genes in cultured myogenic cells. Journal of Cellular Physiology. 2006;207(2):379–388. doi: 10.1002/jcp.20585. [DOI] [PubMed] [Google Scholar]
- 83.Madaro L, Marrocco V, Carnio S, Sandri M, Bouché M. Intracellular signaling in ER stress-induced autophagy in skeletal muscle cells. The FASEB Journal. 2013;27(5):1990–2000. doi: 10.1096/fj.12-215475. [DOI] [PubMed] [Google Scholar]
- 84.Valenzuela JO, Iclozan C, Hossain MS, et al. PKCθ is required for alloreactivity and GVHD but not for immune responses toward leukemia and infection in mice. Journal of Clinical Investigation. 2009;119(12):3774–3786. doi: 10.1172/JCI39692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gruber T, Hermann-Kleiter N, Pfeifhofer-Obermair C, et al. PKCθ cooperates with PKCα in alloimmune responses of T cells in vivo. Molecular Immunology. 2009;46(10):2071–2079. doi: 10.1016/j.molimm.2009.02.030. [DOI] [PubMed] [Google Scholar]
- 86.Marsland BJ, Soos TJ, Späth G, Littman DR, Kopf M. Protein kinase C θ is critical for the development of in vivo T helper (Th)2 cell but not Th-1 cell responses. Journal of Experimental Medicine. 2004;200(2):181–189. doi: 10.1084/jem.20032229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Healy AM, Izmailova E, Fitzgerald M, et al. PKC-θ-deficient mice are protected from th1-dependent antigen-induced arthritis. Journal of Immunology. 2006;177(3):1886–1893. doi: 10.4049/jimmunol.177.3.1886. [DOI] [PubMed] [Google Scholar]
- 88.Tan S-L, Zhao J, Bi C, et al. Resistance to experimental autoimmune encephalomyelitis and impaired IL-17 production in protein kinase Cθ-deficient mice. Journal of Immunology. 2006;176(5):2872–2879. doi: 10.4049/jimmunol.176.5.2872. [DOI] [PubMed] [Google Scholar]
- 89.Nagahama K, Ogawa A, Shirane K, Shimomura Y, Sugimoto K, Mizoguchi A. Protein kinase C theta plays a fundamental role in different types of chronic colitis. Gastroenterology. 2008;134(2):459–469. doi: 10.1053/j.gastro.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 90.Zanin-Zhorov A, Ding Y, Kumari S, et al. Protein kinase C-θ mediates negative feedback on regulatory T cell function. Science. 2010;328(5976):372–376. doi: 10.1126/science.1186068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ma J, Ding Y, Fang X, Wang R, Sun Z. Protein kinase C-θ inhibits inducible regulatory T cell differentiation via an AKT-Foxo1/3a-dependent pathway. Journal of Immunology. 2012;188(11):5337–5347. doi: 10.4049/jimmunol.1102979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Farini A, Sitzia C, Navarro C, et al. Absence of T and B lymphocytes modulates dystrophic features in dysferlin deficient animal model. Experimental Cell Research. 2012;318(10):1160–1174. doi: 10.1016/j.yexcr.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 93.Han R, Campbell KP. Dysferlin and muscle membrane repair. Current Opinion in Cell Biology. 2007;19(4):409–416. doi: 10.1016/j.ceb.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liao H, Franck E, Fréret M, et al. Myoinjury transiently activates muscle antigen-specific CD8+ T cells in lymph nodes in a mouse model. Arthritis and Rheumatism. 2012;64(10):3441–3451. doi: 10.1002/art.34551. [DOI] [PubMed] [Google Scholar]
- 95.Young NA, Sharma R, Friedman AK, Kaffenberger BH, Bolon B, Jarjour WN. Aberrant muscle antigen exposure in mice is sufficient to cause myositis in a Treg cell-deficient milieu. Arthritis & Rheumatism. 2013;65(12):3259–3270. doi: 10.1002/art.38184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chakrabarti S, Kobayashi KS, Flavell RA, et al. Impaired membrane resealing and autoimmune myositis in synaptotagmin VII-deficient mice. Journal of Cell Biology. 2003;162(4):543–549. doi: 10.1083/jcb.200305131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Josefowicz SZ. Regulators of chromatin state and transcription in CD4 T-cell polarization. Immunology. 2013;139(3):299–308. doi: 10.1111/imm.12115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Golding JP, Calderbank E, Partridge TA, Beauchamp JR. Skeletal muscle stem cells express anti-apoptotic ErbB receptors during activation from quiescence. Experimental Cell Research. 2007;313(2):341–356. doi: 10.1016/j.yexcr.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 99.Andrechek ER, Hardy WR, Girgis-Gabardo AA, et al. ErbB2 is required for muscle spindle and myoblast cell survival. Molecular and Cellular Biology. 2002;22(13):4714–4722. doi: 10.1128/MCB.22.13.4714-4722.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wiendl H, Hohlfeld R, Kieseier BC. Immunobiology of muscle: advances in understanding an immunological microenvironment. Trends in Immunology. 2005;26(7):373–380. doi: 10.1016/j.it.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 101.Srinivasan M, Frauwirth KA. Peripheral tolerance in CD8+ T cells. Cytokine. 2009;46(2):147–159. doi: 10.1016/j.cyto.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 102.Spencer MJ, Walsh CM, Dorshkind KA, Rodriguez EM, Tidball JG. Myonuclear apoptosis in dystrophic mdx muscle occurs by perforin-mediated cytotoxicity. Journal of Clinical Investigation. 1997;99(11):2745–2751. doi: 10.1172/JCI119464. [DOI] [PMC free article] [PubMed] [Google Scholar]