

Abstract
Pompe disease is due to mutations in the gene encoding the lysosomal enzyme acid α-glucosidase (GAA). Absence of functional GAA typically results in cardiorespiratory failure in the first year; reduced GAA activity is associated with progressive respiratory failure later in life. While skeletal muscle pathology contributes to respiratory insufficiency in Pompe disease, emerging evidence indicates that respiratory neuron dysfunction is also a significant part of dysfunction in motor units. Animal models show profound glycogen accumulation in spinal and medullary respiratory neurons and altered neural activity. Tissues from Pompe patients show central nervous system glycogen accumulation and motoneuron pathology. A neural mechanism raises considerations about the current clinical approach of enzyme replacement since the recombinant protein does not cross the blood-brain-barrier. Indeed, clinical data suggest that enzyme replacement therapy delays symptom progression, but many patients eventually require ventilatory assistance, especially during sleep. We propose that treatments which restore GAA activity to respiratory muscles, neurons and networks will be required to fully correct ventilatory insufficiency in Pompe disease.
Keywords: Pompe, Respiratory, Motoneurons, Plasticity, Therapy, Pathology
1. Overview of Pompe disease
The clinical features of Pompe disease were originally described by J.C. Pompe (1932) and subsequently the disease pathophysiology is considered the prototypical lyosomal storage disease (Cori, 1954; Hers, 1963). This neuromuscular disorder results from mutations in the GAA gene which has been mapped to the long arm of chromosome 17 (17q25.2–q25.3). More than 350 different mutations have been described, and the genotype–phenoype relationship is a subject of active investigation (Kroos et al., 2012a,b). The gene encodes a lysosomal enzyme – acid α-glucosidase or GAA – that is required for glycogen degradation. It is estimated that approximately 10% of total intracellular glycogen is normally present within lysosomes (Calder and Geddes, 1989; Geddes and Stratton, 1977). The glycogen enters the lysosome via incorporation into an autophagic vacuole or by invagination of the lysosomal membrane (i.e. microautophagy) (Geddes and Stratton, 1977). The 952 amino acid GAA enzyme is synthesized and processed via an intracellular pathway that enables post-translational modifications (Hirschhorn, 2001). After synthesis, GAA is glycosylated in the endoplasmic reticulum producing a 110 kDa precursor molecule (Hirschhorn, 2001). The molecule then acquires mannose 6-phosphate residues in a post-endoplasmic reticulum compartment and ultimately enters the lysosome via receptor-mediated transport (Raben et al., 2002). It appears that the most relevant receptor is the mannose 6-phosphate receptor, although a mannose 6-phosphate-independent pathway has been described (Klumperman et al., 1991; Tsuji and Suzuki, 1987). Once inside the lysosome, the GAA precursor molecule is cleaved to produce catalytically active 95-, 76-, and 70-kDa forms of GAA. Approximately 10% of glycosylated GAA precursor molecules are not cleaved in the lysosomes, but rather are secreted into the cytoplasm (Raben et al., 2002).
Pompe disease is associated with an absence or reduction of functional GAA which results in extensive glycogen accumulation in skeletal muscle, visceral organs and the central nervous system (CNS) (DeRuisseau et al., 2009; Raben et al., 2002; Sidman et al., 2008). The disease occurs in approximately 1 per 40,000 births, and based on appearance of symptoms, patients are typically classified as either early (infantile) or late-onset (juvenile/adult). These classifications, however, actually represent a continuum that relates to the extent of residual enzyme deficiency (Byrne et al., 2011b). Thus, early-onset Pompe disease results from complete or near complete deficiency of functional GAA protein, while late-onset patients maintain some residual enzyme activity (Hirschhorn, 2001; Raben et al., 2002). Heterogeneity of symptoms in Pompe disease is primarily explained by the specific gene mutation (Kroos et al., 2012a,b). For example, the most severely affected Pompe patients have a mutation in both GAA alleles that severely blunts or even eliminates the formation of functional GAA protein. Other mutations may result in variable levels of functional GAA protein and later onset of symptoms.
2. Respiratory insufficiency in Pompe disease
Respiratory insufficiency is extremely common in both the infantile and late-onset forms of Pompe disease (Burghaus et al., 2006; Mellies and Lofaso, 2009; Mellies et al., 2005; Pellegrini et al., 2005). Infants typically present at 4–6 months of age, and “respiratory difficulty” is often noted as the first symptom (van den Hout et al., 2003). Considerable CO2 retention (e.g. PaCO2 > 60 mmHg) can be present during spontaneous breathing in Pompe infants (Hogan et al., 1969), and cardiorespiratory failure is the leading cause of mortality (van den Hout et al., 2003). Late-onset patients show progressive respiratory muscle weakness and approximately 75% of children and adolescents with Pompe disease eventually require mechanical ventilation (Haley et al., 2003; Marsden, 2005). Subtle symptoms of night-time respiratory difficulty can include daytime somnolence or morning headache as well as laboratory data including polycythemia or elevated CO2. Of adults with Pompe disease, roughly 33% require mechanical ventilator support, and respiratory-related problems (e.g. pneumonia, bronchitis) are prevalent (Hagemans et al., 2005). Hypoventilation during sleep may occur even if the patient is still fully mobile and commonly precedes daytime respiratory failure. Respiratory-related symptoms also include restrictive alveolar disease and impaired cough. Impaired cough results in retained secretions and an inability to clear both the normal volume of pulmonary secretions as well as those associated with acute infections. Many adult patients present initially with respiratory insufficiency, and acute respiratory failure is often precipitated by pulmonary infections.
Approximately 60% of patients with late-onset Pompe disease have a mild reduction in vital capacity (<80% predicted), and 30–40% have moderate reduction (<60% predicted) (Hirschhorn and Huie, 1999; Mellies et al., 2001). In one sample of 8 adult-onset Pompe patients, vital capacity and peak inspiratory pressure averaged 31% and 26% of predicted values. In this patient cohort, daytime hypoventilation was evident by arterial blood gas values (PaO2: 56, PaCO2: 67 mmHg) (Mellies et al., 2005). Interestingly, severe respiratory insufficiency can occur in Pompe disease without evidence for significant limb muscle weakness. For example, there is only a weak relationship between indices of respiratory and locomotor function in adults with Pompe disease, and severe respiratory insufficiency can be present without any evidence of limb girdle muscle weakness (Pellegrini et al., 2005). The physiological reasons for this observation are not clear, but could relate to increases in the metabolic activity of respiratory muscles and neurons as compared to other skeletal motor systems. In any case, it appears that the respiratory neuromuscular system is particularly susceptible to dysfunction in Pompe disease which is a unique aspect compared to other forms of muscular dystrophy in which loss of ambulation precedes ventilatory insuf-ficiency.
3. Respiratory muscle function in Pompe disease
It is well accepted that skeletal muscle weakness is prominent in Pompe disease (Mellies and Lofaso, 2009; Mellies et al., 2001; Prigent et al., 2012). Muscular pathology is evident on histological exam, and electron microscopy reveals extensive accumulation of glycogen in muscle cell lysosomes in Pompe patients (Baudhuin et al., 1964; Hudgson and Fulthorpe, 1975). In the early phases of the disease, glycogen is also found dispersed in the cytoplasm and intrafibrillary spaces. In advanced Pompe disease, ruptured lysosomal fragments can be seen in skeletal muscle, and some myofibrils are nearly completely replaced by glycogen (Griffin, 1984). The end result of striated muscle glycogen accumulation is a loss of myofibrils and weakness (Hirschhorn, 2001).
Clinical and animal data both indicate that respiratory muscle function is impaired in Pompe disease. Prigent and colleagues (2012) evaluated trans-diaphragmatic pressure in a large sample of adults with Pompe disease using gastric and esophageal manometry. Magnetic stimulation of the phrenic nerve was used to evoke trans-diaphragmatic twitch pressure, which provides an indicator of diaphragm strength. The study confirmed diaphragmatic weakness, but it should be emphasized that diaphragmatic twitch pressures could also be influenced by conduction impairments along the motor nerve, as well as alterations in the neuromuscular junction. Reductions in expiratory pressures were also observed, and thus expiratory muscle function may also be impaired in Pompe disease (Prigent et al., 2012). This suggestion is strengthened by whole body MRI imaging data showing apparent pathology in lumbar extensor and abdominal muscles of Pompe patients (Carlier et al., 2011). That study also revealed that patients with the most impaired respiratory function had the most substantial alterations in the intercostal muscles. Respiratory muscle dysfunction and histopathology is also prominent in Pompe animal models (Mah et al., 2007, 2010). For example, the in vitro contractile force generated by the Gaa−/− mouse diaphragm is substantially blunted compared to wild-type control mice, and histological and biochemical evaluation shows profound glycogen accumulation. Thus, the literature has unequivocally established that diaphragm weakness is a hallmark feature of Pompe disease, and it is likely that the accessory respiratory muscles are also impaired. In this review, however, we emphasize that ventilatory failure in Pompe disease reflects a complex interplay between neural and muscular function (see Section 8).
4. The upper airway and Pompe disease
In addition to the primary and accessory respiratory “pump” muscles which actively change the volume of the thoracic or abdominal cavities, breathing also involves activation of pharyngeal and laryngeal muscles (Feldman and Del Negro, 2006). Hypoglossal (XII) motoneurons are of particular importance to upper airway patency since they regulate the shape, stiffness and position of the tongue (Bailey and Fregosi, 2004; Fregosi and Fuller, 1997; Gestreau et al., 2005; Remmers, 1978). Contraction of the extrinsic tongue muscles can dilate and/or stiffen the pharyngeal lumen, thereby minimizing airway narrowing and/or collapse in the face of negative inspiratory pressures (Fuller et al., 1999). Importantly, the tongue muscles appear to be particularly susceptible to pathology in Pompe disease. For example, Carlier et al. (2011) evaluated MRI images of Pompe patients, and concluded that while the majority of facial muscles were unaffected in Pompe, the tongue was always affected. Specifically, T1 weighted images showed “massive fat content” in the tongue, but the facial muscles were “systematically spared”. Moreover, the appearance of tongue pathology was unrelated to overall disease severity (i.e., the tongue pathology could be present prior to other symptoms) (Carlier et al., 2011). The first comprehensive evaluation of tongue motor function in Pompe patients was provided by Dubrovsky et al. (2011). These authors directly measured the force produced by tongue movements, and found that significant tongue weakness was present in all subjects, even in those individuals who were otherwise asymptomatic (Dubrovsky et al., 2011). Other published work is also consistent with the presence of tongue and/or upper airway dysfunction (Byrne et al., 2011a; Jones et al., 2010; Margolis et al., 1994; Muller et al., 2009). For example, feeding difficulties are present in >70% of Pompe infants and approximately 50% of infants are never fed by mouth (Byrne et al., 2011a). A videofluoroscopic study of 13 infants and children with Pompe disease showed that dysphagia was always present, and swallowing difficulty did not correlate with gross motor function (Jones et al., 2010). Speech problems can also occur in Pompe disease, with disorders of articulation and/or nasal resonance occurring in approximately 80% of the subjects in a recent report (Muller et al., 2009). Of note, the authors speculated that speech deficits in Pompe disease likely occurred due to “disturbance in muscular control of the speech mechanisms resulting in weakness secondary to lower motor neuron involvement”.
Our clinical experience also suggests upper airway dysfunction in Pompe disease, and we have consistently noted macroglossia and obvious impairments in tongue motor control. Pompe infants typically have a protruded tongue that does not withdraw into the mouth during breathing. Parents have described to us that their child has an “inability to control the tongue” which interferes with or prevents feeding. Indeed, all the Pompe infants at our hospital are fed via gastrostomy. Fig. 1 shows a mid-sagittal MRI image of a 5 mo old Pompe infant treated with enzyme replacement therapy (ERT; see Section 6) at Shands Hospital at the University of Florida. Macroglossia is indicated by the “fullness” of the tongue, particularly at the base. The enlarged tongue causes the epiglottis to move dorsally, and this can potentially obstruct the upper airway.
Fig. 1.
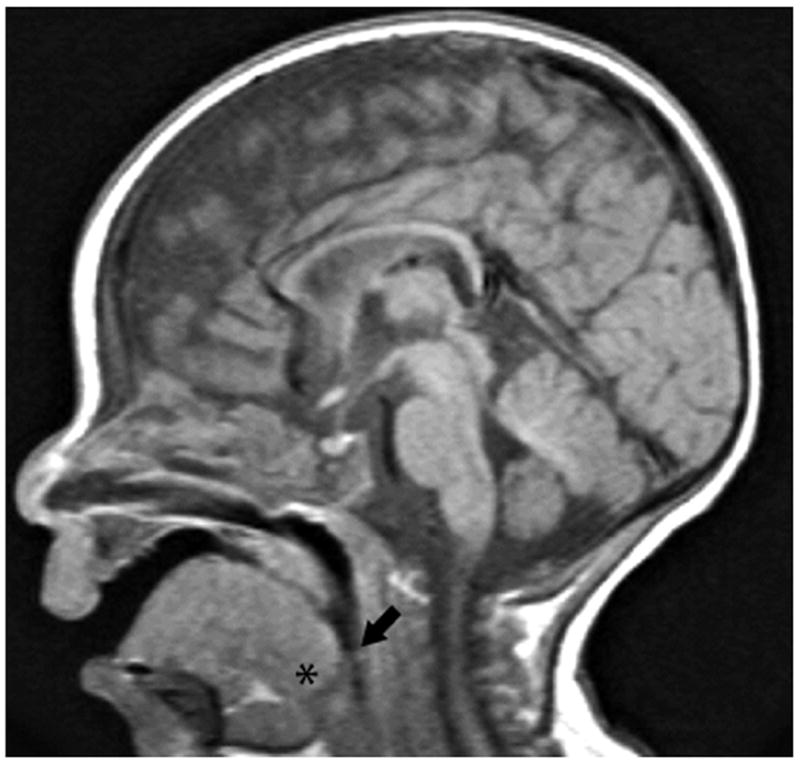
A midsagittal T1-weighted MRI image of a five month old male with Pompe disease. Macroglossia is suggested by enlargement of the tongue, particularly at the base (*). The enlarged tongue is displacing the epiglottis posteriorly (arrow) which partially occludes the airway.
The prominence of tongue pathology and macroglossia in Pompe disease suggests that sleep disordered breathing, and in particular obstructive sleep apnea (OSA), could be a substantial problem (Margolis et al., 1994). OSA is widely accepted to represent an impaired ability of the respiratory neuromuscular system to maintain pharyngeal airway patency during sleep. Most investigators agree that inadequate neural drive to the tongue and pharyngeal muscles is one of the primary factors leading to collapse of the airway and apnea during sleep (Horner, 2008). Pharyngeal muscle pathology may also be a contributing factor, and alterations in airway anatomy may predispose patients to OSA (Mannarino et al., 2012). Margolis et al. (1994) were the first to comment on OSA in Pompe disease. In a case report, they described severe OSA in an adult patient, and emphasized that macroglossia and tongue pathology could be the underlying cause. Subsequently, Mellies and colleagues evaluated the prevalence of sleep disordered breathing in 27 subjects with juvenile or adult onset Pompe disease. Substantial disturbances in breathing during sleep were present in 13 of the subjects, however, only 1 individual showed evidence for OSA (Mellies et al., 2001). In another study, five adult Pompe patients underwent overnight polysomnography prior to initiation of mechanical ventilator support (Pellegrini et al., 2005). The apnea–hypopnea index (AHI) ranged from 7 to 28 events per hour and the duration of significant desaturation events (<90%) ranged from 1–28% of total sleep time. An overnight sleep study conducted by our group on a 2 yr old with Pompe disease revealed an AHI of 5 with the occurrence of both obstructive and central apneic events (Byrne, unpublished). We noted that central apneas were associated with more profound desaturations compared to the obstructive events. Collectively, the available studies indicate that sleep disordered breathing is prevalent in Pompe disease, and centrally mediated apneic events may be common, particularly in the adult population. We suggest that further study of this topic is warranted.
5. The central nervous system, breathing, and Pompe disease
Motor problems in Pompe disease, including impaired breathing, have historically been attributed to muscular pathology (Raben et al., 2002). We emphasize that respiratory muscle pathology and dysfunction are prominent features of Pompe disease (see above) that contribute to respiratory impairments as the disease progresses. However, the genetic mutation in Pompe disease is not restricted to muscle tissues, and accordingly CNS pathology must be considered. Indeed, neural pathology is prominent in the majority of lysosomal storage disorders, and there is evidence that lysosomal dysfunction can lead to neuronal cell death (Bellettato and Scarpa, 2010). Substantial glycogen accumulation is present in the central nervous system of Pompe patients (DeRuisseau et al., 2009; Gambetti et al., 1971; Mancall et al., 1965; Martin et al., 1973; Martini et al., 2001; Teng et al., 2004; Thurberg et al., 2006) as well as animal models including mice and quail (DeRuisseau et al., 2009; Matsui et al., 1983; Sidman et al., 2008). Clinical case reports have also described extensive neuropathology in Pompe tissues obtained on autopsy. For example, Gambetti and colleagues histologically evaluated spinal cords of two Pompe infants (age 5 and 9 months) within hours of death. The anterior cervical spinal cord had massive glycogen accumulation and anterior horn neurons showed marked swelling (Gambetti et al., 1971). Martin et al. reported “very severe” glycogen accumulation in spinal anterior horn neurons (presumably motoneurons), and also in brainstem neurons of a Pompe infant. The authors stated that “the most prominent signs of neuronal storage are found in the spinal ganglia, the anterior horns and in all the motor nuclei of the brain stem” (Martin et al., 1973). Additional Pompe case reports confirm glycogen accumulation in the anterior spinal cord with “swelling” of neurons in this region (i.e. soma size 2–3× normal) being a consistent finding (Hogan et al., 1969; Mancall et al., 1965; Teng et al., 2004). Diminished or even absent spinal reflexes have also been described in Pompe patients (Clement and Godman, 1950; Gambetti et al., 1971; Teng et al., 2004; Willemsen et al., 1998; Zellweger et al., 1955), a finding that strongly implicates alterations in neuronal function in this population.
We recently conducted a histological and biochemical evaluation of cervical spinal cord tissue from a Pompe patient whom had been treated with recombinant GAA ERT (see Section 6) since six months of age (DeRuisseau et al., 2009). Histological evaluation of putative phrenic motoneurons in the C4 spinal cord revealed a swollen cell body and chromatolysis-like appearance. Fig. 2 shows new histological images from the cervical spinal cord of the same patient. In addition, biochemical evaluation of glycogen content indicated substantial accumulation in the cervical spinal cord (DeRuisseau et al., 2009).
Fig. 2.
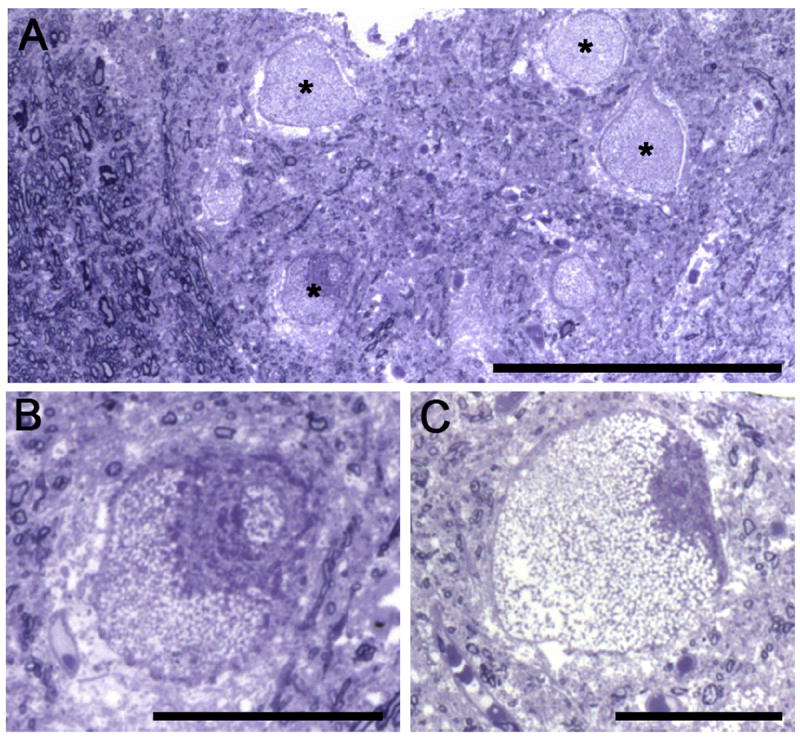
Semi-thin plastic embedded sections from the mid-cervical spinal cord of an 18-month-old child with Pompe disease. Cervical spinal tissue was obtained on autopsy, plastic embedded, cut at 2μm thickness and stained with toluidine blue. The patient had been treated with recombinant enzyme replacement therapy for the preceding 12 months but experienced respiratory failure. Panel A depicts the ventrolateral cervical spinal cord at the junction of the white and gray matter. Myelinated axons are clearly discerned in the left part of the image, and the asterisks highlight four ventral horn neurons (putative motoneurons). Panel B shows a higher magnification of the neuron indicated by the asterisk in A. Panel C shows another ventral horn neuron from an adjacent tissue section. Note that the neurons are swollen and nuclei (when visible) were displaced from the center of the cell – histopathological features characteristic of central chromatolysis. Scale bars: (A) 200 μm; (B–C) 50 μm.
A comprehensive evaluation of CNS pathology in a murine Pompe model (the 6neo/neo mouse) was published by Sidman and colleagues in 2008. Glycogen began to accumulate throughout the brain and spinal cord of Pompe mice as early as one month of age. Accumulation was particularly evident in motoneurons, and electron microscopy revealed that enlarged lysosomes filled more than 50% of the motoneuron cell volume. While neurons were most prominently affected, as Pompe mice reached old age (15–22 months) they began to show signs of astrogliosis in white matter tracks. Behavioral testing using a variety of methods including the “rotorod” balancing test revealed that deficits began to emerge at approximately six months of age (Sidman et al., 2008). It is difficult, however, to ascertain the mechanistic basis of the behavioral changes, and the authors stated that “further studies are needed to dissect the role of muscle versus nervous system in motor deficits”.
Our group has examined behavioral and neurophysiological aspects of breathing in the Gaa−/− Pompe mouse model (Raben et al., 1998) and also in a transgenic strain which expresses GAA activity in skeletal muscle but not in the central nervous system (via muscle specific expression of human GAA (Raben et al., 2001)). The Gaa−/− mice had extensive cervical spinal glycogen accumulation including in phrenic motoneurons identified by cholera toxin labeling (DeRuisseau et al., 2009). Neurophysiological studies, as well as measurements of ventilation, indicated blunted inspiratory motor activity in Gaa−/− as compared to wild-type mice (DeRuisseau et al., 2009; Mah et al., 2007, 2010). For example, unanesthetized and unrestrained Gaa−/− mice showed reductions in inspiratory tidal volume as assessed via whole body plethysmography (DeRuisseau et al., 2009). Another important observation was that during a respiratory challenge with hypercapnia, the muscle specific-GAA mice showed ventilation values that fell between the full knockout (Gaa−/−), and the wild type mice. Thus, while they performed better than the Gaa−/− mice, the muscle specific group still had an impaired ventilatory response when compared to the wild type controls. Lastly, in an anesethetized and ventilated preparation which enables more careful regulation of arterial blood gases, reductions in phrenic nerve inspiratory bursting were noted in Gaa−/− mice (DeRuisseau et al., 2009).
We next tested the hypothesis that restoring GAA enzyme activity in the region of the phrenic motor nucleus could lead to improved breathing in Gaa−/− mice (Qiu et al., 2012). Direct spinal injection of adeno-associated virus (AAV) encoding GAA (i.e., AAV–GAA) restored spinal GAA enzyme activity, and GAA immunostaining was clearly evident in the cervical ventral horn. Biodistribution studies showed a high number of AAV copies (i.e., >1 × 107 genome copies per μg of genomic DNA) in cervical spinal cord at or near the site of injection (C3–C8). However, AAV vector genomes could not be detected in the diaphragm muscle. This latter point is particularly important since plethysmography revealed that inspiratory tidal volume was increased in mice that had received AAV–GAA spinal injections. We concluded that the increased ventilation reflected “correction” of phrenic motoneuron pathology due to restoration of GAA protein in only the cervical spinal cord (Qiu et al., 2012). Accordingly, spinal motoneuron pathology, and in particular phrenic motoneuron pathology, is likely to make a substantial contribution to diaphragm motor deficits in Pompe disease (Byrne et al., 2011a; DeRuisseau et al., 2009; Mah et al., 2007).
The brainstem is also of substantial interest in the context of Pompe disease and respiratory control. While it is well known that the neurons and networks that control and shape the pattern of respiratory motor output reside in the medulla and pons (Feldman et al., 2003), brainstem respiratory networks have not been evaluated in the context of Pompe disease (or, to our knowledge, any other lysosomal storage disorder). Brainstem pathology has been noted on autopsy (Hogan et al., 1969; Mancall et al., 1965; Teng et al., 2004), brainstem dysfunction may be indicated by clinical reports of auditory dysfunction in Pompe disease (Musumeci et al., 2012; van Capelle et al., 2010). Interestingly, Musumeci and colleagues found that respiratory dysfunction was statistically more likely to be present in patients that also showed hearing loss. Since both auditory and respiratory neural circuits are found in the brainstem, one logical, albeit speculative suggestion is that brainstem pathology occurring in both networks explains the link between hearing and breathing dysfunction.
There have been two published studies of brainstem pathology in Pompe animal models. Sidman and colleagues described brainstem glycogen accumulation in the 6neo/6neo murine Pompe model but did not comment on the appearance of XII motoneurons or brainstem respiratory control regions (Sidman et al., 2008). We recently reported that Gaa−/− mouse XII motoneurons show extensive glycogen accumulation with a large, swollen appearance (Lee et al., 2011). This histopathology was accompanied by an increase in the variability of XII motor output as assessed in neurophysiological studies of anesthetized and ventilated mice. Fig. 3 provides a comparison of the histological appearance of XII motoneurons in a control wild-type mouse and a Gaa−/− mouse. Note the complete absence of GAA immunostaining in Gaa−/− motoneurons as well as the larger, swollen appearance when compared to the wild-type motoneurons. Overall, these initial findings from animal models coupled with clinical observations (see Section 4) suggest that brainstem respiratory activity and the motor output of the tongue muscles may be impaired in Pompe disease.
Fig. 3.
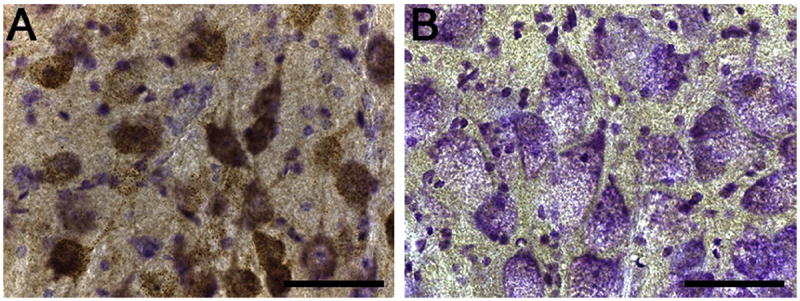
Hypoglossal motoneurons in wild type (129) and Pompe (Gaa−/−) mice. Tissues were processed with an antibody against GAA protein, and were counter-stained with cresyl violet. Tissue from a wild type 129 mouse shows positive GAA immunostaining staining (brown) in hypoglossal motoneurons, as expected. In contrast, motoneurons from the Gaa−/− mouse show a complete absence of positive GAA staining. Note the difference in the histological appearance of cells in panel A vs. B. Specifically, Gaa−/− motoneurons are larger with a swollen appearance. Scale bars: 50 μm.
6. Enzyme replacement therapy (ERT) – impact on breathing in Pompe disease
Therapies aimed at altering glycogen synthesis (e.g. high-protein diet, steroids) have failed to reduce glycogen accumulation in Pompe disease (Isaacs et al., 1986). Other treatment approaches with limited or no clinical impact include bone marrow transplantation and administration of unphosphorylated GAA (de Barsy et al., 1973). The only currently FDA-approved treatment for Pompe disease involves bi-weekly intravenous (i.v.) infusion of recombinant GAA enzyme (Beck, 2009; Byrne et al., 2011a). This method aims to supplement endogenous GAA through delivery of purified, exogenous GAA (i.e., ERT) (Bijvoet et al., 1999). ERT was made feasible by large-scale production of recombinant human GAA (rhGAA) in transgenic rabbits (Bijvoet et al., 1999) and Chinese Hampster Ovary (CHO) cells ((Kishnani et al., 2006). Pre-clinical experiments showed that i.v. rhGAA therapy leads to correction of the biochemical phenotype in cardiac, skeletal and smooth muscle in the Gaa−/− mouse model (Bijvoet et al., 1999; Kikuchi et al., 1998).
An initial test of ERT in four infants was published in 2000 (Van den Hout et al., 2000) After twelve weeks of ERT (15–20 mg/kg/week), skeletal muscle GAA activity remained below normal but reached normal values when the dose was increased to 40 mg/kg for an additional 12 weeks. Histological assessments of muscle tissue showed reductions in glycogen staining after the dose increase, but total tissue glycogen content was not changed. Most importantly, all patients survived beyond the age of 1 year, which exceeds the mean survival age of untreated Pompe patients (Byrne et al., 2011a). In a second trial, three infants received 5 mg/kg of GAA two times per week (Amalfitano et al., 2001). All subjects had no detectable GAA protein prior to ERT, but they developed an anti-GAA antibody titer after treatment. Initial improvements in muscle and pulmonary function were noted, but functional declines occurred coincident with the rise of GAA antibody titer (Amalfitano et al., 2001). In 2004, Van den Hout published a long-term follow up on the 4 patients from the earlier 2001 report. All patients survived beyond 4 years of age, and the two youngest patients demonstrated improvements in locomotor function that were well beyond what could be expected without treatment. However, 3 of the 4 patients developed severe respiratory insufficiency, and thus were completely ventilator dependent at 4 years of age (Van den Hout et al., 2004).
A larger scale randomized, blinded study of ERT in late onset Pompe disease was published in 2010 (van der Ploeg et al., 2010). Ninety patients who were at least 8 years of age, able to ambulate 40 meters in 6 min, and free of invasive ventilator support at study onset were randomly assigned to either placebo or ERT groups. Invasive ventilator support was defined as any support that was applied via an endotracheal tube. Thus, patients who required nocturnal ventilation via a facemask or nose piece were not excluded from the study. Small increases (<4%) in maximal inspiratory and expiratory pressure were noted in the ERT group, and ERT prevented the declines in forced vital capacity over the duration of the study that occurred in the placebo group. Since the 2010 report by van der Ploeg, several additional evaluations of pulmonary function after ERT have been published. Schneider and colleagues evaluated pulmonary function in late-onset Pompe disease and concluded that while ERT can stabilize pulmonary function, it only delays the eventual requirement for mechanical ventilation. Interestingly, in their study the mouth pressure generated in the first 100 ms of inspiration (i.e., P0.1) showed substantial declines over the course of ERT in 5 of the 6 patients (Schneider et al., 2013). This is note-worthy since P0.1 has been suggested to correlate with respiratory neural drive (Milic-Emili et al., 1975). Accordingly, alterations in the neural input to the respiratory muscles may have occurred over the two-year period in which the study was conducted. The authors concluded that “a close collaboration of neurologists and pneumologists” should be an essential component of any treatment strategy for Pompe disease (Schneider et al., 2013). A longitudinal evaluation of 368 late onset Pompe patients found that over the course of ERT, forced vital capacity (FVC) improved in approximately ½ of patients, was unchanged in 14%, and declined in 35% (Toscano and Schoser, 2013). Moreover, no association was seen between the duration of ERT treatment and changes in FVC in a recent study that comprehensively evaluated pulmonary function in 5 adults with “severe Pompe disease” (Orlikowski et al., 2011). All subjects in the study required ventilator support, and were evaluated over the course of 12 months of ERT. Three of the patients showed increases in the sustainable duration of independent breathing (i.e. without ventilator support). Two patients were able to modestly decrease ventilator support by 1–2 h per day, two had no change in their ventilator support, and the final patient required an increase in the amount of ventilator support by 5 h per day. Overall, we conclude that the literature indicates that ERT provides a modest and stabilizing impact on respiratory function with a possibility of slight improvements. Indeed, ERT improves the overall survival in early onset Pompe disease (Beck, 2010; Byrne et al., 2011b). However, variability in the success of ERT is prevalent (Van den Hout et al., 2004), and ventilator-free survival is observed in only 1/3 of early onset patients (Byrne et al., 2011b). The limited success of ERT, along with the aforementioned animal studies and case reports, has led to our overall hypothesis that neuropathology in respiratory neurons and networks makes a substantial contribution to respiratory insufficiency in Pompe disease (DeRuisseau et al., 2009). ERT could not be expected to effectively treat neuropathology since the recombinant GAA enzyme does not cross the blood-brain-barrier where the protein might directly influence the CNS (Kikuchi et al., 1998; Raben et al., 2003).
Several other research groups have reached similar conclusions. Muller and colleagues reported that even with enzyme replacement therapy, children with Pompe Disease are still at high risk for developing speech disorders (Muller et al., 2009). These authors speculated that this could reflect lower motoneuron involvement which will not be effectively targeted by ERT. Similarly, Burrow et al. (Burrow et al., 2010) recently described a 2-year-old Pompe child who after 19 months of ERT showed “acute worsening of weakness, particularly of the extremities and diaphragm”. These authors noted that their Pompe case report “offers strong evidence that storage in the nervous system can lead to a loss of motor neurons and to neuropathic weakness”. Rohrbach and colleagues reported that ERT effectively delayed the muscular progression of a Pompe infant over a 44 month period, but neurological symptoms including impaired language development remained (Rohrbach et al., 2010).
While an accumulating body of evidence indicates that neuropathology contributes to respiratory insufficiency in Pompe disease, this does not rule out potential for recovery with appropriate interventions. For example, in a rodent model of amyotrophic lateral sclerosis (ALS), relatively robust phrenic motor recovery can be induced even after a profound loss of phrenic motoneurons (Nichols et al., 2013). In that work, a novel hypoxic treatment paradigm was used to induce respiratory neural plasticity and motor recovery. Similar paradigms for targeted neural plasticity and recovery have not been examined in Pompe disease, or to our knowledge any other lysosomal storage disorder. In the Gaa−/− mouse model, functional recovery has been noted even when therapies have been initiated in older mice that would be expected to have considerably advanced neural and muscular pathology. For example, delivery of AAV–GAA to the diaphragm of 21-mo-old mice resulted in an increase in ventilation (Mah et al., 2010). Recent work from our group has confirmed intramuscular delivery of AAV encoding the GAA gene can cause persistent expression of GAA as well as reduction of glycogen accumulation in Gaa−/− mouse motoneurons (Elmallah, Fuller, Byrne, unpublished).
7. The future of Pompe respiratory therapy
The current standard of care in Pompe disease therapy is muscle-directed ERT and other supportive measures, including ventilatory assistance. Aside from the problems related to the feasibility of lifelong ERT and managing complications of immune responses to the recombinant protein, it is important to emphasize that the long-term treated population of patients continue to lose ventilatory function. The substantial effort needed for bi-weekly ERT treatment, the associated high cost (>$500,000 per year in an adult), and its limited effectiveness warrant an improved approach. One encouraging strategy is to enhance the effectiveness of the GAA molecule to penetrate the lysosome. For example, a recent study described a modified form of recombinant GAA in which human GAA was fused to the ligand of the insulin-like growth factor II receptor (IGF-IIR) (Maga et al., 2013). The resultant fusion protein had full catalytic activity for glycogen and each molecule of the recombinant protein was able to bind to the cation-independent mannose 6-phosphate – IGF-II co-receptor thereby enhancing GAA uptake into cells (Maga et al., 2013).
Since the underlying pathology results from mutations in a single gene, Pompe disease is an ideal candidate for gene therapy approaches. The overall rationale and preclinical history of gene transfer approaches for Pompe disease was recently reviewed in detail (Byrne et al., 2011a). An advantage of gene therapy, in contrast to intravenously delivered ERT, is that viral vectors will be more effective at targeting the entire motor unit. For example, a recent report from our group showed that a single injection of AAV serotype 9 (AAV9) could effectively drive transgene expression in both tongue myofibers and XII motoneurons in Gaa−/− mice (Elmallah et al., 2012). Studies designed to test the functional impact of AAV9–GAA on Gaa−/− motoneurons following intramuscular delivery are ongoing. However, sustained improvement in both cardiac and respiratory function occur following i.v. delivery of AAV1–GAA to young Gaa−/− mice (Mah et al., 2007). Specifically, plethysmography studies revealed an increase in inspiratory tidal volume in adult mice that were treated with AAV1–GAA. Restoration of GAA activity via application of AAV1–GAA to the diaphragm also improves diaphragm contractility, ventilation, and possibly phrenic neural outflow in Gaa−/− mice (Mah et al., 2010).
An accumulation of preclinical work, including the studies mentioned above, has led to an ongoing open label, Phase I/II study in which rAAV2/1-CMV-hGAA (1–5 × 1012 vg) is being delivered to the diaphragm via direct intramuscular injection in children with Pompe disease (ClinicalTrials.gov Identifier: NCT00976352). The study is designed to target those on assisted ventilation despite ongoing treatment with ERT. The primary purpose is to determine if diaphragm delivery of rAAV1–hGAA is safe based on a number of parameters. A secondary outcome is to test the effects of gene transfer on ventilatory function. All subjects in the study required full-time mechanical ventilation due to respiratory failure that was unresponsive to either ERT or a pre-operative respiratory muscle conditioning paradigm. Subjects were evaluated over a 180-day period, and the initial results have been published (Smith et al., 2013). To date, the results indicate that diaphragm delivery of rAAV1–hGAA is safe and does have functional benefits. Specifically, the unassisted tidal volume showed a statistically significant increase after rAAV1–hGAA, and on average the duration of unassisted breathing that could be tolerated (i.e., breathing with no ventilator support) increased by 425% (Smith et al., 2013). The next phases of this ongoing trial will determine if intervention earlier in the disease progression and/or higher rAAV1–hGAA doses produce a greater functional impact.
8. Conclusion
Respiratory dysfunction is prominent in both early and late onset Pompe disease patients. While respiratory muscular pathology and dysfunction are prominent, a growing basic science and clinical literature supports the hypothesis that neural dysfunction also contributes to respiratory insufficiency. The relative contribution of muscular vs. neural pathology to respiratory dysfunction in Pompe disease is difficult to ascertain because both components of the motor unit are affected by the cellular effects of glycogen accumulation. In Fig. 4, we present a conceptual model of the relationship between declines in muscular and neural function in Pompe disease, and the associated impact on respiratory function. In this model, we propose the existence of three critical periods in the etiology of respiratory dysfunction. The first critical period represents the asymptomatic stage. During this time frame, glycogen accumulation begins throughout respiratory motor units (e.g., myofibers, motoneuron soma, axons, etc.) and possibly in premotor respiratory neurons and networks. Although pathological processes are beginning during this phase, the pathology has not reached a level which will cause motor impairments. The duration of this period will be highly variable across the patient population (e.g., early vs. late-onset), and will be determined largely by the specific gene mutation (Kroos et al., 2012b) and the amount of functional GAA enzyme. During the second critical period, myofibril contractile dysfunction begins to develop. The development of diaphragm and accessory muscle weakness will gradually lead to an impaired ability to generate inspiratory tidal volume. We suggest that during this period, respiratory muscle dysfunction will initially trigger compensatory increases in respiratory neuromotor drive. This concept derives from studies of respiratory muscle failure during weaning from ventilator support – there is often an increase in respiratory drive when the respiratory “pump” begins to fail (Liu et al., 2012). During the third critical period, the combination of neural and muscular pathology reaches a threshold and respiratory failure ensues. Therapies which improve muscle function (e.g., ERT, gene therapy) or neural function (e.g., gene therapy) should prevent or minimize the declines in respiratory function. An important conceptual point from this model is that even if muscle function is improved (i.e., via ERT), declines in neuromotor output may still eventually lead to respiratory failure. We therefore suggest that effective respiratory therapies that have long lasting functional benefits will need to target the entire respiratory motor unit (e.g. myofibers and associated motoneurons). Gene therapy approaches show promise for addressing this goal, and may ultimately be paired with other respiratory management and/or rehabilitation approaches such as diaphragm pacing, respiratory rehabilitation exercises (Jones et al., 2011), and ERT with modified forms of recombinant GAA (Maga et al., 2013). A more thorough understanding of the mechanisms that underlie respiratory weakness in Pompe disease will enable optimization of future therapeutic strategies.
Fig. 4.
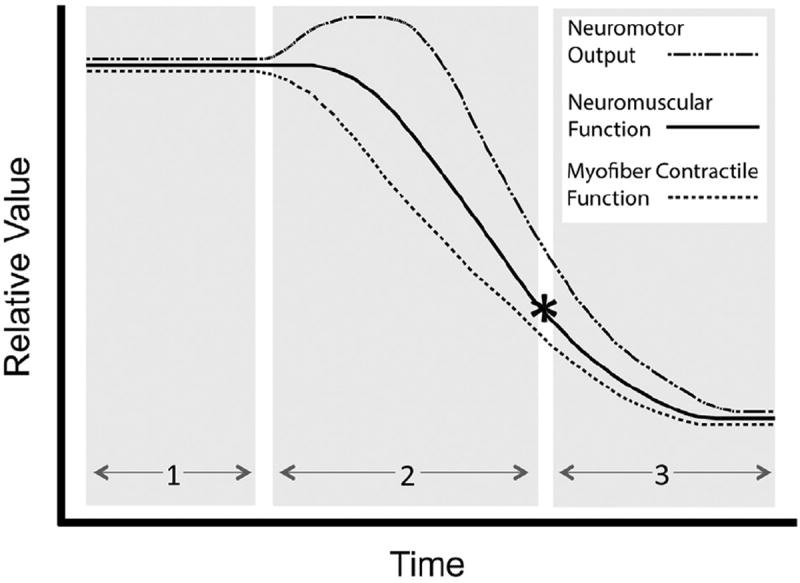
A conceptual model of the mechanisms leading to respiratory failure in Pompe disease. In this model, we hypothesize three critical periods in the development of respiratory failure in Pompe disease. Importantly, the model recognizes the contribution of both skeletal muscle contractile dysfunction and neuropathology to the development of respiratory insufficiency. Critical period 1: Glycogen accumulation begins in respiratory motor units (e.g., myofibers, motoneuron soma, axons, etc.). Pathological processes are beginning during this phase, but no symptoms are apparent. Critical period 2: Myofiber contractile dysfunction begins to develop. Impaired respiratory muscle function is initially met with compensatory increases in respiratory drive. Critical period 3: Neural and muscular pathology reach a critical threshold, and neural compensation for respiratory muscle dysfunction is no longer possible. The resulting respiratory failure (*) necessitates mechanical ventilation. Therapies which improve muscle function (e.g., ERT, gene therapy) or neural function (e.g., gene therapy) will prevent or minimize the declines in neuromotor function, and delay the onset of respiratory failure. Please see the text for a more detailed discussed of the hypothesized critical periods.
Acknowledgments
We thank Dr. Reordan O DeJesus for comments on Fig. 1, Dr. William H. Donnelly Jr. for processing the tissues shown in Fig. 2, Dr. Michael A. Lane for discussion of Figs. 2 and 3, and Dr. Elisa Gonzalez-Rothi for assistance with Fig. 4. This work was supported by the Parker B. Francis Foundation (MKE) and the NIH: 201HD052682-06A1 (DDF, BJB), MDA 216676 (DJF), K12HD055929 (BKS), and PO1 HL59412 (BJB)
Footnotes
This paper is part of a special issue entitled “Clinical Challenges to Ventilatory Control”, guest-edited by Dr. Gordon Mitchell, Dr. Jan-Marino Ramirez, Dr. Tracy Baker-Herman and Dr. Dr. David Paydarfar.
References
- Amalfitano A, Bengur AR, Morse RP, Majure JM, Case LE, Veerling DL, Mackey J, Kishnani P, Smith W, McVie-Wylie A, Sullivan JA, Hoganson GE, Phillips JA, 3rd, Schaefer GB, Charrow J, Ware RE, Bossen EH, Chen YT. Recombinant human acid alpha-glucosidase enzyme therapy for infantile glycogen storage disease type II: results of a phase I/II clinical trial. Genetics in Medicine: Official Journal of the American College of Medical Genetics. 2001;3:132–138. doi: 10.109700125817-200103000-00007. [DOI] [PubMed] [Google Scholar]
- Bailey EF, Fregosi RF. Coordination of intrinsic and extrinsic tongue muscles during spontaneous breathing in the rat. Journal of Applied Physiology. 2004;96:440–449. doi: 10.1152/japplphysiol.00733.2003. [DOI] [PubMed] [Google Scholar]
- Baudhuin P, Hers HG, Loeb H. An electron microscopic and biochemical study of type Ii glycogenosis. Laboratory Investigation. 1964;13:1139–1152. [PubMed] [Google Scholar]
- Beck M. Alglucosidase alfa: Long term use in the treatment of patients with Pompe disease. Therapeutics and Clinical Risk Management. 2009;5:767–772. doi: 10.2147/tcrm.s5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck M. Therapy for lysosomal storage disorders. IUBMB Life. 2010;62:33–40. doi: 10.1002/iub.284. [DOI] [PubMed] [Google Scholar]
- Bellettato CM, Scarpa M. Pathophysiology of neuropathic lysosomal storage disorders. Journal of Inherited Metabolic Disease. 2010;33:347–362. doi: 10.1007/s10545-010-9075-9. [DOI] [PubMed] [Google Scholar]
- Bijvoet AG, Van Hirtum H, Kroos MA, Van de Kamp EH, Schoneveld O, Visser P, Brakenhoff JP, Weggeman M, van Corven EJ, Van der Ploeg AT, Reuser AJ. Human acid alpha-glucosidase from rabbit milk has therapeutic effect in mice with glycogen storage disease type II. Human Molecular Genetics. 1999;8:2145–2153. doi: 10.1093/hmg/8.12.2145. [DOI] [PubMed] [Google Scholar]
- Burghaus L, Liu W, Neuen-Jacob E, Gempel K, Haupt WF. Glycogenesis type II (M. Pompe). Selective failure of the respiratory musculature – a rare first symptom. Der Nervenarzt. 2006;77:181–182. 185–186. doi: 10.1007/s00115-005-2005-7. [DOI] [PubMed] [Google Scholar]
- Burrow TA, Bailey LA, Kinnett DG, Hopkin RJ. Acute progression of neuromuscular findings in infantile Pompe disease. Pediatric Neurology. 2010;42:455–458. doi: 10.1016/j.pediatrneurol.2010.02.006. [DOI] [PubMed] [Google Scholar]
- Byrne BJ, Falk DJ, Pacak CA, Nayak S, Herzog RW, Elder ME, Collins SW, Conlon TJ, Clement N, Cleaver BD, Cloutier DA, Porvasnik SL, Islam S, Elmallah MK, Martin A, Smith BK, Fuller DD, Lawson LA, Mah CS. Pompe disease gene therapy. Human Molecular Genetics. 2011a;20:R61–R68. doi: 10.1093/hmg/ddr174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne BJ, Kishnani PS, Case LE, Merlini L, Muller-Felber W, Prasad S, van der Ploeg A. Pompe disease: design, methodology, and early findings from the Pompe registry. Molecular Genetics and Metabolism. 2011b;103:1–11. doi: 10.1016/j.ymgme.2011.02.004. [DOI] [PubMed] [Google Scholar]
- Calder PC, Geddes R. Regulation of lysosomal glycogen metabolism: studies of the actions of mammalian acid alpha-glucosidases. International Journal of Biochemistry. 1989;21:569–576. doi: 10.1016/0020-711x(89)90139-0. [DOI] [PubMed] [Google Scholar]
- Carlier RY, Laforet P, Wary C, Mompoint D, Laloui K, Pellegrini N, Annane D, Carlier PG, Orlikowski D. Whole-body muscle MRI in 20 patients suffering from late onset Pompe disease: Involvement patterns. Neuromuscular Disorders: NMD. 2011;21:791–799. doi: 10.1016/j.nmd.2011.06.748. [DOI] [PubMed] [Google Scholar]
- Clement DH, Godman GC. Glycogen disease resembling mongolism, cretinism, and amytonia congenita; case report and review of literature. Journal of Pediatrics. 1950;36:11–30. doi: 10.1016/s0022-3476(50)80174-9. illust. [DOI] [PubMed] [Google Scholar]
- Cori GT. Enzymes and glycogen structure in glycogenosis. Osterreichische Zeitschrift fur Kinderheilkunde und Kinderfursorge. 1954;10:38–42. [PubMed] [Google Scholar]
- de Barsy T, Jacquemin P, Van Hoof F, Hers HG. Enzyme replacement in Pompe disease: an attempt with purified human acid alpha-glucosidase. Birth Defects Original Article Series. 1973;9:184–190. [PubMed] [Google Scholar]
- DeRuisseau LR, Fuller DD, Qiu K, DeRuisseau KC, Donnelly WH, Jr, Mah C, Reier PJ, Byrne BJ. Neural deficits contribute to respiratory insufficiency in Pompe disease. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:9419–9424. doi: 10.1073/pnas.0902534106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubrovsky A, Corderi J, Lin M, Kishnani PS, Jones HN. Expanding the phenotype of late-onset Pompe disease: tongue weakness: a new clinical observation. Muscle & Nerve. 2011;44:897–901. doi: 10.1002/mus.22202. [DOI] [PubMed] [Google Scholar]
- Elmallah MK, Falk D, Lane MA, Lee KZ, Conlon TJ, Shafi NI, Reier PJ, Byrne B, Fuller DD. Retrograde gene delivery to hypoglossal motoneurons using AAV9. Human Gene Therapy Methods. 2012;23:148–156. doi: 10.1089/hgtb.2012.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman JL, Del Negro CA. Looking for inspiration: new perspectives on respiratory rhythm. Nature Reviews Neuroscience. 2006;7:232–242. doi: 10.1038/nrn1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman JL, Mitchell GS, Nattie EE. Breathing: rhythmicity, plasticity, chemosensitivity. Annual Review of Neuroscience. 2003;26:239–266. doi: 10.1146/annurev.neuro.26.041002.131103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fregosi RF, Fuller DD. Respiratory-related control of extrinsic tongue muscle activity. Respiration Physiology. 1997;110:295–306. doi: 10.1016/s0034-5687(97)00095-9. [DOI] [PubMed] [Google Scholar]
- Fuller DD, Williams JS, Janssen PL, Fregosi RF. Effect of co-activation of tongue protrudor and retractor muscles on tongue movements and pharyngeal airflow mechanics in the rat. Journal of Physiology. 1999;519(Pt 2):601–613. doi: 10.1111/j.1469-7793.1999.0601m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambetti P, DiMauro S, Baker L. Nervous system in Pompe’s disease. Ultrastructure and biochemistry. Journal of Neuropathology and Experimental Neurology. 1971;30:412–430. doi: 10.1097/00005072-197107000-00008. [DOI] [PubMed] [Google Scholar]
- Geddes R, Stratton GC. The influence of lysosomes on glycogen metabolism. Biochemical Journal. 1977;163:193–200. doi: 10.1042/bj1630193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gestreau C, Dutschmann M, Obled S, Bianchi AL. Activation of XII motoneurons and premotor neurons during various oropharyngeal behaviors. Respiratory Physiology & Neurobiology. 2005;147:159–176. doi: 10.1016/j.resp.2005.03.015. [DOI] [PubMed] [Google Scholar]
- Griffin JL. Infantile acid maltase deficiency. III. Ultrastructure of metachromatic material and glycogen in muscle fibers. Virchows Archiv B Cell Pathology Including Molecular Pathology. 1984;45:51–61. [PubMed] [Google Scholar]
- Hagemans ML, Winkel LP, Hop WC, Reuser AJ, Van Doorn PA, Van der Ploeg AT. Disease severity in children and adults with Pompe disease related to age and disease duration. Neurology. 2005;64:2139–2141. doi: 10.1212/01.WNL.0000165979.46537.56. [DOI] [PubMed] [Google Scholar]
- Haley SM, Fragala MA, Skrinar AM. Pompe disease and physical disability. Developmental Medicine and Child Neurology. 2003;45:618–623. doi: 10.1017/s0012162203001129. [DOI] [PubMed] [Google Scholar]
- Hers HG. Alpha-Glucosidase deficiency in generalized glycogenstorage disease (Pompe’s disease) Biochemical Journal. 1963;86:11–16. doi: 10.1042/bj0860011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschhorn R, Huie ML. Frequency of mutations for glycogen storage disease type II in different populations: the delta525T and deltaexon 18 mutations are not generally common in white populations. Journal of Medical Genetics. 1999;36:85–86. [PMC free article] [PubMed] [Google Scholar]
- Hirschhorn RRAJ. Glycogen storage disease type II: acid alpha-glucosidase (acid Maltase) deficiency. In: Scriver CRALB, Sly WS, Valle D, editors. Metabolic Basis of Inherited Disease. McGraw Hill; New York: 2001. [Google Scholar]
- Hogan GR, Gutmann L, Schmidt R, Gilbert E. Pompe’s disease. Neurology. 1969;19:894–900. doi: 10.1212/wnl.19.9.894. [DOI] [PubMed] [Google Scholar]
- Horner RL. Pathophysiology of obstructive sleep apnea. Journal of Cardiopulmonary Rehabilitation and Prevention. 2008;28:289–298. doi: 10.1097/01.HCR.0000336138.71569.a2. [DOI] [PubMed] [Google Scholar]
- Hudgson P, Fulthorpe JJ. The pathology of type II skeletal muscle glycogenosis. A light and electron-microscopic study. The Journal of pathology. 1975;116:139–147. doi: 10.1002/path.1711160303. [DOI] [PubMed] [Google Scholar]
- Isaacs H, Savage N, Badenhorst M, Whistler T. Acid maltase deficiency: a case study and review of the pathophysiological changes and proposed therapeutic measures. Journal of Neurology, Neurosurgery & Psychiatry. 1986;49:1011–1018. doi: 10.1136/jnnp.49.9.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones HN, Moss T, Edwards L, Kishnani PS. Increased inspiratory and expiratory muscle strength following respiratory muscle strength training (RMST) in two patients with late-onset Pompe disease. Molecular Genetics and Metabolism. 2011;104:417–420. doi: 10.1016/j.ymgme.2011.05.006. [DOI] [PubMed] [Google Scholar]
- Jones HN, Muller CW, Lin M, Banugaria SG, Case LE, Li JS, O’Grady G, Heller JH, Kishnani PS. Oropharyngeal dysphagia in infants and children with infantile Pompe disease. Dysphagia. 2010;25:277–283. doi: 10.1007/s00455-009-9252-x. [DOI] [PubMed] [Google Scholar]
- Kikuchi T, Yang HW, Pennybacker M, Ichihara N, Mizutani M, Van Hove JL, Chen YT. Clinical and metabolic correction of Pompe disease by enzyme therapy in acid maltase-deficient quail. The Journal of Clinical Investigation. 1998;101:827–833. doi: 10.1172/JCI1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishnani PS, Nicolino M, Voit T, Rogers RC, Tsai AC, Waterson J, Herman GE, Amalfitano A, Thurberg BL, Richards S, Davison M, Corzo D, Chen YT. Chinese hamster ovary cell-derived recombinant human acid alpha-glucosidase in infantile-onset Pompe disease. The Journal of Pediatrics. 2006;149:89–97. doi: 10.1016/j.jpeds.2006.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klumperman J, Fransen JA, Boekestijn TC, Oude Elferink RP, Matter K, Hauri HP, Tager JM, Ginsel LA. Biosynthesis and transport of lysosomal alpha-glucosidase in the human colon carcinoma cell line Caco-2: secretion from the apical surface. Journal of Cell Science. 1991;100(Pt 2):339–347. doi: 10.1242/jcs.100.2.339. [DOI] [PubMed] [Google Scholar]
- Kroos M, Hoogeveen-Westerveld M, Michelakakis H, Pomponio R, Van der Ploeg A, Halley D, Reuser A, Consortium GAAD. Update of the pompe disease mutation database with 60 novel GAA sequence variants and additional studies on the functional effect of 34 previously reported variants. Human Mutation. 2012a;33:1161–1165. doi: 10.1002/humu.22108. [DOI] [PubMed] [Google Scholar]
- Kroos M, Hoogeveen-Westerveld M, van der Ploeg A, Reuser AJ. The genotype-phenotype correlation in Pompe disease. American journal of medical genetics. Part C. Seminars in Medical Genetics. 2012b;160:59–68. doi: 10.1002/ajmg.c.31318. [DOI] [PubMed] [Google Scholar]
- Lee KZ, Qiu K, Sandhu MS, Elmallah MK, Falk DJ, Lane MA, Reier PJ, Byrne BJ, Fuller DD. Hypoglossal neuropathology and respiratory activity in Pompe mice. Frontiers in Physiology. 2011;2:31. doi: 10.3389/fphys.2011.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Liu H, Yang Y, Huang Y, Liu S, Beck J, Slutsky AS, Sinderby C, Qiu H. Neuroventilatory efficiency and extubation readiness in critically ill patients. Critical Care. 2012;16:R143. doi: 10.1186/cc11451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maga JA, Zhou J, Kambampati R, Peng S, Wang X, Bohnsack RN, Thomm A, Golata S, Tom P, Dahms NM, Byrne BJ, LeBowitz JH. Glycosylation-independent lysosomal targeting of acid alpha-glucosidase enhances muscle glycogen clearance in pompe mice. The Journal of Biological Chemistry. 2013;288:1428–1438. doi: 10.1074/jbc.M112.438663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mah C, Pacak CA, Cresawn KO, Deruisseau LR, Germain S, Lewis MA, Cloutier DA, Fuller DD, Byrne BJ. Physiological correction of Pompe disease by systemic delivery of adeno-associated virus serotype 1 vectors. Molecular Therapy: The Journal of the American Society of Gene Therapy. 2007;15:501–507. doi: 10.1038/sj.mt.6300100. [DOI] [PubMed] [Google Scholar]
- Mah CS, Falk DJ, Germain SA, Kelley JS, Lewis MA, Cloutier DA, DeRuisseau LR, Conlon TJ, Cresawn KO, Fraites TJ, Jr, Campbell-Thompson M, Fuller DD, Byrne BJ. Gel-mediated delivery of AAV1 vectors corrects ventilatory function in Pompe mice with established disease. Molecular Therapy. 2010;18:502–510. doi: 10.1038/mt.2009.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancall EL, Aponte GE, Berry RG. Pompe’s disease (diffuse glycogenosis) with neuronal storage. Journal of Neuropathology & Experimental Neurology. 1965;24:85–96. doi: 10.1097/00005072-196501000-00008. [DOI] [PubMed] [Google Scholar]
- Mannarino MR, Di Filippo F, Pirro M. Obstructive sleep apnea syndrome. European Journal of Internal Medicine. 2012;23:586–593. doi: 10.1016/j.ejim.2012.05.013. [DOI] [PubMed] [Google Scholar]
- Margolis ML, Howlett P, Goldberg R, Eftychiadis A, Levine S. Obstructive sleep apnea syndrome in acid maltase deficiency. Chest. 1994;105:947–949. doi: 10.1378/chest.105.3.947. [DOI] [PubMed] [Google Scholar]
- Marsden D. Infantile onset Pompe disease: a report of physician narratives from an epidemiologic study. Genetics in Medicine: Official Journal of the American College of Medical Genetics. 2005;7:147–150. doi: 10.1097/01.gim.0000154301.76619.5c. [DOI] [PubMed] [Google Scholar]
- Martin JJ, de Barsy T, van Hoof F, Palladini G. Pompe’s disease: an inborn lysosomal disorder with storage of glycogen. A study of brain and striated muscle. Acta Neuropathologica. 1973;23:229–244. doi: 10.1007/BF00687878. [DOI] [PubMed] [Google Scholar]
- Martini C, Ciana G, Benettoni A, Katouzian F, Severini GM, Bussani R, Bembi B. Intractable fever and cortical neuronal glycogen storage in glycogenosis type 2. Neurology. 2001;57:906–908. doi: 10.1212/wnl.57.5.906. [DOI] [PubMed] [Google Scholar]
- Matsui T, Kuroda S, Mizutani M, Kiuchi Y, Suzuki K, Ono T. Generalized glycogen storage disease in Japanese quail (Coturnix coturnix japonica) Veterinary Pathology. 1983;20:312–321. doi: 10.1177/030098588302000307. [DOI] [PubMed] [Google Scholar]
- Mellies U, Lofaso F. Pompe disease: a neuromuscular disease with respiratory muscle involvement. Respiratory Medicine. 2009;103:477–484. doi: 10.1016/j.rmed.2008.12.009. [DOI] [PubMed] [Google Scholar]
- Mellies U, Ragette R, Schwake C, Baethmann M, Voit T, Teschler H. Sleep-disordered breathing and respiratory failure in acid maltase deficiency. Neurology. 2001;57:1290–1295. doi: 10.1212/wnl.57.7.1290. [DOI] [PubMed] [Google Scholar]
- Mellies U, Stehling F, Dohna-Schwake C, Ragette R, Teschler H, Voit T. Respiratory failure in Pompe disease: treatment with noninvasive ventilation. Neurology. 2005;64:1465–1467. doi: 10.1212/01.WNL.0000158682.85052.C0. [DOI] [PubMed] [Google Scholar]
- Milic-Emili J, Whitelaw WA, Derenne JP. New tests to assess lung function: occlusion pressure–a simple measure of the respiratory center’s output. New England Journal of Medicine. 1975;293:1029–1030. doi: 10.1056/NEJM197511132932006. [DOI] [PubMed] [Google Scholar]
- Muller CW, Jones HN, Grady O, Suarez G, Heller AH, Kishnani JHPS. Language and speech function in children with infantile Pompe disease. Journal of Pediatric Neurology. 2009;7:147–156. [Google Scholar]
- Musumeci O, Catalano N, Barca E, Ravaglia S, Fiumara A, Gangemi G, Rodolico C, Sorge G, Vita G, Galletti F, Toscano A. Auditory system involvement in late onset Pompe disease: a study of 20 Italian patients. Molecular Genetics and Metabolism. 2012;107:480–484. doi: 10.1016/j.ymgme.2012.07.024. [DOI] [PubMed] [Google Scholar]
- Nichols NL, Gowing G, Satriotomo I, Nashold LJ, Dale EA, Suzuki M, Avalos P, Mulcrone PL, McHugh J, Svendsen CN, Mitchell GS. Intermittent hypoxia and stem cell implants preserve breathing capacity in a rodent model of amyotrophic lateral sclerosis. American Journal of Respiratory and Critical Care Medicine. 2013;187:535–542. doi: 10.1164/rccm.201206-1072OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlikowski D, Pellegrini N, Prigent H, Laforet P, Carlier R, Carlier P, Eymard B, Lofaso F, Annane D. Recombinant human acid alpha-glucosidase (rhGAA) in adult patients with severe respiratory failure due to Pompe disease. Neuromuscular Disorders: NMD. 2011;21:477–482. doi: 10.1016/j.nmd.2011.04.001. [DOI] [PubMed] [Google Scholar]
- Pellegrini N, Laforet P, Orlikowski D, Pellegrini M, Caillaud C, Eymard B, Raphael JC, Lofaso F. Respiratory insufficiency and limb muscle weakness in adults with Pompe’s disease. European Respiratory Journal. 2005;26:1024–1031. doi: 10.1183/09031936.05.00020005. [DOI] [PubMed] [Google Scholar]
- Prigent H, Orlikowski D, Laforet P, Letilly N, Falaize L, Pellegrini N, Annane D, Raphael JC, Lofaso F. Supine volume drop and diaphragmatic function in adults with Pompe disease. The European Respiratory Journal: Official Journal of the European Society for Clinical Respiratory Physiology. 2012;39:1545–1546. doi: 10.1183/09031936.00169011. [DOI] [PubMed] [Google Scholar]
- Qiu K, Falk DJ, Reier PJ, Byrne BJ, Fuller DD. Spinal delivery of AAV5 vector restores enzyme activity and increases ventilation in Pompe mice. Molecular Therapy. 2012;20:21–27. doi: 10.1038/mt.2011.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raben N, Danon M, Gilbert AL, Dwivedi S, Collins B, Thurberg BL, Mattaliano RJ, Nagaraju K, Plotz PH. Enzyme replacement therapy in the mouse model of Pompe disease. Molecular Genetics and Metabolism. 2003;80:159–169. doi: 10.1016/j.ymgme.2003.08.022. [DOI] [PubMed] [Google Scholar]
- Raben N, Lu N, Nagaraju K, Rivera Y, Lee A, Yan B, Byrne B, Meikle PJ, Umapathysivam K, Hopwood JJ, Plotz PH. Conditional tissue-specific expression of the acid alpha-glucosidase (GAA) gene in the GAA knockout mice: implications for therapy. Human Molecular Genetics. 2001;10:2039–2047. doi: 10.1093/hmg/10.19.2039. [DOI] [PubMed] [Google Scholar]
- Raben N, Nagaraju K, Lee E, Kessler P, Byrne B, Lee L, LaMarca M, King C, Ward J, Sauer B, Plotz P. Targeted disruption of the acid alpha-glucosidase gene in mice causes an illness with critical features of both infantile and adult human glycogen storage disease type II. The Journal of Biological Chemistry. 1998;273:19086–19092. doi: 10.1074/jbc.273.30.19086. [DOI] [PubMed] [Google Scholar]
- Raben N, Plotz P, Byrne BJ. Acid alpha-glucosidase deficiency (glycogenosis type II, Pompe disease) Current Molecular Medicine. 2002;2:145–166. doi: 10.2174/1566524024605789. [DOI] [PubMed] [Google Scholar]
- Remmers JE, deGroot WJ, Sauerland EK, Anch AM. Pathogenesis of upper airway occlusion during sleep. Journal of Applied Physiology. 1978;44:931–938. doi: 10.1152/jappl.1978.44.6.931. [DOI] [PubMed] [Google Scholar]
- Rohrbach M, Klein A, Kohli-Wiesner A, Veraguth D, Scheer I, Balmer C, Lauener R, Baumgartner MR. CRIM-negative infantile Pompe disease: 42-month treatment outcome. Journal of Inherited Metabolic Disease. 2010;33:751–757. doi: 10.1007/s10545-010-9209-0. [DOI] [PubMed] [Google Scholar]
- Schneider I, Hanisch F, Muller T, Schmidt B, Zierz S. Respiratory function in late-onset Pompe disease patients receiving long-term enzyme replacement therapy for more than 48 months. Wiener medizinische Wochenschrift. 2013;163:40–44. doi: 10.1007/s10354-012-0153-5. [DOI] [PubMed] [Google Scholar]
- Sidman RL, Taksir T, Fidler J, Zhao M, Dodge JC, Passini MA, Raben N, Thurberg BL, Cheng SH, Shihabuddin LS. Temporal neuropathologic and behavioral phenotype of 6neo/6neo Pompe disease mice. Journal of Neuropathology and Experimental Neurology. 2008;67:803–818. doi: 10.1097/NEN.0b013e3181815994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith BK, Collins S, Conlon T, Mah C, Lawson LA, Martin D, Fuller DD, Cleaver B, Clement N, Phillips D, Islam S, Dobjia N, Byrne B. Phase I/II trial of AAV1–GAA gene therapy to the diaphragm for chronic respiratory failure in Pompe disease: initial safety and ventilatory outcomes. Human Gene Therapy. 2013;24:630–640. doi: 10.1089/hum.2012.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng YT, Su WJ, Hou JW, Huang SF. Infantile-onset glycogen storage disease type II (Pompe disease): report of a case with genetic diagnosis and pathological findings. Chang Gung Medical Journal. 2004;27:379–384. [PubMed] [Google Scholar]
- Thurberg BL, Lynch Maloney C, Vaccaro C, Afonso K, Tsai AC, Bossen E, Kishnani PS, O’Callaghan M. Characterization of pre- and post-treatment pathology after enzyme replacement therapy for Pompe disease. Laboratory Investigations. 2006;86:1208–1220. doi: 10.1038/labinvest.3700484. [DOI] [PubMed] [Google Scholar]
- Toscano A, Schoser B. Enzyme replacement therapy in late-onset Pompe disease: a systematic literature review. Journal of Neurology. 2013;260:951–959. doi: 10.1007/s00415-012-6636-x. [DOI] [PubMed] [Google Scholar]
- Tsuji A, Suzuki Y. The precursor of acid alpha-glucosidase is synthesized as a membrane-bound enzyme. Biochemistry International. 1987;15:945–952. [PubMed] [Google Scholar]
- van Capelle CI, Goedegebure A, Homans NC, Hoeve HL, Reuser AJ, van der Ploeg AT. Hearing loss in Pompe disease revisited: results from a study of 24 children. Journal of Inherited Metabolic Disease. 2010;33:597–602. doi: 10.1007/s10545-010-9144-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Hout H, Reuser AJ, Vulto AG, Loonen MC, Cromme-Dijkhuis A, Van der Ploeg AT. Recombinant human alpha-glucosidase from rabbit milk in Pompe patients. Lancet. 2000;356:397–398. doi: 10.1016/s0140-6736(00)02533-2. [DOI] [PubMed] [Google Scholar]
- van den Hout HM, Hop W, van Diggelen OP, Smeitink JA, Smit GP, Poll-The BT, Bakker HD, Loonen MC, de Klerk JB, Reuser AJ, van der Ploeg AT. The natural course of infantile Pompe’s disease: 20 original cases compared with 133 cases from the literature. Pediatrics. 2003;112:332–340. doi: 10.1542/peds.112.2.332. [DOI] [PubMed] [Google Scholar]
- Van den Hout JM, Kamphoven JH, Winkel LP, Arts WF, De Klerk JB, Loonen MC, Vulto AG, Cromme-Dijkhuis A, Weisglas-Kuperus N, Hop W, Van Hirtum H, Van Diggelen OP, Boer M, Kroos MA, Van Doorn PA, Van der Voort E, Sibbles B, Van Corven EJ, Brakenhoff JP, Van Hove J, Smeitink JA, de Jong G, Reuser AJ, Van der Ploeg AT. Long-term intravenous treatment of Pompe disease with recombinant human alpha-glucosidase from milk. Pediatrics. 2004;113:e448–e457. doi: 10.1542/peds.113.5.e448. [DOI] [PubMed] [Google Scholar]
- van der Ploeg AT, Clemens PR, Corzo D, Escolar DM, Florence J, Groeneveld GJ, Herson S, Kishnani PS, Laforet P, Lake SL, Lange DJ, Leshner RT, Mayhew JE, Morgan C, Nozaki K, Park DJ, Pestronk A, Rosenbloom B, Skrinar A, van Capelle CI, van der Beek NA, Wasserstein M, Zivkovic SA. A randomized study of alglucosidase alfa in late-onset Pompe’s disease. New England Journal of Medicine. 2010;362:1396–1406. doi: 10.1056/NEJMoa0909859. [DOI] [PubMed] [Google Scholar]
- Willemsen MA, Jira PE, Gabreels FJ, van der Ploeg AT, Smeitink JA. Three hypotonic neonates with hypertrophic cardiomyopathy: Pompe’s disease. Ned Tijdschr Geneeskd. 1998;142:1388–1392. [PubMed] [Google Scholar]
- Zellweger H, Dark A, Abu-Haidar GA. Glycogen disease of skeletal muscle; report of two cases and review of literature. Pediatrics. 1955;5:715–732. [PubMed] [Google Scholar]