

Abstract
Importance
Because deposition of cerebral beta amyloid (Aβ) appears to be a key initiating event in Alzheimer’s disease, factors associated with increased deposition are of great interest. Whether or not elevated serum cholesterol acts as such a factor is unknown.
Objective
To investigate the relationship between serum cholesterol levels and cerebral Aβ during life, early in the AD process.
Design
Cross sectional analysis of potential associations between contemporaneously measured total serum cholesterol, HDL cholesterol, LDL cholesterol and cerebral Aβ, measured using PIB PET.
Setting
Multi-site, university medical center based study of vascular contributions to dementia.
Participants
74 persons, mean age 78, recruited via direct outreach in stroke clinics and community senior facilities following a protocol designed to obtain a cohort enriched for cerebrovascular disease and elevated vascular risk. Three cases had mild dementia. All others were clinically normal (33 cases) or had mild cognitive impairment (38 cases).
Results
Cerebral Aβ was quantified using a global PIB index, which averages PIB retention in cortical areas prone to amyloidosis. Statistical models that controlled for age and the apoE ε4 allele showed independent associations between LDL cholesterol, HDL cholesterol and PIB index. Higher LDL and lower HDL were both associated with higher PIB index. No association was found between total cholesterol and PIB index. No association was found between statin use and PIB index, nor did controlling for cholesterol treatment in the statistical models alter the basic findings.
Conclusions and Relevance
Elevated cerebral Aβ was associated with cholesterol fractions in a pattern analogous to that found in coronary artery disease. This finding, in living, non-demented humans, is consistent with prior autopsy reports, with epidemiological findings, and with both animal and in vitro work suggesting an important role for cholesterol in Aβ processing. Because cholesterol levels are modifiable, understanding their link to amyloid deposition could potentially and eventually have impact in retarding the pathological cascade of AD. These findings suggest that understanding the mechanisms through which serum lipids modulate Aβ could offer new approaches to slowing Aβ deposition and thus to reducing the incidence of AD.
Cholesterol, vital to neuronal structure and function, has been shown to play potentially important roles in the synthesis, deposition, and clearance of beta amyloid (Aβ) (1–3), thus suggesting that it may play a pathogenic role in AD. Interest in the potential role of cholesterol is heightened by a series of epidemiological reports generally demonstrating a correspondence between the atherogenic risk factors for cardiovascular disease and clinically diagnosed AD (4–6). In addition, observational studies have found that statin use is associated with a reduced odds of AD (7). There are also important connections between apolipoprotein E, Aβ (8) and cholesterol. A strong genetic risk factor for AD, the ε4 allele is associated with earlier and higher deposition of Aβ. ApoE is the primary transporter of cholesterol in brain and its isoforms differentially modulate brain cholesterol levels (8). Thus, the potential role of peripheral cholesterol in cerebral Aβ deposition is of considerable interest.
The epidemiological evidence on serum cholesterol and AD is, however, complex. Higher midlife total cholesterol has been associated with elevated risk of AD in late life (5, 6) and higher late-life HDL was associated with reduced risk of incident AD (9). Other studies have found no association (10) and some have reported higher total cholesterol levels (typically measured in late life) to be associated with reduced risk of AD (11, 12). In contrast to the observational studies on statin use and AD, randomized controlled treatment trials of statins in AD have been uniformly negative (13, 14).
It is also possible any association between cholesterol and AD reflect processes that have nothing to do with amyloid. For example, community-based autopsy studies have demonstrated an important role for cerebrovascular lesions in the mix of pathologies that generally underlie clinical AD (15). The association between lipids and AD may therefore alternatively reflect the role of dyslipidemia as a risk factor for cerebral ischemia. We investigated whether or not there is an in vivo association between serum lipids and cerebral Aβ deposition.
Methods
Participants were 74 elderly persons (mean age 78) from the Aging Brain study. Persons were recruited via stroke clinics, support groups, community senior facilities and a university memory clinic using criteria designed to obtain a cohort with cognitive function between normal and mildly impaired, and representing a wide-range of atherosclerotic disease and vascular risk. Inclusion criteria included age 60 or older with cognitive function in the normal to mild dementia range; recruitment emphasized non-demented persons over age 70. Exclusion criteria were severe or unstable medical illness, Axis I psychopathology other than depression, head injury with significant loss of consciousness and/or cognitive sequelae, sensory or physical limitations that would preclude cognitive testing, diagnosis of dementia due to causes other than Alzheimer’s disease or vascular disease or the combination thereof, use of medications likely to impair cognitive function (anti-dementia medications accepted), excessive alcohol use, history of drug or alcohol abuse within the past 5 years. Data from 43 of these participants were included in our prior report on cardiovascular risk and Aβ (16). Clinical diagnostic evaluations appropriate for memory disorders and dementia were done using uniform diagnostic criteria and clinical protocols. Three cases had mild dementia, 33 were cognitively normal and 38 had mild cognitive impairment. Participant characteristics are given in Table 1.
Table 1.
Participant characteristics
| N | 74 |
|---|---|
| Gender (Ns) | 52 male, 22 female |
| Age | Mean 77.8, SD 6.1, range 68–91 |
| Education | Mean 14.5, SD 2.8 |
| Clinical status (Ns) | 33 CDR 0; 38 CDR 0.5; 3 CDR 1 |
| apoE genotypes | 40 ε3/ε3; 14 ε2/ε3; 18 ε3/ε4; 1 ε2/ε4; 1 ε4/ε4 |
| Vascular disease (Ns) | 40 stroke/TIA; 15 MI; 21 CABG; 48 one or more of these |
| Anti-cholesterol drugs | 48 cases on statins; 4 on other agents |
While cholesterol levels were heterogeneous, the distributions appear to reflect the effects of treatment as mean levels for total and LDL cholesterol were within the current “optimal levels” established by the American Heart Association. The mean fasting total cholesterol for the group was 171mg/dL and mean levels for the LDL and HDL fractions were 92mg/dL and 54 mg/dL, respectively. See Figure 1.
Figure 1.
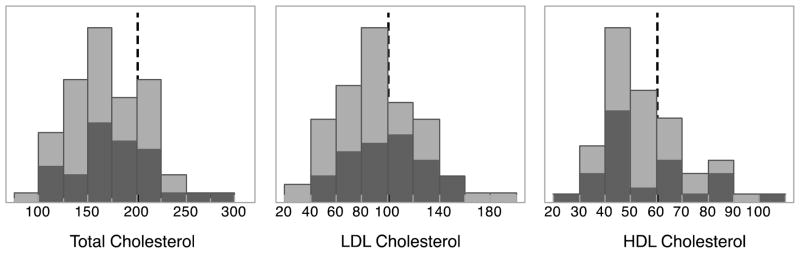
Distributions of cholesterol values. Light gray indicates total sample and dark gray indicates subset of cases that were PIB positive. Dashed vertical lines mark the target guideline values given by the American Heart Association.
PET imaging
Cerebral Aβ was measured with PET using the tracer 11C-labeled Pittsburgh Compound-B (PIB), which specifically binds fibrilar Aβ plaques. The PIB radiotracer was synthesized at Lawrence Berkeley National Laboratory using a previously published protocol(17) PIB-PET imaging was conducted using a Siemens ECAT HR scanner in 3-D acquisition mode. PIB (10–15 mCi) was injected as a bolus into an antecubital vein after which dynamic acquisition frames were obtained for a total of 90 minutes over progressively longer intervals (18).
Image analysis
PIB data were preprocessed using Statistical Parametric Mapping 8 (SPM8; www.fil.ion.ucl.ac.uk/spm/). Frames 6 through 35, as well as the sum of frames 1 to 5, were realigned to frame 17. Realigned frames reflecting the first 20 minutes of acquisition (frames 1–23) were then averaged and used to guide coregistration with the T1- weighted magnetic resonance image (MRI). Distribution volume ratio (DVR) images were generated from PIB frames corresponding to 35–90 minutes post-injection, and quantified using Logan graphical analysis and the participant’s gray matter cerebellar reference region.
DVR values were extracted from regions of interest (ROIs) vulnerable to early AB deposition, which include the frontal cortex (anterior to the precentral gyrus), lateral parietal cortex, lateral temporal cortex, posterior cingulate, and precuneus. ROIs were defined using the Desikan-Killiany atlas and the semi-automated FreeSurfer processing stream (19).
A global measure of PIB uptake (Global PIB Index) was generated in each subject native space by averaging the mean DVR value of these ROIs (20). The occipital cortex was also examined due to its susceptibility to cerebral amyloid angiopathy. This Global PIB Index served as the primary dependent variable. For purposes of describing the sample (but not for purposes of data analysis) PIB positivity was defined. Eleven young adults (mean age = 24.5, SD = 3.4) underwent PIB-PET imaging using the same acquisition and processing procedures described above. PIB uptake was determined using DVR values from the Global PIB index. Values 2 SDs above the young average for these 2 regions were established as defining values of PIB positivity. Therefore participants with a Global PIB Index ≥1.08 were determined to be PIB-positive (32 cases).
Other measures
Fasting HDL, LDL cholesterol, and triglycerides were assayed at a central laboratory under standardized research protocols from blood samples obtained at study entry. ApoE genotyping was done using TaqMan probes (21). Fluorescence was detected using an ABI 7900HT Sequence Detection System and the alleles are scored using Sequence Detector Software (ABI). For APOE 2 assays for SNPs in Exon 4 were run on each sample. These include a C/T SNP at amino acid 112 and a C/T SNP at amino acid 158. The APOE alleles (e2, e3, e4) were determined according to (22). Quantitative levels of apoA and apoB were measured.
Medications to treat cholesterol levels were recorded by a physician or nurse during a clinic visit and recorded using the National Alzheimer’s Coordinating Center Uniform Data Set (2.0) form A4.
Standard Protocol Approvals
All study activities were conducted under protocols approved by the institutional review boards at University of California Berkeley, University of California, Davis, University of California, San Francisco, and University of Southern California. All participants provided written informed consent.
Results
Associations between lipids and Aβ were tested in a series of multiple linear regression models that covaried age, sex, and apoE ε4 status. In this data set LDL is uncorrelated with HDL (r = 0.06) and triglycerides (r = 0.08); HDL and tricylcertides were negatively correlated (r = −0.24, p = 0.045). Initial models evaluated total cholesterol, HDL, LDL, and triglycerides as individual predictors of Aβ. In those separate models HDL was negatively associated (p = 0.022) and LDL was positively associated at a trend level (p =0.052) with PIB index, whereas total cholesterol, and triglycerides had no association with PIB index (p values = 0.56 and 0.71). Modeled jointly, and covarying age, sex and apoE ε4, lower HDL (p = 0.018) and higher LDL (p =0.033) were both associated with higher PIB index. These associations were independent of apoE ε4, which was positively associated (p = .002) with PIB index. See Figure 2. The increase in R2 gives a measure of effect size. Compared to age and sex alone, the addition of apoE ε4 to the model explained an additional 11% of variance in PIB index and adding HDL and LDL to the model explained an additional 11% beyond the effect of apoE ε4. Table 2 provides shows the parameter estimates for the joint HDL and LDL model. Models testing the effect of triglycerides in combination with HDL, LDL or both found no significant effect of triglycerides on PIB index.
Figure 2.
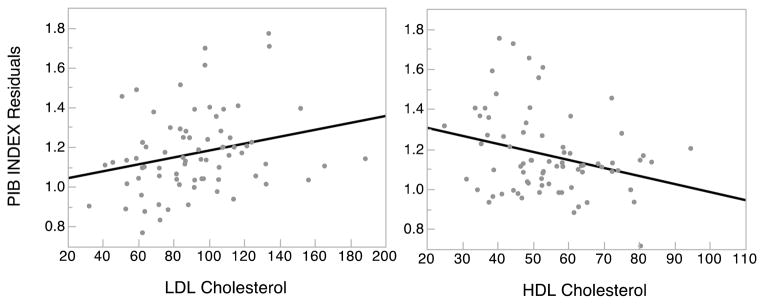
Independent effects of LDL and HDL cholesterol on cerebral Aβ. Individual data points and the slope of the regression of Aβ on LDL cholesterol (left) and HDL cholesterol (right). PIB index values are residual values from the model that evaluated HDL and LDL simultaneously while covarying age, sex and apoE ε4 status.
Table 2.
Parameter estimates and significance values for multiple regression of PIB index on sex, age, apoE ε4, HDL and LDL cholesterol.
| Variable | Estimate | Std Error | t ratio | p |
|---|---|---|---|---|
| SEX[1] | 0.021 | 0.028 | 0.75 | 0.4546 |
| age | 0.012 | 0.004 | 2.82 | 0.0062 |
| apoE ε4 status[0] | −0.088 | 0.027 | −3.23 | 0.0019 |
| HDL cholesterol | −0.004 | 0.002 | −2.43 | 0.0177 |
| LDL cholesterol | 0.002 | 0.001 | 2.17 | 0.0335 |
ApoA1 is moderately strongly correlated with HDL, and apoB is strongly correlated with LDL. When modeled jointly with the covariates age, sex and apoE ε4, lower apoA1 (p = 0.015) and higher apoB (p = 0.047) are both significant predictors of PIB index. However, neither has any effect on PIB index that is independent of HDL and LDL.
The role of current cholesterol treatment was investigated. We found little evidence of drug effects on PIB index in either simple or fully adjusted models regardless of how treatment indicator was used (e.g. any drug v. none, statins v. other v. none, or lipophilic statins v. hydrophilic v. none). No treatment indicator was significantly associated with PIB index on a bivariate basis (p values between 0.41 and 0.75) or in models controlling for age, sex, and apoE ε4. As expected statin treatment was associated with lower LDL levels. Adding statin treatment as a covariate to either the unadjusted (bivariate) or fully adjusted models described above did not reduce the parameter estimates for either HDL or LDL and the pattern of p values produced by the models remained the same.
Discussion
Higher fasting levels of LDL and lower levels of HDL cholesterol were both associated with greater brain amyloid independently of ApoE genotype. We believe this to be the first human experimental evidence of a direct relationship between cholesterol fractions in blood and amyloid deposition in brain. There are two prior reports linking elevated LDL to Aβ in AD patients at autopsy(23, 24) but to our knowledge the present report is the first to show a correlation in vivo, early in the disease process.
The brain is rich in cholesterol, accounting for nearly 25% of all cholesterol in the body (25). There is substantial in vitro and animal evidence that cholesterol levels in brain affect the synthesis, clearance, and toxicity of Aβ. The Aβ peptide results from the dual cleavage of APP, a transmembrane protein, by BACE1 in extracellular space and by γ secretase within the transmembrane domain. Thus, the characteristics of the cellular lipid bilayer may influence trafficking and proteolysis. Membrane bound cholesterol is concentrated in “lipid rafts”, microdomains which also carry APP, β and γ secretases, and where most of the metabolism of APP occurs (26). In vitro studies report that lower cholesterol levels shift APP processing to non-raft regions of the membrane where the benign α secretase cleavage pathway is favored (27). Additional in vitro work has demonstrated that modulation of local cholesterol levels modifies Aβ production. For example, adding cholesterol to cultured HEK 293 cells increased Aβ production four-fold, while adding a statin to these cultures markedly reduced Aβ (28). Similarly, reduction of cellular cholesterol inhibited the production of Aβ in hippocampal cells (29).
Consideration of possible mechanisms linking cholesterol fractions and Aβ must account for the fact that essentially all CNS cholesterol is locally synthesized and that there is minimal exchange of HDL particles and essentially no exchange of LDL/VLDL particles across the intact BBB (30). At the same time, animal work demonstrates a potential role for serum cholesterol in Aβ deposition. For example, in transgenic APP mice a high cholesterol diet was reversibly associated with greater Aβ plaque formation (31, 32). Similarly, feeding rabbits a high cholesterol diet doubled the number of Aβ plaques (33).
Oxysterols, which do efficiently pass the BBB, have gained considerable recent attention as a potential link between serum cholesterol, altered brain cholesterol metabolism and Aβ (2, 34). The oxysterol 24-hydroxycholesterol (24-OH, cerebrosterol), is produced almost entirely in brain where it plays an important role in cholesterol homeostasis (2). The brain also synthesizes a small amount of 27-hydroxycholesterol (27-OH). However, most 27-OH is produced by peripheral tissues and serum hypercholesterolemia is associated with increased brain levels of 27-OH(35). Thus, the balance between 27-OH and 24-OH may affect amyloidosis (36) perhaps through cholesterol metabolism, or perhaps by more directly affecting Aβ production (2).
In addition, Aβ degradation is less efficient in a high cholesterol environment. Cholesterol efflux from brain and microglia is achieved by apoE-dependent mechanisms involving HDL particles and it may be that HDL function associated with apolipoproteins or other factors affecting cholesterol efflux are important (37). Interestingly, it was recently reported that genetic variation in the ATP-binding cassette transporter (ABCA7) -- known to affect cellular cholesterol efflux-- modifies the risk of Alzheimer’s disease in African Americans (38).
Alternatively, systemic hyperlipidemia may damage the BBB via inflammatory and other mechanisms, with consequent leakage of serum cholesterol, inflammatory cytokines, and other amyloidogenic factors (39). Or, it may be that HDL and LDL are themselves unimportant but correlate with some other factor(s) that is the mechanistic driver. Cholesterol fractions may reflect unmeasured genetic factors or dietary patterns(40). For example, there is evidence that a high fat diet affects the conformational state of apoE (41), perhaps reducing its ability to clear Aβ.
These data do not convincingly exclude the possibility that statin treatment is associated with lowered Aβ production, which is an effect that has been observed in vitro (28, 42). A larger sample, and better treatment history would be needed to address this definitively.
In conclusion, in this relatively small, “high vascular risk” cohort, HDL and LDL cholesterol levels showed the same pattern of association with Aβ levels as they do with coronary artery disease. However, the mean lipid profile for this cohort falls within the range considered “desirable” from the standpoint of cardiac health. The regulation of cholesterol both in periphery and brain as well as the processing of APP are processes that change with age. Thus, a major limitation of our study is that the findings are cross sectional and obtained late in life. Cholesterol levels were measured at a single time point, contemporaneously with the amyloid measure. Our cohort is relatively small and the findings need replication. An important factor in any replication effort may be the level of vascular risk in the study cohort. Our cohort was enriched for vascular risk, while many existing AD clinical trials cohorts use selection criteria that minimize cerebrovascular disease. Investigation of the potential modulatory role of serum cholesterol on amyloidosis is of great importance because a substantial amount is known about manipulating lipid metabolism, and any factor that could reduce Aβ deposition could have major impact on the incidence of AD.
Acknowledgments
This work was supported by grants P01 AG12435 (Chui), AG034570 (Jagust) and P30 AG10129 (DeCarli). We thank the recruiters for the Aging Brain project, especially Nancy Gubbins and Gwendolyn Gates for their efforts to enroll participants.
Footnotes
Authorship
Dr. Reed: Contributed to the design and conceptualization of the study, analyzed the data, helped to interpret the data, and drafted the manuscript
Dr. Villenueve: contributed data analysis and revised the manuscript
Dr. Mack: contributed data analysis and revised the manuscript
Dr. DeCarli: Contributed to the design and conceptualization of the study, helped to interpret the data and revised the manuscript
Dr. Jagust: Contributed to the design and conceptualization of the study, helped to interpret the data and revised the manuscript
Dr. Chui: Contributed to the design and conceptualization of the study, helped to interpret the data and revised the manuscript
Dr. Reed had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Disclosures
Dr. Reed: Has been supported by the NIH.
Dr. Villenueve: receives support from the Canadian Institutes of Health
Dr. Mack: has been supported by the NIH
Dr. DeCarli: serves as editor of Alzheimer’s Disease and Associated Disorders. He has been supported by the NIH.
Dr. Jagust: has served as a consultant to Genentech, Synarc, F. Hoffman La Roche, Janssen Alzheimer Immunotherapy. His research has been supported by the NIH.
Dr. Chui: has received support from the NIH.
Contributor Information
Bruce Reed, Email: brreed@ucdavis.edu.
Sylvia Villeneuve, Email: villeneuve.sylvia@gmail.com.
Wendy Mack, Email: wmack@usc.edu.
Charles DeCarli, Email: charles.decarli@ucdmc.ucdavis.edu.
Helena C. Chui, Email: Helena.Chui@med.usc.edu.
William Jagust, Email: jagust@berkeley.edu.
References
- 1.Reitz C. Dyslipidemia and dementia: current epidemiology, genetic evidence, and mechanisms behind the associations. J Alzheimers Dis. 2012;30( Suppl 2):S127–45. doi: 10.3233/JAD-2011-110599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gamba P, Testa G, Sottero B, Gargiulo S, Poli G, Leonarduzzi G. The link between altered cholesterol metabolism and Alzheimer’s disease. Ann N Y Acad Sci. 2012 Jul;1259:54–64. doi: 10.1111/j.1749-6632.2012.06513.x. [DOI] [PubMed] [Google Scholar]
- 3.Di Paolo G, Kim T-W. Linking lipids to Alzheimer’s disease: cholesterol and beyond. Nat Rev Neurosci. 2011;12(5):284–96. doi: 10.1038/nrn3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luchsinger JA, Reitz C, Honig LS, Tang MX, Shea S, Mayeux R. Aggregation of vascular risk factors and risk of incident Alzheimer disease. Neurology. 2005 Aug 23;65(4):545–51. doi: 10.1212/01.wnl.0000172914.08967.dc. Epub 2005/08/24. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kivipelto M, Helkala EL, Laakso MP, Hanninen T, Hallikainen M, Alhainen K, et al. Apolipoprotein E epsilon4 allele, elevated midlife total cholesterol level, and high midlife systolic blood pressure are independent risk factors for late-life Alzheimer disease. Ann Intern Med. 2002 Aug 6;137(3):149–55. doi: 10.7326/0003-4819-137-3-200208060-00006. Epub 2002/08/06. eng. [DOI] [PubMed] [Google Scholar]
- 6.Whitmer RA, Sidney S, Selby J, Johnston SC, Yaffe K. Midlife cardiovascular risk factors and risk of dementia in late life. Neurology. 2005 Jan 25;64(2):277–81. doi: 10.1212/01.WNL.0000149519.47454.F2. Epub 2005/01/26. eng. [DOI] [PubMed] [Google Scholar]
- 7.Jick H, Zornberg GL, Jick SS, Seshadri S, Drachman DA. Statins and the risk of dementia. Lancet. 2000 Nov 11;356(9242):1627–31. doi: 10.1016/s0140-6736(00)03155-x. [DOI] [PubMed] [Google Scholar]
- 8.Holtzman DM, Herz J, Bu G. Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harbor perspectives in medicine. 2012 Mar;2(3):a006312. doi: 10.1101/cshperspect.a006312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reitz C, Tang MX, Schupf N, Manly JJ, Mayeux R, Luchsinger JA. Association of higher levels of high-density lipoprotein cholesterol in elderly individuals and lower risk of late-onset Alzheimer disease. Arch Neurol. 2010 Dec;67(12):1491–7. doi: 10.1001/archneurol.2010.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tan ZS, Seshadri S, Beiser A, Wilson PW, Kiel DP, Tocco M, et al. Plasma total cholesterol level as a risk factor for Alzheimer disease: the Framingham Study. Arch Intern Med. 2003 May 12;163(9):1053–7. doi: 10.1001/archinte.163.9.1053. [DOI] [PubMed] [Google Scholar]
- 11.Reitz C, Tang MX, Luchsinger J, Mayeux R. Relation of plasma lipids to Alzheimer disease and vascular dementia. Arch Neurol. 2004 May;61(5):705–14. doi: 10.1001/archneur.61.5.705. Epub 2004/05/19. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mielke MM, Zandi PP, Sjogren M, Gustafson D, Ostling S, Steen B, et al. High total cholesterol levels in late life associated with a reduced risk of dementia. Neurology. 2005 May 24;64(10):1689–95. doi: 10.1212/01.WNL.0000161870.78572.A5. Epub 2005/05/25. eng. [DOI] [PubMed] [Google Scholar]
- 13.Sano M, Bell KL, Galasko D, Galvin JE, Thomas RG, van Dyck CH, et al. A randomized, double-blind, placebo-controlled trial of simvastatin to treat Alzheimer disease. Neurology. 2011 Aug 9;77(6):556–63. doi: 10.1212/WNL.0b013e318228bf11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trompet S, van Vliet P, de Craen AJ, Jolles J, Buckley BM, Murphy MB, et al. Pravastatin and cognitive function in the elderly. Results of the PROSPER study. J Neurol. 2010 Jan;257(1):85–90. doi: 10.1007/s00415-009-5271-7. [DOI] [PubMed] [Google Scholar]
- 15.Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology. 2007 Dec 11;69(24):2197–204. doi: 10.1212/01.wnl.0000271090.28148.24. Epub 2007/06/15. eng. [DOI] [PubMed] [Google Scholar]
- 16.Reed BR, Marchant NL, Jagust WJ, DeCarli CC, Mack W, Chui HC. Coronary risk correlates with cerebral amyloid deposition. Neurobiol Aging. 2012 Sep;33(9):1979–87. doi: 10.1016/j.neurobiolaging.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mathis CA, Wang Y, Holt DP, Huang GF, Debnath ML, Klunk WE. Synthesis and evaluation of 11C-labeled 6-substituted 2-arylbenzothiazoles as amyloid imaging agents. J Med Chem. 2003 Jun 19;46(13):2740–54. doi: 10.1021/jm030026b. [DOI] [PubMed] [Google Scholar]
- 18.Marchant NL, Reed BR, DeCarli CS, Madison CM, Weiner MW, Chui HC, et al. Cerebrovascular disease, beta-amyloid, and cognition in aging. Neurobiol Aging. 2012 May;33(5):1006, e25–36. doi: 10.1016/j.neurobiolaging.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Desikan RS, Ségonne F, Fischl B, Quinn BT, Dickerson BC, Blacker D, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. NeuroImage. 2006;31(3):968– 80. doi: 10.1016/j.neuroimage.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 20.Mormino EC, Kluth JT, Madison CM, Rabinovici GD, Baker SL, Miller BL, et al. Episodic memory loss is related to hippocampal-mediated beta-amyloid deposition in elderly subjects. Brain. 2009 May;132(Pt 5):1310–23. doi: 10.1093/brain/awn320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee LG, Connell CR, Bloch W. Allelic discrimination by nick-translation PCR with fluorogenic probes. Nucleic Acids Res. 1993 Aug 11;21(16):3761–6. doi: 10.1093/nar/21.16.3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu CE, Devlin B, Galloway N, Loomis E, Schellenberg GD. ADLAPH: A molecular haplotyping method based on allele-discriminating long-range PCR. Genomics. 2004 Sep;84(3):600–12. doi: 10.1016/j.ygeno.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 23.Kuo YM, Emmerling MR, Bisgaier CL, Essenburg AD, Lampert HC, Drumm D, et al. Elevated low-density lipoprotein in Alzheimer’s disease correlates with brain abeta 1–42 levels. Biochem Biophys Res Commun. 1998 Nov 27;252(3):711–5. doi: 10.1006/bbrc.1998.9652. [DOI] [PubMed] [Google Scholar]
- 24.Lesser GT, Beeri MS, Schmeidler J, Purohit DP, Haroutunian V. Cholesterol and LDL relate to neuritic plaques and to APOE4 presence but not to neurofibrillary tangles. Curr Alzheimer Res. 2011 May;8(3):303–12. doi: 10.2174/156720511795563755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reiss AB, Siller KA, Rahman MM, Chan ES, Ghiso J, de Leon MJ. Cholesterol in neurologic disorders of the elderly: stroke and Alzheimer’s disease. Neurobiol Aging. 2004 Sep;25(8):977–89. doi: 10.1016/j.neurobiolaging.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 26.Cordy JM, Hooper NM, Turner AJ. The involvement of lipid rafts in Alzheimer’s disease. Molecular membrane biology. 2006 Jan-Feb;23(1):111–22. doi: 10.1080/09687860500496417. [DOI] [PubMed] [Google Scholar]
- 27.Ehehalt R, Keller P, Haass C, Thiele C, Simons K. Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. The Journal of cell biology. 2003 Jan 6;160(1):113–23. doi: 10.1083/jcb.200207113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frears ER, Stephens DJ, Walters CE, Davies H, Austen BM. The role of cholesterol in the biosynthesis of beta-amyloid. Neuroreport. 1999 Jun 3;10(8):1699–705. doi: 10.1097/00001756-199906030-00014. [DOI] [PubMed] [Google Scholar]
- 29.Simons M, Keller P, De Strooper B, Beyreuther K, Dotti CG, Simons K. Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc Natl Acad Sci U S A. 1998 May 26;95(11):6460–4. doi: 10.1073/pnas.95.11.6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bjorkhem I, Meaney S. Brain cholesterol: long secret life behind a barrier. Arteriosclerosis, thrombosis, and vascular biology. 2004 May;24(5):806–15. doi: 10.1161/01.ATV.0000120374.59826.1b. [DOI] [PubMed] [Google Scholar]
- 31.Refolo LM, Malester B, LaFrancois J, Bryant-Thomas T, Wang R, Tint GS, et al. Hypercholesterolemia accelerates the Alzheimer’s amyloid pathology in a transgenic mouse model. Neurobiol Dis. 2000 Aug;7(4):321–31. doi: 10.1006/nbdi.2000.0304. [DOI] [PubMed] [Google Scholar]
- 32.Shie FS, Jin LW, Cook DG, Leverenz JB, LeBoeuf RC. Diet-induced hypercholesterolemia enhances brain A beta accumulation in transgenic mice. Neuroreport. 2002 Mar 25;13(4):455–9. doi: 10.1097/00001756-200203250-00019. [DOI] [PubMed] [Google Scholar]
- 33.Sparks DL, Kuo YM, Roher A, Martin T, Lukas RJ. Alterations of Alzheimer’s disease in the cholesterol-fed rabbit, including vascular inflammation. Preliminary observations. Ann N Y Acad Sci. 2000 Apr;903:335–44. doi: 10.1111/j.1749-6632.2000.tb06384.x. [DOI] [PubMed] [Google Scholar]
- 34.Hughes TM, Rosano C, Evans RW, Kuller LH. Brain cholesterol metabolism, oxysterols, and dementia. J Alzheimers Dis. 2013;33(4):891–911. doi: 10.3233/JAD-2012-121585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heverin M, Meaney S, Lutjohann D, Diczfalusy U, Wahren J, Bjorkhem I. Crossing the barrier: net flux of 27-hydroxycholesterol into the human brain. Journal of lipid research. 2005 May;46(5):1047–52. doi: 10.1194/jlr.M500024-JLR200. [DOI] [PubMed] [Google Scholar]
- 36.Bjorkhem I, Cedazo-Minguez A, Leoni V, Meaney S. Oxysterols and neurodegenerative diseases. Molecular aspects of medicine. 2009 Jun;30(3):171–9. doi: 10.1016/j.mam.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 37.Lee CY, Tse W, Smith JD, Landreth GE. Apolipoprotein E promotes beta-amyloid trafficking and degradation by modulating microglial cholesterol levels. J Biol Chem. 2012 Jan 13;287(3):2032–44. doi: 10.1074/jbc.M111.295451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reitz C, Jun G, Naj A, Rajbhandary R, Vardarajan BN, Wang LS, et al. Variants in the ATP-binding cassette transporter (ABCA7), apolipoprotein E 4, and the risk of late-onset Alzheimer disease in African Americans. JAMA. 2013 Apr 10;309(14):1483–92. doi: 10.1001/jama.2013.2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Altman R, Rutledge JC. The vascular contribution to Alzheimer’s disease. Clin Sci (Lond) 2010 Nov;119(10):407–21. doi: 10.1042/CS20100094. Epub 2010/08/06. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Corsinovi L, Biasi F, Poli G, Leonarduzzi G, Isaia G. Dietary lipids and their oxidized products in Alzheimer’s disease. Molecular nutrition & food research. 2011 Sep;55( Suppl 2):S161–72. doi: 10.1002/mnfr.201100208. [DOI] [PubMed] [Google Scholar]
- 41.den Hartigh LJ, Altman R, Hutchinson R, Petrlova J, Budamagunta MS, Tetali SD, et al. Postprandial apoE isoform and conformational changes associated with VLDL lipolysis products modulate monocyte inflammation. PLoS One. 2012;7(11):e50513. doi: 10.1371/journal.pone.0050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buxbaum JD, Geoghagen NS, Friedhoff LT. Cholesterol depletion with physiological concentrations of a statin decreases the formation of the Alzheimer amyloid Abeta peptide. J Alzheimers Dis. 2001 Apr;3(2):221–9. doi: 10.3233/jad-2001-3207. [DOI] [PubMed] [Google Scholar]