

Abstract
Chromosome translocations involving antigen receptor loci are a genetic hallmark of non-Hodgkin’s lymphomas in humans. Most commonly, these translocations result in juxtaposition of the immunoglobulin heavy-chain (IgH) locus with one of several cellular proto-oncogenes, leading to deregulated oncogene expression. The V(D)J recombinase, which mediates physiologic rearrangements of antigen receptor genes, may play a mechanistic role in some lymphoma translocations, although evidence is indirect. A high incidence of B-lineage lymphomas has been observed in mice with severe combined immunodeficiency (SCID) and p53-null mutations. We show that these tumors are characteristic of the pro–B-cell stage of development and that they harbor recurrent translocations involving chromosomes 12 and 15. Fluorescence in situ hybridization (FISH) shows retention of IgH sequences on the derivative chromosome 12, implying that breakpoints involve the IgH locus. Pro–B-cell lymphomas were suppressed in SCID p53–/– mice by a Rag-2–null mutation, demonstrating that DNA breaks generated during V(D)J recombination are required for oncogenic transformation, and suggesting that t(12;15) arise during attempted IgH rearrangement in pro-B cells. These studies indicate that the oncogenic potential inherent in antigen receptor diversification is controlled in vivo by efficient rejoining of DNA ends generated during V(D)J recombination and an intact cellular response to DNA damage.
J. Clin. Invest. 103:1669–1675 (1999).
Introduction
Chromosome translocations involving antigen receptor genes represent a central genetic abnormality in the pathogenesis of lymphoid malignancies in humans, particularly the non-Hodgkin’s lymphomas (1–4). Transposition of a cellular proto-oncogene in proximity to the regulatory elements of antigen receptor loci leads to deregulation of oncogene expression, with subsequent effects on cell growth, differentiation, or apoptosis. The involvement of antigen receptor genes in lymphoma translocations parallels physiologic mechanisms of genome rearrangement in lymphocytes. V(D)J recombination joins immunoglobulin and T-cell receptor (TCR) variable-region gene segments to generate the vast diversity of antigen specificities characteristic of vertebrate immunity (reviewed in refs. 5, 6). Peripheral B cells may also undergo isotype switch recombination within the IgH locus, substituting production of IgG, IgE, or IgA for IgM and IgD (7). Circumstantial evidence suggests that in some cases translocations may be catalyzed by the V(D)J or switch recombinases, based on similarities between breakpoint sequences and physiologic V(D)J or switch joints (8–13). However, the structure of many breakpoints is not characteristic of a particular rearrangement process, and a thorough understanding of the cause of lymphoma translocations is lacking.
Much has been learned in the past few years regarding the mechanism of V(D)J recombination. DNA scission is catalyzed by the Rag-1 and Rag-2 proteins at recognition signals (RSS) bordering antigen receptor variable-region gene segments, creating blunt signal ends and sealed hairpins at coding ends (14–17). Mice and humans with induced null mutations of Rag-1 or Rag-2 are unable to initiate rearrangement of endogenous Ig or TCR loci and consequently lack mature B or T cells (18–20). Rejoining of coding ends requires the DNA-dependent protein kinase (DNA-PK) complex, including the catalytic subunit (DNA-PKcs), Ku70 and Ku86, whereas signal ends are resolved by a distinct pathway, also involving the Ku proteins (reviewed in refs. 6, 21). Severe combined immunodeficiency (SCID) mice, which have a mutation affecting the DNA-PKcs, are capable of initiating Rag-mediated DNA cleavage but are severely limited in their ability to efficiently rejoin coding ends. As a result, there is a near-complete arrest of T- and B-cell development at an early progenitor stage, with accumulation of unresolved DNA breaks at coding ends (22–26).
The p53 tumor suppressor gene is mutated in a wide variety of human cancers and plays an important role in the cellular response to DNA damage. DNA breaks lead to rapid upregulation of p53 expression, which leads to 2 major cellular effects: arrest of the cell cycle at the G1 phase and induction of apoptosis (27–29). Mice lacking p53 are susceptible to several types of malignancies; of these, up to two thirds are lymphomas, which are predominantly localized thymic tumors (30, 31). SCID mice are also susceptible to thymic lymphomas, which may occur in up to 15% of animals (23). Mice with combined SCID and p53-null mutations develop disseminated B-cell lymphomas with an incidence approaching 100%; these lymphomas occur at a younger age than do lymphoid tumors in either parental strain (31–33). In this report we show that pro–B-cell lymphomas in SCID p53–/– mice harbor recurrent translocations involving the telomere of chromosome 12, at or near the IgH locus, and that tumor development requires initiation of V(D)J recombination. This genetic pathway to recurrent lymphoma translocations indicates that mitigation of the oncogenic potential inherent in antigen receptor gene rearrangement requires efficient rejoining of cleaved DNA and an intact cellular response to DNA damage.
Methods
Mice.
SCID p53+/– mice (ICR and C57Bl/6 backgrounds; Taconic Farms, Germantown, New York, USA) were intercrossed to generate SCID p53–/– mice. Rag-2–/– mice (C57Bl/6 and 129 backgrounds; obtained from Frederick Alt, Children’s Hospital, Boston, Massachusetts, USA) were crossed with SCID–/– p53+/– mice, and offspring were bred to generate the following 3 experimental cohorts: p53–/– (SCIDwt/wt, Rag-2+/–, or Rag-2+/+); SCID p53–/– (Rag-2+/– or Rag-2+/+); and SCID p53–/– Rag-2–/–. The genotype of all mice was confirmed by PCR for each allele. Mice were housed under specific pathogen-free conditions and sacrificed upon recognition of tumor masses or ill health, which marked the endpoint for calculating tumor incidence. Kaplan-Meier analysis was performed using the SPSS software package, and separate analyses were performed for mortality due to disseminated pro–B-cell lymphomas and for all tumors. Statistical analysis of survival differences was performed using the Wilcoxon log rank test.
Analysis of tumor phenotype.
Tumor tissue was fixed in formalin for standard histology. For flow cytometry, cell suspensions were stained using FITC-, phycoerythrin-, biotin-, or allophycocyanin-conjugated antibodies to the indicated markers, with secondary staining by streptavidin-CyChrome (PharMingen, San Diego, California, USA). Analysis was performed using a dual-laser FACScalibur (Becton Dickinson Immunocytometry Systems, San Jose, California, USA). Cytospin preparations from primary tumors were fixed in ice-cold acetone and stained using horseradish peroxidase–conjugated goat anti-mouse IgM, μ-chain specific, followed by colorimetric detection using 3, 3′-diaminobenzidine (Sigma Chemical Co., St. Louis, Missouri, USA) according to the manufacturer’s protocol. Controls included normal mouse spleen cells, murine cell lines p5424 (pro-T cell), and WEHI 231.1 (immature B cell).
Southern blot analysis.
Genomic DNA was digested with EcoRI, BamHI, or BglII and resolved by agarose gel electrophoresis prior to transfer to nylon membranes. Probes were used to detect rearrangements involving JH (34), Cμ (600-bp cDNA fragment starting near the 5′ end of the constant region), and Cδ (500-bp genomic DNA fragment encompassing the first exon). A genomic library was made from the SP04 tumor by ligation of partial Sau3A-digested genomic DNA into the sCOS1 vector, followed by packaging with lambda extracts, according to the manufacturer’s instructions (Stratagene, La Jolla, California, USA). The library was grown on replica filters and screened with the JH probe using standard techniques.
Fluorescence in situ hybridization analysis.
Metaphase spreads were prepared from primary lymphoma cell suspensions incubated for 2–6 hours with colchicine, and G-band analysis was performed using standard techniques. Chromosome 12 and 15 DNA paints (Oncor Inc., Gaithersburg, Maryland, USA) were labeled with digoxigenin and biotin, respectively, and were hybridized using the manufacturer’s protocol. Fluorescence in situ hybridization (FISH) probes included a BAC clone covering the constant regions of the IgH locus (generous gift of Roy Riblet, Medical Biology Institute, La Jolla, California, USA), a c-myc cosmid probe generated in our laboratory, and a P1 clone identifying the centromeric region of mouse chromosome 12 (35). FISH was performed as previously described (36), after labeling probes with biotin or digoxigenin by nick translation (GIBCO BRL, Rockville, Maryland, USA). Detection used fluorescein- or Texas red–conjugated avidin and fluorescein- or rhodamine-conjugated anti-digoxigenin (Vector Laboratories, Burlingame, California, USA). Chromosomes were identified with 4′,6′-diamidino-2-phenylindole (DAPI) staining.
Results
SCID p53–/– lymphomas have a pro–B-cell phenotype.
In agreement with prior reports in the literature (31–33), we found that 24 of 24 SCID p53–/– mice developed lymphomas at 6–13 weeks of age, with lymphomas observed in 6 of 15 p53–/– animals at later times. SCID mice had a significantly lower tumor incidence. Lymphomas in p53–/– mice included 5 thymic lymphomas and 1 localized neck mass. In contrast, SCID p53–/– tumors were disseminated throughout lymphoid tissue and other organs, including kidney and liver. Peripheral blood films demonstrated that lymphomas in 6 of 6 SCID p53–/– mice were associated with frank leukemia, a finding that was absent in 2 of 2 p53–/– lymphomas studied. Histologic examination of SCID p53–/– tumor tissue revealed near-total replacement of normal lymphoid architecture with high-grade lymphoma.
Flow cytometric analysis of 24 SCID p53–/– lymphomas revealed expression of B-cell markers B220 and CD19, but not T-cell markers CD4, CD8, or CD90 (Figure 1). These tumors were negative for surface IgM and showed low to moderate expression of CD43. Six p53–/– tumors were similarly characterized and included 5 thymic tumors expressing T-cell markers CD4, CD8, and CD90, which had either negative (1 tumor), low (3 tumors), or high (1 tumor) expression of TCR-αβ. One cervical lymphoma in a p53–/– mouse was positive for B220, CD19, and surface IgM, indicating a mature stage of B-cell development (Figure 1). Six SCID p53–/– tumors were also characterized for expression of cytoplasmic μH chain by immunohistochemistry on cytospin preparations, which was negative in all cases. Thus, SCID p53–/– lymphomas most resemble the pro–B-cell developmental stage.
Figure 1.

Flow cytometric analysis of primary tumor cells from lymphomas arising in p53–/– and SCID p53–/– mice. The majority of p53–/– tumors were thymic lymphomas with T-cell markers (top row, representative of 5 tumors), whereas 1 peripheral B-cell lymphoma expressing IgM was observed (middle row). SCID p53–/– lymphomas (bottom row, representative of 24 tumors) expressed B-lineage markers but lacked IgM expression. Single-cell suspensions from primary tumors were stained with antibodies to the markers indicated. Fluorescence intensity is expressed on a logarithmic scale. Control (unstained) plots are shown in broken lines.
Bone marrow cells in the pro–B-cell compartment undergo sequential D to J rearrangements, followed by V-to-DJ rearrangements. If the latter are in frame, production of μH chain drives developmental progression to the pre–B-cell stage (37). To better characterize the stage of B-lineage development represented by SCID p53–/– lymphomas, rearrangements of the IgH locus were analyzed by genomic Southern blots of primary tumor DNA using a JH-region probe (Table 1). In addition, because rearrangements in SCID mice may exhibit large deletions (38), rearrangements within Cμ and Cδ were analyzed. Overall, IgH locus rearrangements were found in 6 of 7 SCID p53–/– tumors. In 4 of these, at least 1 JH allele was rearranged. Tumor SP04 demonstrated rearrangement of 1 JH allele and deletion of JH on the second allele, with rearrangement of Cμ. Using a cosmid genomic library, the JH rearrangement from SP04 was cloned and analyzed by DNA sequencing, revealing a nonproductive rearrangement involving a DH pseudogene joined to JH4. Rearrangement of the JH probe was not detected with 2 different digests in 3 of 7 SCID p53–/– tumors. Two of these tumors exhibited rearrangement of Cμ, indicating a large deletion involving JH on the second allele. Thus, in 4 of 4 SCID p53–/– tumors in which rearrangements could be definitively characterized, we were able to exclude productive V(D)J rearrangements. In p53–/– tumors we found JH rearrangements in 1 thymic lymphoma, likely representing DJ rearrangement, which may occur at some frequency in T cells, and in 1 IgM-expressing B-cell lymphoma, which reflects a productive V(D)J rearrangement. These data indicate that the majority of lymphomas arising in SCID p53–/– mice are of a relatively uniform type of high-grade lymphoma representing the pro–B-cell stage of development, and they differ with respect to lineage or developmental stage from the thymic or mature B-cell lymphomas observed in our study or reported for the parental p53–/– strain (39).
Table 1.
Southern blot analysis of IgH rearrangements in SCID p53–/– and p53–/– lymphomas

SCID p53–/– lymphomas carry recurrent chromosome 12 translocations.
In humans, clinical and histologic subtypes of non-Hodgkin’s lymphoma correlate with specific chromosome translocations (40). To determine whether SCID p53–/– pro–B-cell lymphomas were associated with recurrent cytogenetic abnormalities, we performed G-band karyotype studies on primary tumors from 7 SCID p53–/– mice and 3 p53–/– mice. In 6 of 7 SCID p53–/– lymphomas, additional genetic material was observed at the telomere of chromosome 12, indicating a translocation (Figure 2 and Table 2). The band pattern suggested that this material was derived from chromosome 15. To identify the translocation partner, dual-color FISH was used with DNA paints specific for chromosomes 12 and 15 (Figure 3a and Table 2). In 5 of 6 tumors containing a chromosome 12 translocation, the partner was identified as chromosome 15, whereas in the sixth tumor the chromosome 12 translocation involved a different partner. In contrast to these results, 3 lymphomas from p53–/– mice exhibited a hyperdiploid karyotype and lacked t(12;15), as determined by dual-color FISH analysis.
Figure 2.
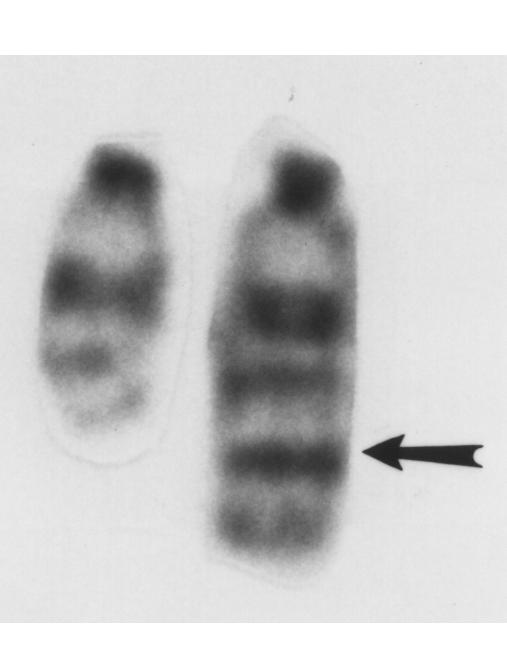
G-band karyotype showing chromosome 12 from a representative SCID p53–/– lymphoma. The normal chromosome is on the left. Arrow indicates a translocation.
Table 2.
Cytogenetic analysis of SCID p53–/– and p53–/– lymphomas

Figure 3.

FISH analysis of representative SCID p53–/– primary tumor metaphase spreads. (a) Whole-chromosome paints specific for chromosomes 12 (red) and 15 (green). Red arrow shows t(12;15), which was present in the majority of metaphases. Several tumors, including this one, demonstrated a large chromosome 15, possibly representing a duplication or amplification event. (b) IgH BAC probe. Hybridization is seen at the telomere on the normal chromosome 12 (red arrow) and at the breakpoint region on der12 (yellow arrow). Retention of the BAC signal on der12 indicates that the breakpoint lies within or telomeric to the region spanned by the probe. (c) Dual-color FISH analysis using chromosome 12 centromeric-region probe (red arrow) and c-myc probe (green arrow), demonstrating absence of c-myc on the der12 chromosome.
The IgH locus extends to the telomere on human chromosome 14 (41) and is similarly positioned on mouse chromosome 12, although continuity with the telomere has not yet been shown. By G-band analysis, the chromosome 12 breakpoint is consistent with the murine IgH complex. To further localize the breakpoint region, primary tumor metaphase spreads were hybridized with a BAC probe containing the 3′ part of the IgH locus, extending from the proximal VH segments to include Cα. In 4 of 6 tumors analyzed, the IgH BAC signal was retained on the derivative chromosome 12 (der12; Figure 3b and Table 2), implying that the breakpoint was within the IgH locus. Lack of an IgH signal in 2 t(12;15) tumors could indicate that the breakpoint is located toward the 3′ end of the IgH locus (where the BAC probe may have diminished sensitivity) or centromeric to the probe. We did not detect a reciprocal t(15;12), either with chromosome paints or with the IgH BAC probe. Taken together, these data show that SCID and p53-null mutations define a genetic pathway to pro–B-cell lymphomas that carry recurrent translocations involving chromosome 12, probably within the IgH locus.
Review of the genetic map of chromosome 15 suggested c-myc as a candidate oncogene for involvement in t(12;15) translocation. The position of c-myc in t(12;15) pro–B-cell lymphomas was analyzed using dual-color FISH with probes for c-myc and the centromeric region of chromosome 12 (Figure 3c). Whereas c-myc signals were readily recognized on chromosome 15, they were absent from the der12 chromosome. Similar results were obtained in 4 additional t(12;15) tumors, indicating that c-myc is not deregulated directly by the t(12;15) translocations, and suggesting the involvement of another oncogene on chromosome 15.
Initiation of V(D)J recombination is required for t(12;15) pro–B-cell lymphomas in SCID p53–/– mice.
Predisposition to t(12;15) pro–B-cell lymphomas in SCID 53–/– mice involves a lesion in the V(D)J recombinase, suggesting that translocations may arise as a result of aberrant IgH rearrangement in pro-B cells. By introducing a Rag-2–null mutation into the SCID p53–/– strain, we tested whether initiation of V(D)J recombination was a required step in oncogenesis. To exclude background effects, 3 cohorts of mice were derived from this interbreeding, including p53–/– (SCIDwt/wt, Rag-2+/–, or Rag-2+/+; n = 13), SCID p53–/– (Rag-2+/+ or Rag-2+/–; n = 17), and SCID p53–/– Rag-2–/– (n = 14). Mice were observed for tumor development, and tumor phenotype was characterized by flow cytometry as described above. Two separate analyses were performed using survival data for each cohort, considering mortality due to disseminated pro–B-cell lymphoma as well as mortality from all tumor types (Figure 4).
Figure 4.

Rag-2–null mutation suppresses pro–B-cell lymphomas in SCID p53–/– mice. (a) Kaplan-Meier survival analysis of mortality due to disseminated pro–B-cell lymphomas in mice derived from a common background. Disseminated pro–B-cell tumors occurred in all of the SCID p53–/– mice (n = 17) but were not observed in either the p53–/– cohort (n = 13) or in the SCID p53–/– Rag-2–/– cohort (n = 14). Follow-up intervals for individual mice are indicated for the p53–/– (circles) and SCID p53–/– Rag-2–/– (triangles) cohorts. Differences in pro–B-cell lymphoma mortality between SCID p53–/– and either p53–/– or SCID p53–/– Rag-2–/– cohorts were highly statistically significant (P < 0.001) using the Wilcoxon log rank test. (b) Survival analysis for overall tumor mortality. Differences between the SCID p53–/– and either the p53–/– or SCID p53–/– Rag-2–/– cohorts were statistically significant using the log rank test (P < 0.001). Differences in overall tumor mortality between the p53–/– and SCID p53–/– Rag-2–/– cohorts were not statistically significant (P > 0.40).
Consistent with previous reports and our earlier experiments, lymphomas developed in 100% of SCID p53–/– mice at a median of 8 weeks. All of these tumors were disseminated B220+CD19+IgM– lymphomas. In the p53–/– cohort, no disseminated pro–B-cell lymphomas were observed during a period of follow-up ranging from 6 to 33 weeks, although 1 animal developed a localized enlargement of a cervical lymph node containing a mature B-cell tumor (B220+CD19+IgM+). In the SCID p53–/– Rag-2–/– cohort, which was observed for a period ranging from 6 to 22 weeks, no disseminated B-lineage lymphomas were identified. These differences were statistically significant (P < 0.001). Analysis of overall tumor mortality demonstrated that the SCID p53–/– cohort had the shortest median survival (8 weeks), whereas the median survival of the p53–/– and SCID p53–/– Rag-2–/– cohorts was greater (23 and 18 weeks, respectively). Statistical comparisons indicated that the overall survival of the SCID p53–/– cohort was significantly less than either of the other groups of mice (P < 0.001), whereas survival differences between the p53–/– and SCID p53–/– Rag-2–/– cohorts were not statistically significant (P > 0.40).
The occurrence of localized thymic lymphomas was similar in the p53–/– and SCID p53–/– Rag-2–/– cohorts (Table 3). Phenotypically, the majority of SCID p53–/– Rag-2–/– lymphomas expressed CD90, CD4, and CD8, and lacked TCR-αβ, CD3, and B-cell markers. One thymic tumor expressed CD90 as well as the B-cell markers B220 and CD19. Sarcomas were also seen in both the p53–/– and SCID p53–/– Rag-2–/– mice. Thymic lymphomas and sarcomas were not observed in SCID p53–/– mice, likely reflecting the early mortality from disseminated pro–B-cell tumors (Figure 4b). These data indicate that initiation of V(D)J recombination is a required element in the oncogenic pathway leading to disseminated t(12;15) pro–B-cell lymphomas in SCID p53–/– mice, but not to the thymic lymphomas and sarcomas characteristic of the p53–/– background.
Table 3.
Comparison of tumor types developing in 3 cohorts of mice derived from a common background

Discussion
Chromosome translocations involving antigen receptor genes represent a common pathway for lymphoid oncogenesis and likely represent an early and critical genetic change in the evolution of these malignancies (1, 3, 4). The hypothesis that these translocations involve V(D)J recombination has been inferred from the sequences of cloned breakpoints. The evidence appears most consistent in the t(14;18) translocations found in most cases of follicular B-cell lymphomas, which juxtapose the Bcl-2 proto-oncogene with 1 of the JH segments, and in t(7;9) of T-cell lymphoblastic lymphoma/leukemia, which involves breaks flanking the D and J segments of the TCR-β gene on chromosome 7 (4, 8, 9, 12, 42). Other features of V(D)J recombination, including cryptic RSS on partner chromosomes and N-nucleotide additions, have also been reported at translocation breakpoints (reviewed in ref. 4). Despite the resemblance of some breakpoints to V(D)J joints, much of the available data on breakpoint sequences do not strictly conform to the attributes of V(D)J recombination, indicating that most translocations do not arise from a normal V(D)J reaction. Nevertheless, the consistent participation of antigen receptor loci in lymphoma translocations suggests that components of the V(D)J recombinase may participate in creating the conditions for aberrant chromosome joining.
The recurrent involvement of t(12;15) in pro–B-cell lymphomas arising in SCID p53–/– mice defines a genetic pathway leading reproducibly to antigen receptor–locus translocations. The fact that a Rag-2–null mutation suppressed the development of t(12;15) lymphomas in the SCID p53–/– background suggests that the translocations occur as a result of aberrant rejoining of IgH loci cleaved during attempted V(D)J recombination at the pro–B-cell stage. An alternative interpretation is that oncogenesis is developmentally specific and requires Rag-2 for maturation of B cells to a susceptible stage. B-cell progression from the pro-B to pre-B stage is signaled by productive rearrangement of Cμ and expression in the context of other components of the pre–B-cell receptor (37). A block in B-cell development at the pro-B stage occurs in both Rag-2–/– and SCID mice, although in the latter strain, there is some leakiness in T- and B-cell development because of the infrequent assembly of V(D)J joints. In all the SCID p53–/– tumors tested, we found no evidence for surface or cytoplasmic μ-chain expression. Furthermore, in all 4 t(12;15) lymphomas in which IgH rearrangements could be characterized, we definitively ruled out productive V(D)J rearrangement on both IgH alleles. Thus, the cellular target for oncogenic transformation in SCID p53–/– mice could not have passed the pro–B-cell stage.
Following cleavage of antigen receptor DNA in SCID mice, the inability to efficiently rejoin DNA ends could potentially lead to illegitimate recombination. In SCID thymocytes, p53 is activated by DNA breaks, leading to early apoptosis (33, 43). In the absence of p53 function, SCID pro-B cells may survive longer, permitting Rag-mediated DNA breaks at the IgH locus to be rejoined by activities other than the DNA-PK complex. Such alternative DNA repair pathways may be less stringent in preferentially rejoining antigen receptor DNA ends to each other than in involving another locus, leading to a higher frequency of chromosome translocations. In p53-deficient mice, which have normal lymphoid development, presence of the DNA-PK complex may ensure efficient rejoining of DNA breaks during V(D)J recombination so that unresolved DNA breaks are relatively rare. Our data confirm previous observations that thymic lymphomas commonly occurring in p53–/– mice are not associated with recurrent chromosome translocations (44). The finding that thymic lymphomas occurred in SCID p53–/– Rag-2–/– mice with frequency similar to that of p53–/– mice also agrees with previous work, showing that p53–/– thymic tumors are not suppressed by Rag-1 or Rag-2 mutations (44–46). Thus, for some types of lymphoma, p53 contributes to oncogenesis in a manner that is independent of chromosome translocations involving antigen receptor genes. In SCID p53–/– mice, the p53 lesion may, therefore, contribute to the high incidence of pro–B-cell lymphoma by failing to prevent chromosome translocations, by facilitating downstream genetic changes, or both.
The hypothesis that cellular responses to DNA damage suppress oncogenic side reactions of the V(D)J recombinase is supported by the association between genetic conditions affecting these pathways and predisposition to lymphoid malignancies. Patients with ataxia-telangiectasia, the Nijmegen breakage syndrome, and Bloom’s syndrome all exhibit abnormal cellular responses to DNA damage and have a high incidence of non-Hodgkin’s lymphomas (47–50). Lymphomas in ataxia-telangiectasia patients are most commonly T-cell derived and involve translocations with TCR loci on chromosomes 7 or 14 (47). This observation, and the fact that translocation-associated lymphomas in SCID p53–/– mice exclusively involved the B-cell lineage, suggests that the spectrum of critical elements for tumor suppression during antigen receptor rearrangement may differ in the various lymphoid tissues. Advances in understanding the cellular functions of lymphoma tumor suppressors may shed more light on this issue.
Targeted genomic instability is an essential process in diversification of the antigen receptor repertoire and occurs in 3 distinct forms in the B-cell lineage. In addition to VDJ recombination, immune responses induce germinal-center B cells to undergo isotype class-switch recombination within the IgH locus, as well as somatic hypermutation of immunoglobulin variable regions. Class-switch recombination has been implicated in generating t(8;14) in nonendemic Burkitt’s lymphoma, juxtaposing switch regions lying upstream of constant-region exons with c-myc. In mice, plasmacytomas induced by mineral oil are associated with t(12;15), placing the c-myc locus on chromosome 15 into the switch segments of 3′ IgH constant-region genes (13, 51, 52). This mechanism is unlikely to be involved in mediating the t(12;15) in SCID p53–/– lymphomas, given that Rag-2 is required for generation of these tumors but is not needed for class switching (53, 54). Recent data indicate that somatic hypermutation also involves DNA breaks and may contribute to some IgH translocations that occur within the variable region (55, 56). It has also been determined that Rag-1 and Rag-2 are reactivated in germinal-center B cells, where immunoglobulin genes may undergo additional V(D)J recombination events (57–60). The apparent genetic plasticity of immunoglobulin genes in the germinal center correlates with a postulated germinal-center origin for the majority of B-cell lymphomas and suggests that, in many cases, chromosome translocations may arise in the course of immune responses (61, 62).
Genetic instability at antigen receptor loci represents the distinguishing feature of vertebrate immunity and conflicts with the cellular paradigm of preserving genomic integrity. An important question regarding the mechanism of V(D)J recombination and other forms of antigen receptor revision is how these processes are normally confined to intended targets. Our data suggest that this confinement involves 2 essential components. First, efficient rejoining of DNA breaks may minimize cellular exposure to free DNA ends. This is likely facilitated by the coupling of Rag-mediated cleavage events and the formation of a synaptic complex that activates rejoining (63). Second, activation of DNA damage–sensing pathways by unresolved DNA breaks may either kill cells or otherwise prevent the activity of nonspecific repair pathways. Genetic factors or environmental exposures that interfere with either of these processes may increase the frequency of chromosome translocations and contribute to the development of lymphomas.
Acknowledgments
We thank Roy Riblet for generously providing us with the IgH BAC, Barbara Malynn for the IgH locus probes, and Frederick Alt for supplying Rag-2–/– mice. We are grateful to Mark Groudine and Patrick Concannon for reviewing the manuscript. This work was supported in part by grants from the National Institutes of Health (to G.J. Vanasse, D.M. Willerford, and C.M. Disteche) and the University of Washington Royalty Research Fund (to D.M. Willerford).
References
- 1.Rabbitts TH. Chromosomal translocations in human cancer. Nature. 1994;372:143–149. doi: 10.1038/372143a0. [DOI] [PubMed] [Google Scholar]
- 2.Showe LC, Croce CM. The role of chromosomal translocations in B- and T-cell neoplasia. Annu Rev Immunol. 1987;5:253–277. doi: 10.1146/annurev.iy.05.040187.001345. [DOI] [PubMed] [Google Scholar]
- 3.Korsmeyer SJ. Chromosomal translocations in lymphoid malignancies reveal novel proto-oncogenes. Annu Rev Immunol. 1992;10:785–807. doi: 10.1146/annurev.iy.10.040192.004033. [DOI] [PubMed] [Google Scholar]
- 4.Tycko B, Sklar J. Chromosomal translocations in lymphoid neoplasia: a reappraisal of the recombinase model. Cancer Cells. 1990;2:1–8. [PubMed] [Google Scholar]
- 5.Alt FW, et al. VDJ recombination. Immunol Today. 1992;13:306–314. doi: 10.1016/0167-5699(92)90043-7. [DOI] [PubMed] [Google Scholar]
- 6.Roth DB, Lindahl T, Gellert M. Repair and recombination. How to make ends meet. Curr Biol. 1995;5:496–499. doi: 10.1016/s0960-9822(95)00101-1. [DOI] [PubMed] [Google Scholar]
- 7.Harriman W, Volk H, Defranoux N, Wabl M. Immunoglobulin class switch recombination. Annu Rev Immunol. 1993;11:361–384. doi: 10.1146/annurev.iy.11.040193.002045. [DOI] [PubMed] [Google Scholar]
- 8.Bakhshi A, et al. Cloning the chromosomal breakpoint of t(14;18) human lymphomas: clustering around JH on chromosome 14 and near a transcriptional unit on 18. Cell. 1985;41:899–906. doi: 10.1016/s0092-8674(85)80070-2. [DOI] [PubMed] [Google Scholar]
- 9.Tsujimoto Y, Gorham J, Cossman J, Jaffe E, Croce CM. The t(14;18) chromosome translocations involved in B-cell neoplasms result from mistakes in VDJ joining. Science. 1985;22:1390–1393. doi: 10.1126/science.3929382. [DOI] [PubMed] [Google Scholar]
- 10.Taub R, et al. Translocation of the c-myc gene into the immunoglobulin heavy chain locus in human Burkitt lymphoma and murine plasmacytoma cells. Proc Natl Acad Sci USA. 1982;79:7837–7841. doi: 10.1073/pnas.79.24.7837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haluska FG, Finver S, Tsujimoto Y, Croce CM. The t(8;14) chromosomal translocation occurring in B-cell malignancies results from mistakes in V-D-J joining. Nature. 1986;325:158–161. doi: 10.1038/324158a0. [DOI] [PubMed] [Google Scholar]
- 12.Cleary ML, Sklar J. Nucleotide sequence of a t(14;18) chromosomal breakpoint in follicular lymphoma and demonstration of a breakpoint cluster region near a transcriptionally active locus on chromosome 18. Proc Natl Acad Sci USA. 1985;82:7439–7443. doi: 10.1073/pnas.82.21.7439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cory S, Gerondakis S, Adams JM. Interchromosomal recombination of the cellular oncogene c-myc with the immunoglobulin heavy chain locus in murine plasmacytomas is a reciprocal exchange. EMBO J. 1983;2:697–703. doi: 10.1002/j.1460-2075.1983.tb01487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schatz DG, Oettinger MA, Schlissel MS. V(D)J recombination: molecular biology and regulation. Annu Rev Immunol. 1992;10:359–383. doi: 10.1146/annurev.iy.10.040192.002043. [DOI] [PubMed] [Google Scholar]
- 15.Oettinger MA, Schatz DG, Gorka C, Baltimore DG. RAG-1 and RAG-2, adjacent genes that synergistically activate V(D)J recombination. Science. 1990;248:1517–1522. doi: 10.1126/science.2360047. [DOI] [PubMed] [Google Scholar]
- 16.van Gent DC, et al. Initiation of V(D)J recombination in a cell-free system. Cell. 1995;81:925–934. doi: 10.1016/0092-8674(95)90012-8. [DOI] [PubMed] [Google Scholar]
- 17.McBlane JF, et al. Cleavage at a V(D)J recombination signal requires only RAG1 and RAG2 proteins and occurs in two steps. Cell. 1995;83:387–395. doi: 10.1016/0092-8674(95)90116-7. [DOI] [PubMed] [Google Scholar]
- 18.Mombaerts P, et al. RAG-1 deficient mice have no mature B and T lymphocytes. Cell. 1992;68:869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- 19.Shinkai Y, et al. RAG-2 deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell. 1992;68:855–867. doi: 10.1016/0092-8674(92)90029-c. [DOI] [PubMed] [Google Scholar]
- 20.Schwartz K, et al. RAG mutations in human B cell-negative SCID. Science. 1996;274:97–99. doi: 10.1126/science.274.5284.97. [DOI] [PubMed] [Google Scholar]
- 21.Taccioli GE, Alt FW. Potential targets for autosomal SCID mutations. Curr Opin Immunol. 1992;7:436–440. doi: 10.1016/0952-7915(95)80085-9. [DOI] [PubMed] [Google Scholar]
- 22.Roth DB, Menetski JP, Nakajima PB, Bosma MJ, Gellert M. V(D)J recombination: broken DNA molecules with covalently sealed (hairpin) coding ends in scid mouse thymocytes. Cell. 1992;70:983–991. doi: 10.1016/0092-8674(92)90248-b. [DOI] [PubMed] [Google Scholar]
- 23.Bosma MJ, Carroll AM. The SCID mouse mutant: definition, characterization, and potential uses. Annu Rev Immunol. 1991;9:323–350. doi: 10.1146/annurev.iy.09.040191.001543. [DOI] [PubMed] [Google Scholar]
- 24.Blunt T, et al. Defective DNA-dependent protein kinase activity is linked to V(D)J recombination and DNA repair defects associated with the murine scid mutation. Cell. 1995;80:813–823. doi: 10.1016/0092-8674(95)90360-7. [DOI] [PubMed] [Google Scholar]
- 25.Kirchgessner CU, et al. DNA-dependent protein kinase (p350) as a candidate gene for the murine SCID defect. Science. 1995;267:1178–1183. doi: 10.1126/science.7855601. [DOI] [PubMed] [Google Scholar]
- 26.Peterson SR, et al. Loss of the catalytic subunit of the DNA-dependent protein kinase in DNA double-strand-break-repair mutant mammalian cells. Proc Natl Acad Sci USA. 1995;92:3171–3174. doi: 10.1073/pnas.92.8.3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kastan MB, et al. A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell. 1992;71:587–597. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- 28.Clarke AR, et al. Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature. 1993;362:849–852. doi: 10.1038/362849a0. [DOI] [PubMed] [Google Scholar]
- 29.Lowe S, Schmitt EM, Smith SW, Osborne BA, Jacks T. Nature. 1993;362:847–851. doi: 10.1038/362847a0. [DOI] [PubMed] [Google Scholar]
- 30.Donehower LA, et al. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 31.Nacht M, et al. Mutations in the p53 and SCID genes cooperate in tumorigenesis. Genes Dev. 1996;10:2055–2066. doi: 10.1101/gad.10.16.2055. [DOI] [PubMed] [Google Scholar]
- 32.Bogue MA, Zhu C, Aguilar-Cordova E, Donehower LA, Roth DB. p53 is required for both radiation-induced differentiation and rescue of V(D)J rearrangement in scid mouse thymocytes. Genes Dev. 1996;10:553–565. doi: 10.1101/gad.10.5.553. [DOI] [PubMed] [Google Scholar]
- 33.Guidos CJ, et al. V(D)J recombination activates a p53-dependent DNA damage checkpoint in scid lymphocyte precursors. Genes Dev. 1996;10:2038–2054. doi: 10.1101/gad.10.16.2038. [DOI] [PubMed] [Google Scholar]
- 34.Malynn BA, et al. The scid defect affects the final step of the immunoglobulin VDJ recombinase mechanism. Cell. 1988;54:453–460. doi: 10.1016/0092-8674(88)90066-9. [DOI] [PubMed] [Google Scholar]
- 35.Shi YP, et al. FISH probes for mouse chromosome identification. Genomics. 1997;45:42–47. doi: 10.1006/geno.1997.4904. [DOI] [PubMed] [Google Scholar]
- 36.Edelhoff SE, et al. Mapping of two genes encoding members of a distinct subfamily of MAX interacting proteins: MAD to human chromosome 2 and mouse chromosome 6, and MXI1 to human chromosome 10 and mouse chromosome 19. Oncogene. 1994;9:665–668. [PubMed] [Google Scholar]
- 37.Willerford DM, Swat W, Alt FW. Developmental regulation of V(D)J recombination and lymphocyte differentiation. Curr Opin Genet Dev. 1996;6:603–609. doi: 10.1016/s0959-437x(96)80090-6. [DOI] [PubMed] [Google Scholar]
- 38.Schuler W, et al. Rearrangement of antigen receptor genes is defective in mice with severe combined immune deficiency. Cell. 1986;46:963–972. doi: 10.1016/0092-8674(86)90695-1. [DOI] [PubMed] [Google Scholar]
- 39.Donehower LA, et al. Effects of genetic background on tumorigenesis in p53-deficient mice. Mol Carcinog. 1995;14:16–22. doi: 10.1002/mc.2940140105. [DOI] [PubMed] [Google Scholar]
- 40.Yunis JJ, et al. Distinctive chromosomal abnormalities in histologic subtypes of non-Hodgkin’s lymphoma. N Engl J Med. 1982;307:1231–1236. doi: 10.1056/NEJM198211113072002. [DOI] [PubMed] [Google Scholar]
- 41.Cook GP, et al. A map of the human immunoglobulin VH locus completed by analysis of the telomeric region of chromosome 14q. Nat Genet. 1994;7:162–168. doi: 10.1038/ng0694-162. [DOI] [PubMed] [Google Scholar]
- 42.Tycko B, Reynolds TC, Smith SD, Sklar J. Consistent breakage between consensus recombinase heptamers of chromosome 9 DNA in a recurrent chromosomal translocation of human T cell leukemia. J Exp Med. 1989;169:369–377. doi: 10.1084/jem.169.2.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Diamond RA, Ward SB, Owada-Makabe K, Wang H, Rothenberg EV. Different developmental arrest points in Rag-2–/– and SCID thymocytes on two genetic backgrounds. J Immunol. 1997;158:4052–4064. [PubMed] [Google Scholar]
- 44.Liao M-J, et al. No requirement for V(D)J recombination in p53-deficient thymic lymphoma. Mol Cell Biol. 1998;18:3495–3501. doi: 10.1128/mcb.18.6.3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mombaerts P, Terhorst C, Jacks T, Tonegawa S, Sancho J. Characterization of immature thymocyte lines derived from T-cell receptor or recombination activating gene 1 and p53 double mutant mice. Proc Natl Acad Sci USA. 1995;92:7420–7424. doi: 10.1073/pnas.92.16.7420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nacht M, Jacks T. V(D)J recombination is not required for the development of lymphoma in p53-deficient mice. Cell Growth Differ. 1998;9:131–138. [PubMed] [Google Scholar]
- 47.Lavin MF, Shiloh Y. The genetic defect in ataxia-telangiectasia. Annu Rev Immunol. 1997;15:177–202. doi: 10.1146/annurev.immunol.15.1.177. [DOI] [PubMed] [Google Scholar]
- 48.Varon R, et al. Nibrin, a novel DNA double-strand break repair protein is mutated in Nijmegen breakage syndrome. Cell. 1998;93:467–476. doi: 10.1016/s0092-8674(00)81174-5. [DOI] [PubMed] [Google Scholar]
- 49.Ellis NA, et al. The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell. 1995;83:655–666. doi: 10.1016/0092-8674(95)90105-1. [DOI] [PubMed] [Google Scholar]
- 50.German J. Bloom’s syndrome. XX. The first 100 cancers. Cancer Genet Cytogenet. 1997;93:100–106. doi: 10.1016/s0165-4608(96)00336-6. [DOI] [PubMed] [Google Scholar]
- 51.Gerondakis S, Cory S, Adams JM. Translocation of the myc cellular oncogene to the immunoglobulin heavy chain locus in murine plasmacytomas is an imprecise reciprocal exchange. Cell. 1984;36:973–982. doi: 10.1016/0092-8674(84)90047-3. [DOI] [PubMed] [Google Scholar]
- 52.Potter M, Wiener F. Plasmacytomagenesis in mice: model of neoplastic development dependent upon chromosomal translocations. Carcinogenesis. 1992;13:1681–1697. doi: 10.1093/carcin/13.10.1681. [DOI] [PubMed] [Google Scholar]
- 53.Rolink A, Melchers F, Andersson J. The SCID but not the RAG-2 gene product is required for S mu-S epsilon heavy chain class switching. Immunity. 1996;5:319–330. doi: 10.1016/s1074-7613(00)80258-7. [DOI] [PubMed] [Google Scholar]
- 54.Lansford R, Manis JP, Sonoda E, Rajewsky K, Alt FW. Ig heavy chain class switching in Rag-deficient mice. Int Immunol. 1998;10:325–332. doi: 10.1093/intimm/10.3.325. [DOI] [PubMed] [Google Scholar]
- 55.Sale JE, Neuberger MS. TdT-accessible breaks are scattered over the immunoglobulin V domain inn a constitutively hypermutating B cell line. Immunity. 1998;9:859–869. doi: 10.1016/s1074-7613(00)80651-2. [DOI] [PubMed] [Google Scholar]
- 56.Goossens T, Klein U, Kuppers R. Frequent occurrence of deletions and duplications during somatic hypermutation: implications for oncogene translocations and heavy chain disease. Proc Natl Acad Sci USA. 1998;95:2463–2468. doi: 10.1073/pnas.95.5.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Han S, Zheng B, Schatz DG, Spanopoulou E, Kelsoe G. Neoteny in lymphocytes: Rag1 and Rag2 expression in germinal center B cells. Science. 1996;274:2094–2097. doi: 10.1126/science.274.5295.2094. [DOI] [PubMed] [Google Scholar]
- 58.Han S, et al. V(D)J recombinase activity in a subset of germinal center B lymphocytes. Science. 1997;278:301–305. doi: 10.1126/science.278.5336.301. [DOI] [PubMed] [Google Scholar]
- 59.Hikida M, et al. Reexpression of Rag-1 and Rag-2 genes in activated mature mouse B cells. Science. 1996;274:2092–2094. doi: 10.1126/science.274.5295.2092. [DOI] [PubMed] [Google Scholar]
- 60.Papavasiliou F, et al. V(D)J recombination in mature B cells: a mechanism for altering antibody responses. Science. 1997;278:298–301. doi: 10.1126/science.278.5336.298. [DOI] [PubMed] [Google Scholar]
- 61.Harris NL, et al. A revised European-American classification of lymphoid neoplasms: a proposal from the International Lymphoma Study Group. Blood. 1994;84:1361–1392. [PubMed] [Google Scholar]
- 62.Klein U, et al. Somatic hypermutation in normal and transformed B cells. Immunol Rev. 1998;162:261–280. doi: 10.1111/j.1600-065x.1998.tb01447.x. [DOI] [PubMed] [Google Scholar]
- 63.Hiom K, Gellert M. Assembly of a 12/23 paired signal complex: a critical control point in V(D)J recombination. Mol Cell. 1998;1:1011–1019. doi: 10.1016/s1097-2765(00)80101-x. [DOI] [PubMed] [Google Scholar]