

Abstract
Disorders of carbohydrate and lipid metabolism have been reported to cluster in patients with essential hypertension and in spontaneously hypertensive rats (SHRs). A deletion in the Cd36 gene on chromosome 4 has recently been implicated in defective carbohydrate and lipid metabolism in isolated adipocytes from SHRs. However, the role of Cd36 and chromosome 4 in the control of blood pressure and systemic cardiovascular risk factors in SHRs is unknown. In the SHR.BN-Il6/Npy congenic strain, we have found that transfer of a segment of chromosome 4 (including Cd36) from the Brown Norway (BN) rat onto the SHR background induces reductions in blood pressure and ameliorates dietary-induced glucose intolerance, hyperinsulinemia, and hypertriglyceridemia. These results demonstrate that a single chromosome region can influence a broad spectrum of cardiovascular risk factors involved in the hypertension metabolic syndrome. However, analysis of Cd36 genotypes in the SHR and stroke-prone SHR strains indicates that the deletion variant of Cd36 was not critical to the initial selection for hypertension in the SHR model. Thus, the ability of chromosome 4 to influence multiple cardiovascular risk factors, including hypertension, may depend on linkage of Cd36 to other genes trapped within the differential segment of the SHR.BN-Il6/Npy strain.
J. Clin. Invest. 103:1651–1657 (1999).
Introduction
For decades, essential hypertension has been primarily viewed from a hemodynamic, neural, and renal perspective. However, based on mounting evidence from clinical, epidemiological, and experimental studies, it has become increasingly recognized that disturbances in carbohydrate and lipid metabolism often accompany high blood pressure, and that essential hypertension may also represent a disorder of cardiovascular endocrinology and metabolism (1, 2). In patients with essential hypertension, clustering of metabolic cardiovascular risk factors — including glucose intolerance, hyperinsulinemia, and hypertriglyceridemia — may promote susceptibility to target organ damage and partly explain why conventional antihypertensive agents have failed to reduce the risk for coronary heart disease to the extent predicted from epidemiological studies (2).
Recently, a provocative hypothesis has emerged in which inherited disorders of carbohydrate or lipid metabolism are held to be at the core of the hypertension syndrome and to contribute to the primary pathogenesis of increased blood pressure. Studies in nonobese subjects with a family history of hypertension and in a variety of experimental animal models have suggested that alterations in carbohydrate and/or lipid metabolism can influence the regulation of blood pressure and might precede the development of hypertension (3–5). The lack of insulin resistance in patients with secondary forms of hypertension, together with observations of disordered carbohydrate and lipid metabolism in cultured adipocytes from hypertensive animals, indicates that at least some endocrine-metabolic disturbances are not simply a consequence of increased blood pressure (4, 6–8). Hence, there is intense interest in identifying genetic mechanisms that may underlie the association between increased blood pressure and other cardiovascular risk factors in essential hypertension.
The spontaneously hypertensive rat (SHR) is the most widely studied model of essential hypertension and has been reported to exhibit a number of abnormalities in carbohydrate and lipid metabolism (4, 5, 7, 9–11). As with studies of other complex traits, the ability to detect disordered carbohydrate or lipid metabolism in the SHR will depend on the type of normotensive control strain used for comparison, the techniques used to assess the phenotypes of interest, and the environmental and dietary circumstances of the study (5, 11, 12). Under the appropriate experimental conditions, systemic and cellular defects in both carbohydrate and lipid metabolism can be clearly demonstrated in the SHR. Thus, given proper understanding of key study variables that influence these metabolic phenotypes, the SHR can provide a useful tool for investigating the clustering of cardiovascular risk factors in spontaneous hypertension.
In the SHR, a significant advance has recently been made in understanding the molecular basis for disordered carbohydrate and lipid metabolism that could shed light on the clustering of systemic cardiovascular risk factors in essential hypertension. Aitman et al. (13) have identified a deletion in the SHR gene for Cd36 that encodes a fatty acid receptor/transporter involved in the transmembrane transport of long-chain fatty acids in adipose tissue and in cardiac and skeletal muscle. The SHR carries a deletion variant of Cd36 that results in the near-complete lack of expression of normal Cd36 protein in adipocyte plasma membranes (13). Cd36 maps to the telomeric region of rat chromosome 4 that includes peak areas of linkage to SHR defects in insulin-stimulated glucose transport and catecholamine-mediated lipolysis in isolated adipocytes (8, 13). To confirm these linkage studies directly, we developed a congenic strain in which the deletion variant of Cd36 in the SHR was replaced with a wild-type variant from the normotensive Brown Norway (BN) rat. Transfer of the Cd36 gene from the normotensive BN rat onto the genetic background of this SHR congenic strain was associated with significant increases in both insulin-stimulated glucose transport and catecholamine-mediated lipolysis in isolated adipocytes (13). These findings, together with results of metabolic studies in transgenic mice that overexpress Cd36, have provided compelling evidence that at least in isolated adipocytes, the SHR deletion in Cd36 is a major genetic determinant of disordered carbohydrate and lipid metabolism (8, 13).
Based on the foregoing observations, a key question that now arises is whether the region of chromosome 4 that contains Cd36 is also involved in the genetic control of blood pressure and the systemic cardiovascular risk factors that tend to cluster in essential hypertension — namely, impaired glucose tolerance, hyperinsulinemia, and hypertriglyceridemia. Accordingly, we investigated whether replacing this segment of chromosome 4 in the SHR with the corresponding chromosome region from the normotensive BN rat would improve blood pressure, systemic glucose tolerance, serum insulin levels, and serum triglycerides. We have also genotyped multiple strains of SHR to investigate the relationship of the Cd36 deletion variant to the genealogy of hypertension in this model.
Methods
Strain derivation and genetic characterization.
We measured blood pressure and systemic metabolic risk factors in the same SHR progenitor and congenic strains used to investigate the relationship of Cd36 to SHR defects in adipocyte insulin sensitivity and lipolysis (13). The SHR progenitor and congenic strains were derived from a progenitor strain of SHR (SHR/Ola) that descends from inbred SHR originally obtained from the National Institutes of Health (Bethesda, Maryland, USA). This progenitor strain of SHR has been maintained by brother × sister mating in Prague for more than 15 years and was beyond the F90 generation when congenic breeding was initiated. The SHR-chromosome 4 congenic strain was derived by transferring a segment of chromosome 4 from an inbred BN strain (Charles River Laboratories, Kingston, New York, USA) onto the genetic background of the SHR/Ola strain. In previous linkage studies in the SHR, putative quantitative trait loci (QTLs) for increased blood pressure and disordered carbohydrate and lipid metabolism were mapped to relatively broad regions of chromosome 4 (8, 14–16). Accordingly, in deriving the current congenic strain, we selected a large piece of chromosome 4 to increase the chance of capturing the QTL in the transferred chromosome segment. The markers Il6 and Npy were used for selection of heterozygous carriers in each backcross generation. After 8 generations of selective backcrossing to the SHR progenitor strain, the differential segment was fixed by intercrossing heterozygotes and selecting for offspring inheriting the homozygous BN Il6-Npy chromosome segment. The size of the differential segment was estimated by genotyping of microsatellite markers on chromosome 4 (17). The congenic status of the strain was confirmed by analysis of more than 60 widely dispersed microsatellite markers on other chromosomes. The official designation of this congenic strain is SHR.BN-Il6/Npy. For purposes of simplicity, however, we refer to it as the SHR-chr.4 congenic strain. Animals of the N8F3 or greater generation were used in the current studies. Cd36 genotyping was also performed in SHRs obtained from Charles River Laboratories, Taconic Farms (Germantown, New York, USA), the Czech Academy of Sciences, and from a stroke-prone strain of SHR obtained from the colony maintained at the University of Michigan.
Cd36 genotyping.
The SHR deletion variant in Cd36 was detected by Southern blot analysis of PstI-digested genomic DNA probed with a DNA fragment that hybridizes to exon 6 of the published sequence for Cd36 (GenBank accession no. L19658). The probe was prepared by PCR amplification of exon 6 of Cd36 using the following primers: upstream 5′-TCA AGG TGT GCT CAA CAG CC-3′; downstream 5′-AGG ATA AAA CAC ACC AAC TGT-3′. This probe-enzyme combination detects both the normally transcribed wild-type copy of Cd36 and a duplicated nontranscribed version of Cd36 present in a variety of normotensive rat strains tested to date (WKY, BN, Lewis, Dahl R). Southern blot analysis of SHR genomic DNA shows missing bands caused by deletion of the normally transcribed copy of Cd36 (13). We also performed PCR analysis of the HinfI RFLP that distinguishes the SHR deletion variant in Cd36 described previously by Aitman et al. (13).
Hemodynamic studies.
Arterial blood pressures and heart rates were measured continuously in unanesthetized, unrestrained male SHR progenitor rats and SHR-chr.4 congenic rats between 10 and 14 weeks of age, using radiotelemetry. All rats were allowed to recover for at least 10 days after implantation of radiotelemetry transducers before the blood pressure recordings were started. Pulsatile pressures and heart rates were recorded in 5-second bursts every 5 minutes during the day (0600 to 1800 hours) and during the night (1800 to 0600 hours) for 4 weeks. From these data, individual weekly means for daytime and nighttime blood pressures and heart rates were calculated for each rat in each week of the study. The results from the rats were then averaged to obtained group means. Rats were fed standard laboratory chow and tap water ad libitum throughout the study.
Assessment of metabolic cardiovascular risk factors.
We tested for the presence of hyperglycemia, hyperinsulinemia, and dyslipidemia in 8- to 10-week-old male SHR progenitor and congenic rats under 2 different dietary conditions. Baseline blood samples were obtained via tail vein between 0900 and 1200 hours in nonfasted rats maintained on standard laboratory chow. The animals were then given a high-fructose diet (60% fructose) for 15 days, and the blood sampling was repeated. Administration of a high-fructose diet is a well-known method for provoking insulin resistance, and enabled us to test for possible environment-genotype interaction in the regulation of risk factor clustering in the SHR model. An intraperitoneal glucose tolerance test was performed on day 13 of fructose feeding. The fructose diet was taken away from the rats in the morning; 7 hours later, the rats were intraperitoneally injected with a 15% glucose solution (1 mL/100 g body weight, or 1.5 g/kg). Blood was taken from the tail without anesthesia before the glucose injection and then 30, 60, and 120 minutes after the injection. Blood samples were obtained and immediately centrifuged at 4°C, and the serum was stored frozen at –70°C until analysis. Glucose was measured by the glucose oxidase technique; rat insulin was measured by radioimmunoassay (Amersham Life Sciences Inc., Arlington Heights, Illinois, USA); and total cholesterol, triglycerides (without glycerol blanking), HDL cholesterol, and nonesterified fatty acids were measured using standard enzymatic techniques (14, 18, 19).
Statistical analysis.
All data are expressed as mean ± SEM. Daytime and nighttime blood pressures, heart rates, and body weights over the course of the study were separately analyzed by repeated measures ANOVA. Individual means for blood pressures, body weights, and serum levels of glucose, insulin, and lipids were compared by t test. Statistical significance was defined as P < 0.05.
Results
Genotyping results.
Genotype results obtained with more than 60 widely dispersed polymorphic microsatellite markers confirmed that the SHR progenitor strain and the SHR-chr.4 congenic strain were genetically identical except for the differential segment of chromosome 4 transferred from the BN donor strain. The size of the transferred BN chromosome segment is approximately 37 cM, as defined by testing with the following chromosome 4 markers (Figure 1): D4Rat247, Il6, D4Rat142, D4Rat139, D4Rat1, D4Rat149, D4Mgh2, D4Rat26, D4Rat33, D4Rat169, D4Rat108, and D4Rat34. Mapping studies in recombinant inbred strains, and the results of radiation hybrid analysis, have shown that the Cd36 gene is located near Il6 at the telomere of chromosome 4 (Figure 1) (13).
Figure 1.

Linkage map of chromosome 4. Linkage map showing the transferred segment of chromosome 4 in the SHR.BN-Il6/Npy congenic strain. The filled bar denotes the chromosome region from the BN strain that has been fixed in the homozygous state on the SHR background. The cross-hatched bar denotes region in which some residual heterozygosity may exist within the congenic strain. The open region denotes the flanking segment of SHR chromosome. Map distances are adapted from chromosome 4 maps of the WIBR/MIT Rat Genome Center (17), Jacob et al. (30), and Pravenec et al. (31). The position of Cd36 is based on radiation hybrid mapping results of Aitman et al. (13).
Southern blot analysis showed that the SHR strain is missing the Cd36 bands present in both the BN progenitor strain and the SHR-chr.4 congenic strain (Figure 2). Southern blot analysis of DNA from the stroke-prone SHR strain revealed a pattern distinctly different from that observed in the SHR progenitor strain (Figure 2). Analysis of the HinfI RFLP described by Aitman et al. (13) also confirmed that the stroke-prone SHR strain did not inherit the deletion variant of Cd36 carried by the SHR (data not shown). In contrast, SHR obtained from 3 different sources (Charles River Laboratories, Taconic Farms, and the Czech Academy of Sciences) were found to harbor the Cd36 deletion variant reported by Aitman et al. (data not shown).
Figure 2.
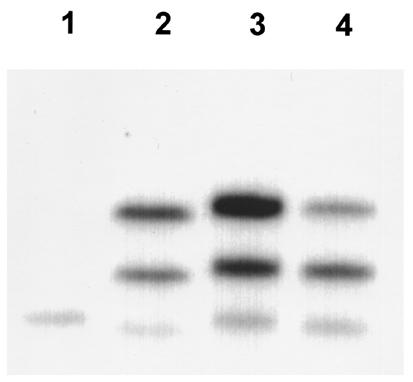
Southern blot analysis of Cd36. The Cd36 deletion variant is present in the SHR strain (note missing bands in lane 1) but not in the BN (lane 2), SHR.BN-Il6/Npy congenic (lane 3), or stroke-prone SHR (lane 4) strains.
Hemodynamic analysis.
The weekly daytime and nighttime systolic blood pressures in the SHR progenitor and SHR-chr. 4 congenic strain are shown in Figure 3a. Both daytime and nighttime systolic blood pressures were significantly lower in the SHR congenic strain than in the progenitor strain. The least square means for daytime systolic blood pressures in the SHR progenitor strain and SHR congenic strain were 170 ± 4 mmHg vs. 157 ± 3 mmHg, respectively (P < 0.025); and for nighttime systolic blood pressures, 173 ± 3 vs. 159 ± 3, respectively (P < 0.005). Diastolic and mean blood pressures were also lower in the congenic strain than in the progenitor strain, but the differences were not statistically significant (data not shown). Heart rates were similar between the SHR progenitor and congenic strains, despite the clear differences in systolic pressure (Figure 3b). The growth rates of the SHR congenic and progenitor strains were similar, although the mean body weight of the congenic strain tended to be slightly lower than that of the progenitor strain (Figure 4). However, after the first week of blood pressure measurements, the differences in body weight did not achieve statistical significance. Thus, the persistent differences in blood pressure between the SHR progenitor and congenic strains cannot be readily attributed to any major strain differences in body weights. The magnitude of the difference in blood pressure between the SHR progenitor strain and SHR congenic strain was not affected by fructose feeding (data not shown).
Figure 3.

Blood pressures and heart rates. Daytime and nighttime average systolic blood pressures (a) and heart rates (b) determined by radiotelemetry over a period of 4 weeks in the SHR progenitor strain and the SHR.BN-Il6/Npy congenic strain. Each data point represents the weekly daytime (open symbols) or nighttime (filled symbols) average blood pressure or heart rate (mean ± SEM) in the SHR progenitor strain (circles; n = 7) and the SHR.BN-Il6/Npy congenic strain (squares; n = 7). The sawtooth pattern reflects the circadian variation in blood pressure and heart rate. Systolic blood pressure was significantly lower in the SHR congenic strain than in the SHR progenitor strain during all daytime and nighttime periods. Heart rates were not significantly different between the 2 strains.
Figure 4.

Body weights. Mean body weights in the SHR progenitor strain (filled circles) and the SHR.BN-Il6/Npy congenic strain (open circles) determined over the 4-week period of blood pressure measurement. Mean body weight of the SHR progenitor strain was significantly lower than that of the SHR congenic strain at 11 weeks of age (*P < 0.05).
Risk-factor phenotyping.
Circulating levels of glucose, insulin, and lipids were similar between SHR progenitor rats and SHR-chr.4 congenic rats fed standard laboratory chow (data not shown). However, in SHR progenitor rats fed the high-fructose diet, serum insulin, triglyceride, and free fatty acid levels were much greater than in SHR congenic rats fed the high-fructose diet (Table 1). Serum glucose values were similar between the 2 groups fed the high-fructose diet. Figure 5 shows the ratios of serum insulin/glucose in SHR and SHR-chr.4 congenic rats before and after administration of the high-fructose diet. After fructose feeding, the insulin/glucose ratios were significantly increased (P < 0.001) in the SHR progenitor strain compared with the SHR-chr.4 congenic strain, owing to the marked hyperinsulinemia in the SHR progenitor strain. These metabolic changes were also accompanied by marked impairment in glucose tolerance in the SHR progenitor strain. Figure 6 shows the results of intraperitoneal glucose tolerance testing in the SHR progenitor and SHR-chr.4 congenic strain. Baseline glucose levels were similar in the 2 strains; however, after glucose loading, marked and prolonged hyperglycemia occurred in the SHR progenitor strain but not in the SHR-chr.4 congenic strain.
Table 1.
Serum levels of glucose, insulin, and lipids in rats fed a high-fructose diet

Figure 5.

Insulin/glucose ratios. Scatter plot showing serum insulin/glucose ratios in SHR progenitor rats (filled symbols; n = 9) and SHR.BN-Il6/Npy congenic rats (open symbols; n = 6) before and after 15 days on a high-fructose diet. Horizontal bars denote group means. After fructose feeding, insulin/glucose ratios were significantly greater in the SHR progenitor strain than in the SHR congenic strain (*P < 0.001).
Figure 6.

Glucose tolerance testing. Glucose tolerance test results in the SHR progenitor strain (circles; n = 9) and the SHR.BN-Il6/Npy congenic strain (squares; n = 6). Serum glucose levels were significantly greater in the SHR progenitor strain than in the SHR congenic strain at 30, 60, and 120 minutes after glucose loading (*P < 0.05).
Discussion
Glucose intolerance and hypertriglyceridemia are metabolic risk factors for cardiovascular disease and are often found in patients with essential hypertension (1, 2, 5). Evidence for disordered carbohydrate and lipid metabolism has also been reported in the SHR model of essential hypertension (4, 5, 9, 11). Despite extensive research, the basis for this clustering of cardiovascular risk factors remains a mystery. In a novel congenic strain of SHR, we have found that transfer of a single segment of chromosome 4 from the normotensive BN rat onto the SHR background can induce significant reductions in systolic blood pressure and ameliorate fructose-induced glucose intolerance, hyperinsulinemia, and hypertriglyceridemia. These findings demonstrate that a single chromosome region has the capacity to influence genetically a broad spectrum of systemic cardiovascular risk factors involved in the hypertension metabolic syndrome. The findings are consistent with the hypothesis that a set of linked genes, or perhaps even a single gene, on chromosome 4 can contribute to the clustering of hypertension and multiple metabolic risk factors for atherosclerotic cardiovascular disease.
Recent studies have established that a deletion in the Cd36 gene underlies defects in insulin sensitivity and catecholamine-stimulated lipolysis in isolated adipocytes from the SHR (13). Cd36 maps within the differential segment of the SHR-chr. 4 congenic strain and encodes a fatty acid receptor/transporter involved in the transmembrane transport of long-chain fatty acids. Transgenic overexpression of Cd36 has also been shown to induce decreases in circulating levels of triglycerides and fatty acids in FVB mice, although the effects of Cd36 overexpression on blood pressure and insulin sensitivity remain to be determined (13). The observations in SHR adipocytes and Cd36 transgenic mice, together with the current results in the SHR-chr. 4 congenic strain, strongly suggest that the SHR defect in Cd36 not only promotes disordered carbohydrate and lipid metabolism at the cellular level but also predisposes to systemic glucose intolerance, hyperinsulinemia, and hypertriglyceridemia. It is possible that the alterations in systemic glucose tolerance might involve mechanisms other than just insulin resistance per se, e.g., alterations in gluconeogenesis, tissue glucose delivery, etc. However, the previous studies in isolated SHR adipocytes strongly suggest that resistance to insulin-stimulated glucose uptake and alterations in cellular fatty acid metabolism might play at least some role in the pathogenesis of impaired glucose tolerance in this model (13). Abnormalities in lipid metabolism have been proposed to contribute to impaired insulin sensitivity and the pathogenesis of insulin-resistance disorders such as non–insulin-dependent diabetes (20). In the SHR, further studies will be required to determine whether the alterations in fatty acid and triglyceride metabolism induced by mutant Cd36 contribute to the pathogenesis of systemic glucose intolerance by promoting insulin resistance in skeletal muscle.
In the current studies, we have found that the capacity of chromosome 4 to influence metabolic risk factors for cardiovascular disease in the SHR is environmentally determined. Circulating levels of glucose, insulin, triglycerides, and fatty acids determined on single serum samples obtained during the day were similar between the SHR progenitor and congenic strains fed standard laboratory chow. In contrast, marked differences in glucose tolerance and circulating levels of insulin, triglycerides, and fatty acids were observed between SHR progenitor and congenic rats fed a high-fructose diet. These observations are consistent with a role for environment-genotype interaction in determining cardiovascular risk, and suggest that Cd36 deficiency can influence susceptibility to dietary-induced exacerbation of common risk factors for atherosclerotic heart disease. Given that Cd36 may serve as a receptor for HDL, LDL, and VLDL, it will be of interest to determine whether changes in the dietary intake of fat can also exert different effects on systemic cardiovascular risk factors in the SHR progenitor strain versus the SHR-chr. 4 congenic strain (21).
Based on the differences in blood pressure between the SHR progenitor and SHR congenic strains, it appears that the segment of chromosome 4 trapped in the SHR.BN-Il6/Npy strain may account for 15–20% of the systolic hypertension in the SHR-BN model. This effect is comparable to the effects of other blood pressure QTLs mapped in the SHR either by linkage analysis or by chromosome transfer studies in congenic strains (16, 22–26). Because multiple genes are involved in the pathogenesis of increased blood pressure in the SHR, the effect of this single chromosome region on systolic blood pressure is relatively substantial. A number of interesting candidate genes for hypertension are linked to Cd36 within the differential chromosome segment of the SHR congenic strain that includes the genes for endothelial nitric oxide synthase, leptin, and neuropeptide Y. By subdividing the target segment of chromosome 4 in multiple congenic sublines, it will be possible to test whether the clustering of metabolic risk factors with increased blood pressure depends on linkage between Cd36 and other genes trapped within the differential segment of the SHR congenic strain.
Although Cd36 deficiency appears to be a likely determinant of disordered carbohydrate and lipid metabolism in the SHR, the role of defective Cd36 in the pathogenesis of spontaneous hypertension is a far more open question. In demonstrating that the SHR congenic strain has significantly lower blood pressure than the SHR progenitor strain, we have clearly established that the transferred segment of chromosome 4 is involved in the genetic control of hypertension in the SHR-BN model. However, in the absence of supporting studies of the hemodynamic effects of Cd36 in transgenic or knockout animals, it is premature to conclude that the SHR deletion in Cd36 contributes to the effects of this region of chromosome 4 on the pathogenesis of hypertension.
In the current studies, we have found that SHR from several different colonies carry the deletion variant in Cd36. However, genotype testing using Southern blot analysis indicates that the stroke-prone strain of SHR did not inherit the SHR deletion variant of Cd36. This finding might suggest that Cd36 itself is not the blood pressure QTL trapped in the differential segment of the SHR-chr. 4 congenic strain. According to reports describing the derivation of the SHR and the stroke-prone SHR strains, the hypertension in the original SHR line was largely fixed by the F5 generation of selective inbreeding (27). However, the stroke-prone subline did not branch off from the original SHR line until after the F10 generation (28). Had the SHR deletion variant in Cd36 been important in the genetic evolution of hypertension in the SHR, it would have been fixed during the initial process of selective inbreeding for hypertension, which was essentially complete by the F5 generation, i.e., before splitting off of the stroke-prone subline. If so, the stroke-prone SHR subline would have inherited the deletion variant in Cd36 from the SHR. However, the fact that the stroke-prone SHR did not inherit the SHR deletion variant of Cd36 suggests that this gene was not particularly important in the original process of selection for hypertension in the SHR. Of course, the subsequent inbreeding of the SHR over multiple generations may have resulted in the fixation of additional genes (e.g., the Cd36 deletion variant) that further increased blood pressure in the SHR strain. That the stroke-prone SHR did not inherit the SHR deletion variant does not exclude the possibility that Cd36 deficiency might contribute to hypertension at some level. Nevertheless, the current findings, together with the published genealogy of the SHR and stroke-prone SHR strains, appear to indicate that the deletion in Cd36 did not play a key role in the selection for increased blood pressure that occurred during the early derivation of the SHR.
Recently, Furukawa et al. (12) reported that SHR (SHR/Izm) obtained from a commercial supplier in Izumo, Japan, displayed insulin resistance when compared with WKY rats (WKY/Izm) obtained from the same source. In contrast, SHR (SHR/NCrj) obtained from a commercial source in Tokyo did not show evidence of insulin resistance when compared with WKY rats (WKY/NCrj) obtained from the Tokyo source (12). That SHRs from both Izumo and Tokyo exhibit hypertension, whereas only SHRs from Izumo exhibit insulin resistance, suggests that spontaneous hypertension per se is not inextricably related to a genetic predisposition to insulin resistance. Although we have found evidence of Cd36 deletions in SHRs from 3 different sources, it is possible that other colonies of SHRs exist that do not harbor deletions in Cd36. Given the well-known genetic heterogeneity among normotensive WKY rats from different sources, it is also conceivable that some WKY sublines exist that carry deletions in Cd36 (29). It remains to be determined whether genetic variation in insulin sensitivity among SHRs from different sources is related to genetic variation in Cd36.
In a novel SHR congenic strain, we have succeeded in isolating a single chromosome region that is involved in the genetic regulation of multiple cardiovascular risk factors, including glucose intolerance, hypertriglyceridemia, and hypertension. This chromosome region includes the SHR deletion variant in Cd36 responsible for defective carbohydrate and lipid metabolism in isolated adipocytes. Given these observations, it will be of interest to determine whether genetic variants in Cd36 are associated with clustering of metabolic cardiovascular risk factors in humans. It may also be of interest to determine whether genes linked to Cd36 on human chromosome 7q are involved in the pathogenesis of human essential hypertension. Finally, the current results indicate that genetic dissection techniques in congenic strains can provide a powerful approach for investigating the basis for risk factor clustering in spontaneous hypertension. These findings should also serve to motivate the development of congenic sublines and knockout models to investigate further the relationship of defective Cd36 to the clustering of systemic cardiovascular risk factors and the pathogenesis of spontaneous hypertension.
Acknowledgments
This work was supported by grants from the Grant Agency of the Czech Republic (306/97/0521 and 204/98/K015 to M. Pravenec) and from the National Institutes of Health (NIH; RO1 HL-56028 and PO1 HL-35018 to T. Kurtz, and HL-18575 to R.C. Webb). M. Pravenec is an International Research Scholar of the Howard Hughes Medical Institute, and E.M. St. Lezin is a recipient of a Mentored Clinical Scientist Award from the NIH National Heart, Lung, and Blood Institute.
References
- 1.Reaven GM, Hoffman BB. Hypertension as a disease of carbohydrate and lipoprotein metabolism. Am J Med. 1989;87(Suppl. 6A):2S–6S. doi: 10.1016/0002-9343(89)90488-9. [DOI] [PubMed] [Google Scholar]
- 2.Reaven GM, Lithell H, Landsberg L. Hypertension and associated metabolic abnormalities—the role of insulin resistance and the sympathoadrenal system. N Engl J Med. 1996;334:374–381. doi: 10.1056/NEJM199602083340607. [DOI] [PubMed] [Google Scholar]
- 3.Facchini F, Chen YD, Clinkingbeard C, Jeppesen J, Reaven GM. Insulin resistance, hyperinsulinemia, and dyslipidemia in nonobese individuals with a family history of hypertension. Am J Hypertens. 1992;5:694–699. doi: 10.1093/ajh/5.10.694. [DOI] [PubMed] [Google Scholar]
- 4.Reaven GM, Chang H. Relationship between blood pressure, plasma insulin and triglyceride concentration, and insulin action in spontaneous hypertensive and Wistar-Kyoto rats. Am J Hypertens. 1991;4:34–38. doi: 10.1093/ajh/4.1.34. [DOI] [PubMed] [Google Scholar]
- 5.Reaven GM. Insulin resistance, hyperinsulinemia, hypertriglyceridemia, and hypertension. Parallels between human disease and rodent models. Diabetes Care. 1991;14:195–202. doi: 10.2337/diacare.14.3.195. [DOI] [PubMed] [Google Scholar]
- 6.Shamiss A, Carroll J, Rosenthal T. Insulin resistance in secondary hypertension. Am J Hypertens. 1992;5:26–28. doi: 10.1093/ajh/5.1.26. [DOI] [PubMed] [Google Scholar]
- 7.Reaven GM, Chang H, Hoffman BB, Azhar S. Resistance to insulin-stimulated glucose uptake in adipocytes isolated from spontaneously hypertensive rats. Diabetes. 1989;38:1155–1160. doi: 10.2337/diab.38.9.1155. [DOI] [PubMed] [Google Scholar]
- 8.Aitman TJ, et al. Quantitative trait loci for cellular defects in glucose and fatty acid metabolism in hypertensive rats. Nat Genet. 1997;16:197–201. doi: 10.1038/ng0697-197. [DOI] [PubMed] [Google Scholar]
- 9.Mondon CE, Reaven GM. Evidence of abnormalities of insulin metabolism in rats with spontaneous hypertension. Metabolism. 1988;37:303–305. doi: 10.1016/0026-0495(88)90127-8. [DOI] [PubMed] [Google Scholar]
- 10.Horl WH, Schaefer RM, Heidland A. Abnormalities of carbohydrate metabolism in spontaneously hypertensive rats. Klin Wochenschr. 1988;66:924–927. doi: 10.1007/BF01728956. [DOI] [PubMed] [Google Scholar]
- 11.Rao RH. Insulin resistance in spontaneously hypertensive rats. Difference in interpretation based on insulin infusion rate or on plasma insulin in glucose clamp studies. Diabetes. 1993;42:1364–1371. doi: 10.2337/diab.42.9.1364. [DOI] [PubMed] [Google Scholar]
- 12.Furukawa LN, et al. Variations in insulin sensitivity in spontaneously hypertensive rats from different sources. Metabolism. 1998;47:493–496. doi: 10.1016/s0026-0495(98)90229-3. [DOI] [PubMed] [Google Scholar]
- 13.Aitman TJ, et al. Identification of Cd36 (Fat) as an insulin-resistance gene causing defective fatty acid and glucose metabolism in hypertensive rats. Nat Genet. 1999;21:76–83. doi: 10.1038/5013. [DOI] [PubMed] [Google Scholar]
- 14.Bottger A, et al. Quantitative trait loci influencing cholesterol and phospholipid phenotypes map to chromosomes that contain genes regulating blood pressure in the spontaneously hypertensive rat. J Clin Invest. 1996;98:856–862. doi: 10.1172/JCI118858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kovacs P, Kloting I. Quantitative trait loci on chromosomes 1 and 4 affect lipid phenotypes in the rat. Arch Biochem Biophys. 1998;354:139–143. doi: 10.1006/abbi.1998.0686. [DOI] [PubMed] [Google Scholar]
- 16.Katsuya T, et al. A neuropeptide Y locus on chromosome 4 cosegregates with blood pressure in the spontaneously hypertensive rat. Biochem Biophys Res Commun. 1993;192:261–267. doi: 10.1006/bbrc.1993.1408. [DOI] [PubMed] [Google Scholar]
- 17.Rat Genome Center. http://www.genome.wi.mit.edu/rat/public/.
- 18.Kazdová L, Zák A, Vrána A. Increased lipoprotein oxidability and aortic lipid peroxidation in an experimental model of insulin resistance syndrome. Ann NY Acad Sci. 1997;827:521–525. doi: 10.1111/j.1749-6632.1997.tb51863.x. [DOI] [PubMed] [Google Scholar]
- 19.Horky K, et al. Metabolic, humoral and haemodynamic characteristics of normotensive offspring from hypertensive families. J Hum Hypertens. 1996;10(Suppl. 3):S85–S87. [PubMed] [Google Scholar]
- 20.McGarry JD. What if Minkowski had been ageusic? An alternative angle on diabetes. Science. 1992;258:766–770. doi: 10.1126/science.1439783. [DOI] [PubMed] [Google Scholar]
- 21.Calvo D, Gomez–Coronado D, Suarez Y, Lasuncion MA, Vega MA. Human CD36 is a high affinity receptor for the native lipoproteins HDL, LDL, and VLDL. J Lipid Res. 1998;39:777–788. [PubMed] [Google Scholar]
- 22.St. Lezin E, et al. Genetic isolation of a chromosome 1 region affecting blood pressure in the spontaneously hypertensive rat. Hypertension. 1997;30:854–859. doi: 10.1161/01.hyp.30.4.854. [DOI] [PubMed] [Google Scholar]
- 23.Kren V, et al. Genetic isolation of a region of chromosome 8 that exerts major effects on blood pressure and cardiac mass in the spontaneously hypertensive rat. J Clin Invest. 1997;99:577–581. doi: 10.1172/JCI119198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pravenec M, et al. Mapping of quantitative trait loci for blood pressure and cardiac mass in the rat by genome scanning of recombinant inbred strains. J Clin Invest. 1995;96:1973–1978. doi: 10.1172/JCI118244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Samani NJ, et al. A gene differentially expressed in the kidney of the spontaneously hypertensive rat cosegregates with increased blood pressure. J Clin Invest. 1993;92:1099–1103. doi: 10.1172/JCI116616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schork NJ, et al. A biometrical genome search in rats reveals the multigenic basis of blood pressure variation. Genome Res. 1995;5:164–172. doi: 10.1101/gr.5.2.164. [DOI] [PubMed] [Google Scholar]
- 27.Okamoto K, Aoki K. Development of a strain of spontaneously hypertensive rats. Jpn Circ J. 1963;27:282–293. doi: 10.1253/jcj.27.282. [DOI] [PubMed] [Google Scholar]
- 28.Okamoto, K., Yamori, Y., and Nagaoka, A. 1974. Establishment of the stroke-prone spontaneously hypertensive rat (SHR). Circ. Res. IV: (Suppl. I):I-143–I-153.
- 29.Kurtz TW, Montano M, Chan L, Kabra P. Molecular evidence of genetic heterogeneity in Wistar-Kyoto rats: implications for research with the spontaneously hypertensive rat. Hypertension. 1989;13:188–192. doi: 10.1161/01.hyp.13.2.188. [DOI] [PubMed] [Google Scholar]
- 30.Jacob HJ, et al. A genetic linkage map of the laboratory rat, Rattus norvegicus. Nat Genet. 1995;9:63–69. doi: 10.1038/ng0195-63. [DOI] [PubMed] [Google Scholar]
- 31.Pravenec M, et al. A genetic linkage map in the rat derived from recombinant inbred strains. Mamm Genome. 1996;7:117–127. doi: 10.1007/s003359900031. [DOI] [PubMed] [Google Scholar]