

Abstract
We have studied complex I (NADH-ubiquinone reductase) defects of the mitochondrial respiratory chain in 2 infants who died in the neonatal period from 2 different neurological forms of severe neonatal lactic acidosis. Specific and marked decrease in complex I activity was documented in muscle, liver, and cultured skin fibroblasts. Biochemical characterization and study of the genetic origin of this defect were performed using cultured fibroblasts. Immunodetection of 6 nuclear DNA–encoded (20, 23, 24, 30, 49, and 51 kDa) and 1 mitochondrial DNA–encoded (ND1) complex I subunits in fibroblast mitochondria revealed 2 distinct patterns. In 1 patient, complex I contained reduced amounts of the 24- and 51-kDa subunits and normal amounts of all the other investigated subunits. In the second patient, amounts of all the investigated subunits were severely decreased. The data suggest partial or extensive impairment of complex I assembly in both patients. Cell fusion experiments between 143B206 ρ° cells, fully depleted of mitochondrial DNA, and fibroblasts from both patients led to phenotypic complementation of the complex I defects in mitochondria of the resulting cybrid cells. These results indicate that the complex I defects in the 2 reported cases are due to nuclear gene mutations.
Introduction
Mitochondrial cytopathies are involved in an increasing number of different clinical phenotypes. These diseases are associated with symptoms that affect various organs or tissues that are particularly dependent on oxidative metabolism, such as the central nervous system, sensory organs, heart and skeletal muscle, and less frequently, kidney, liver, and endocrine glands (1–3). Mitochondrial cytopathies are thought to be associated with reduced energy production through the oxidative phosphorylation process supported by the enzymatic complexes (complexes I–V) located in the inner mitochondrial membrane. Subunits of complexes I, III, IV, and V are encoded either by the mitochondrial DNA (mtDNA) or, for the majority, by the nuclear DNA (nDNA). Complex II subunits are encoded by nuclear genes only. Complex I (NADH-ubiquinone reductase) is a multimeric assembly of 7 mitochondrial-encoded subunits (ND subunits) and at least 36 nuclear-encoded subunits.
Numerous clinical syndromes have been associated with complex I deficiency, ranging from lethal neonatal forms to neurodegenerative disorders in adult life, including Leber’s hereditary optic neuropathy (LHON) and Parkinson’s disease (4–8). In neonates and infants, isolated complex I deficiencies have been sorted into several different clinical phenotypes with initial lactic acidosis (9). Recently, a new phenotype of complex I deficiency was described in infants with fatal progressive macrocephaly, hypertrophic cardiomyopathy, and lactic acidosis (10). In a number of pathological cases, immunochemical analyses have revealed heterogeneity of complex I deficiency with disproportionate loss of 1 or several subunits (5, 11–16).
At the molecular level, complex I defects can originate from mutations in either nuclear or mitochondrial DNA genes. Mutations in the mtDNA genes were first reported in association with LHON (4, 17). Recently, a 5-bp duplication in the gene coding for the 18-kDa subunit and point mutations in the genes encoding the 23- and 51-kDa subunits of complex I were detected in 5 different complex I–deficient patients, presenting either a multisystemic progressive phenotype or Leigh’s syndrome (18–20). However, the consequences of these mutations remain to be characterized. Mutations that are responsible for complex I defects have yet to be identified for a large majority of complex I–deficient patients.
Here, we present 2 different biochemical phenotypes in 2 patients with fatal infantile lactic acidosis (FILA) associated with isolated complex I deficiency, and we demonstrate the genetic origin of both forms of the disease. This was addressed by immunodetecting individual complex I subunits with specific antibodies and by assessing complex I activity in patients’ fibroblasts and in transnuclear cybrid cells obtained after fusion between patients’ fibroblasts and mtDNA-less cells (ρ° cells). Reexpression of complex I subunits and recovery of complex I activity in patients’ mitochondria after transnuclear complementation by ρ°-cell nuclei enabled us to infer the nDNA origin of both defects. This is the first demonstration of the nDNA origin of complex I defects in FILA. Moreover, the 2 distinct patterns of complex I subunit composition in patients’ cells suggest that at least 2 different nuclear gene abnormalities may be responsible for the reported phenotypes.
Methods
Case reports.
Patient no. 1 was a boy born after an uncomplicated full-term pregnancy. He was the second child of healthy, unrelated parents. The first child, a boy, is in good health. There was no past medical history over 3 generations in this family. In the first 24 hours of life, the newborn developed generalized hypotonia with poor gesticulation. At admission, on day 2, he was very floppy with poor response to painful stimuli. He rapidly showed symptoms of respiratory failure and was intubated and ventilated. There was no clinical malformation. Hepatic enlargement was noticed, and the chest x-ray revealed a slight cardiomegaly. Cranial ultrasonography showed a brain edema. The first electroencephalography (EEG) showed status epilepticus with a paroxysmal pattern. A severe lactic acidosis was detected (20 mmol/L; normal <2.2). The lactate/pyruvate ratio (51:1; normal <20:1) and the 3-hydroxybutyrate/acetoacetate ratio (8.3:1 under fed condition; normal <2:1) were high in plasma, suggesting an abnormal oxidoreduction status. Under mechanical ventilation, bicarbonate supplementation, and therapy with L-carnitine, biotine, thiamine, and dichloropropionate, there was a transient improvement in clinical state, ultrasound brain edema, and EEG pattern. At 11 days of life, despite therapy, the child went into a deep coma and died. Muscle, liver, and skin biopsies were obtained with informed parental consent. Light microscopy, only performed on the liver biopsy, showed slight inflammation of portal tracts associated with diffuse microvesicular and macrovesicular steatosis. Autopsy was not performed.
Patient no. 2 was a boy who was the second child of healthy, unrelated parents whose first child, a girl, is in good health. Analysis of the family history over 4 generations revealed that there was no other affected member. This boy was born by normal delivery after an uncomplicated full-term pregnancy. In the first 2 weeks of life, he vomited frequently, and at 5 weeks, when he was admitted, his neurological status deteriorated with hypotonia, weakness, and lethargy. In the first month, the head circumference was noticed to be rapidly increasing from 33 cm to 40 cm (from 10th to 95th percentile). Computer tomographic scan showed a very hypodense brain with increased brain volume and extensive cerebral edema. EEG showed a burst suppression pattern. Laboratory investigations revealed a marked metabolic acidosis with hyperlactacidemia (13 mmol/L; normal <2.2), and a high lactate/pyruvate ratio in plasma (44:1; normal <20:1). Under fed condition, hyperketonemia and a high 3-hydroxy-butyrate/acetoacetate ratio (18.9:1; normal <2:1) were also detected. Cerebrospinal fluid lactate was increased (10.7 mmol/L; normal <1.8). Urinary organic acids showed a marked increase in excretion of lactate and Krebs cycle intermediates (fumarate, succinate, and α-ketoglutarate). Despite intensive care, the neurological state worsened rapidly and brain death occurred at 6 weeks of age. Autopsy showed acute necrotizing encephalopathy, but no hypertrophic cardiomyopathy was noticed. Muscle, liver, and skin biopsies were performed just before death with informed parental consent. Histochemistry examinations were normal in muscle, and electron microscopy only revealed slightly enlarged mitochondria. Light and electron microscopy were normal in liver, except for a mild microvesicular steatosis.
Cell lines and culture conditions.
Fibroblasts from forearm skin explants obtained from both patients and controls were cultured in high-glucose DMEM supplemented with 10% FBS, 100 μg/mL pyruvate, and 50 μg/mL uridine in a 5% CO2 atmosphere at 37°C. The 143B TK– osteosarcoma cell line (CRL 8303) was obtained from the American Type Culture Collection (Rockville, Maryland, USA). The daughter 143B206 ρ° cells, completely depleted of mtDNA by long-term exposure to ethidium bromide (21), were grown in supplemented DMEM, as already described here, with 100 μg/mL 5-bromo-2′-deoxyuridine (BrdU). This cell line was provided by G. Attardi (California Institute of Technology, Pasadena, California, USA). Lactate and pyruvate were measured in cultured fibroblasts after 1 hour of incubation with glucose, as reported (14).
Cell fusion.
Trypsinized cells were collected by low-speed centrifugation, resuspended in DMEM, and counted. Fibroblasts (106 cells) were mixed with an equal number of ρ° cells, and culture medium was carefully eliminated by centrifugation. Cells were resuspended for 1 minute in 0.2 mL of sterile polyethylene-glycol 1500 (50% wt/vol) (Roche Diagnostics, Meylan, France). After cell fusions, screening of transnuclear complemented cells was performed by growing cells in standard DMEM for 24 hours and then in selective medium composed of pyruvate- and uridine-free DMEM supplemented with 10% dialyzed FBS and 100 μg/mL BrdU. After 10 days, about 30 independent colonies were observed.
Preparation of mitochondria.
Mitochondria-enriched preparations were obtained as described (22), with slight modifications. Exponentially growing cells were trypsinized, washed, and resuspended in medium A composed of 0.25 M sucrose, 1 mM EDTA, 10 mM Tris-HCl (pH 7.3), and 1 mg/mL skimmed milk. Cells were gently broken in a glass pestle Dounce homogenizer, and the homogenate was centrifuged for 8 minutes at 800 g. The supernatant was then centrifuged 10 minutes at 11,000 g. Pellets were resuspended in medium A without milk, and the resulting mitochondria-enriched suspension was used for enzymatic measurements and immunodetection. Protein concentrations were determined by the bicinchoninic acid method.
Enzymatic measurements.
Spectrophotometric analysis of the respiratory chain complexes was performed in an 800-g supernatant of crude homogenates from liver or muscle (23) and in mitochondrial preparations from cultured cells. At a protein concentration of 1–2 mg/mL, mitochondria were disrupted by 3 freeze-thaw cycles in liquid nitrogen and were briefly sonicated. In tissue homogenates, this freeze-thaw method was used only for the measurement of complex I activity.
The following enzymatic activities were assayed at 30°C: citrate synthase, NADH-ubiquinone reductase (complex I), succinate-cytochrome c reductase (complex II + III), succinate dehydrogenase (SDH), succinate-ubiquinone reductase (complex II), ubiquinol-cytochrome c reductase (complex III), and cytochrome c oxidase (complex IV). Methodology was as reported previously (23), with slight modifications (24); for complex II measurement, methodology was according to Rustin et al. (22). Complex I activity was assayed in 55 mM phosphate buffer (pH 7.5) containing 2.5 g/L defatted BSA, 1.8 mM NaCN, 2 μg antimycine , and 0.1 mM decylubiquinone. The reaction was initiated with 0.2 mM NADH, and NADH oxidation was monitored at 340 nm. Rotenone-sensitive complex I activity was calculated from the difference between 2 parallel rates measured with and without 5 μM rotenone.
Immunodetection.
Mitochondrial proteins (5 μg) were separated on 12% polyacrylamide gels, electroblotted onto PVDF membranes, and incubated with antibodies as described (25). Decrease in subunits in patients’ mitochondria was estimated by comparing the signal yielded by different dilutions of the patients’ samples to the signal obtained for equivalent samples of control fibroblasts. Antibodies were raised in rabbit against peptides corresponding to the major complex I subunits (26, 27) and the cytochrome c oxidase subunit II (COII). The anti–24 kDa bovine subunit antibody was a gift from M. Skehel and J. Walker (Medical Research Council, Cambridge, United Kingdom).
Electron microscopy.
Confluent cells on glass slides were fixed and embedded as described (28). After staining by uranyl acetate and lead citrate, ultrathin sections (90 nm) were examined with a JEOL 1200EX II electron microscope at 80 kV.
DNA and RNA analysis.
Total DNA prepared from cells (27) was subjected to PCR amplification using appropriate primers and standard protocols. Total RNA was extracted from cultured cells harvested at a nonconfluent state by using the RNAgents kit from Promega Corp. (Madison, Wisconsin, USA). Northern blot analysis and RT-PCR were performed according to standard protocols. cDNA sequences are available through our MitoPick database (29). Nuclear genotypes were characterized by high polymorphic marker analysis. CA repeats from chromosomes 1, 3, 5, 7, and 19 (D1S306, D3S1281, D5S455, D7S849, and D19S220) were amplified using an appropriate set of primers (30) and were sized by electrophoresis. PCR fragments were sequenced by Genome Express SA (Grenoble, France).
Results
Identification of the complex I defects in tissues and cultured cells.
Enzymatic measurements of the respiratory chain complex activities revealed that complex I activity was decreased by more than 83% in muscle and liver of both patients (Table 1). In the muscle, the activities of the other complexes were within the control range, except for complex III and II + III activities, which were at the upper limit for patient no. 1. In the liver, the activities of complexes II, III, and IV were increased for both patients. This may reflect some metabolic adaptation to compensate for decreased energy production as a consequence of complex I deficiency. Citrate synthase activity was increased in the liver of both patients and was at the upper limit of the control range in the muscle of patient no. 1. Increase in citrate synthase activity has already been reported for respiratory chain disorders (31, 32) associated or not with deletion or depletion of mtDNA (33–35). As shown in Table 1, the values of the ratios comparing the activities of the different complexes are consistent with a severe complex I defect in both liver and muscle.
Table 1.
Mitochondrial enzyme activities in muscle and liver from both patients

In cultured fibroblasts from both patients, the lactate/pyruvate ratio was increased, suggesting a defect in electron transport through the respiratory chain (Table 2). Respiratory measurements showed that the rate of rotenone-sensitive oxidation of malate + glutamate (NADH oxidation) was decreased by more than 70% in mitochondria from both patients, whereas the succinate and cytochrome c oxidation rates were comparable to those of controls (not shown). Enzymatic measurements of the respiratory complex activities revealed normal values for complexes II, III, and IV, but a specific defect of complex I (Table 2). Residual rotenone-sensitive complex I activities were 16% and 10% for patients nos. 1 and 2, respectively. The reliability of the complex I activity measurements in our mitochondria-enriched preparations from fibroblasts was confirmed by the 70–86% rotenone sensitivity of the total NADH-ubiquinone reductase activity in control mitochondria. Together, these results demonstrated a multisystemic and severe decrease in complex I activity in both patients.
Table 2.
Biochemical measurements in cultured fibroblasts from both patients

Immunodetection of complex I subunits.
Immunoblotting was performed for 7 complex I subunits in fibroblast mitochondria, using specific antibodies directed against 1 mitochondrial-encoded subunit (ND1) and 6 nuclear-encoded subunits (20, 23, 24, 30, 49, and 51 kDa). In patient no. 1, most of the detected subunits were present in normal or near-normal amounts, except for the 24-kDa subunit, which was present at a very low level in the samples (less than 5% of the control), and the 51-kDa subunit, which consistently amounted to about 50% of the control (Figure 1). In patient no. 2, all the investigated nuclear-encoded subunits and the mitochondrial-encoded ND1 subunit were in trace amounts. In contrast, the levels of the COII subunit, encoded by the mtDNA, were normal compared with the control. This confirmed that comparable amounts of protein had been loaded into the gel lanes, and it highlighted the specific loss of complex I subunits in patient no. 2 mitochondria.
Figure 1.
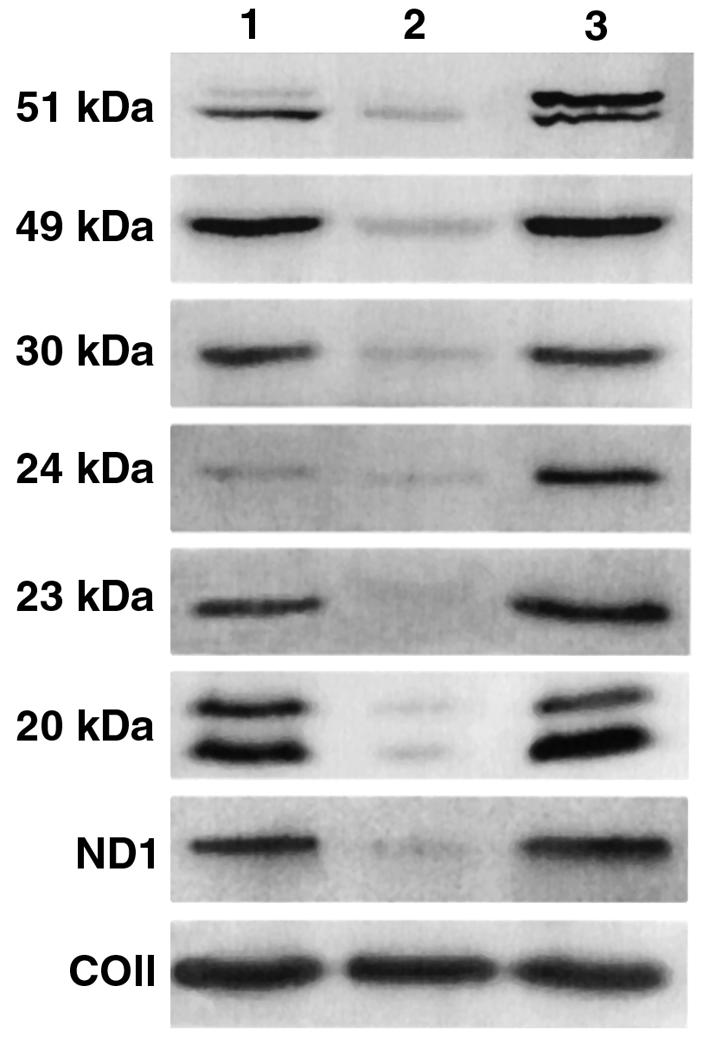
Immunological detection of complex I subunits in mitochondria from cultured fibroblasts. Proteins from mitochondrial preparations were analyzed by Western blot, with antibodies directed against the ND1 and 20-, 23-, 24-, 30-, 49-, and 51-kDa subunits of complex I, and the COII subunit. Note that under our electrophoretic conditions, the 20- and 51-kDa subunits are specifically detectable as doublets with their respective antibodies. Lane 1, mitochondria from patient no. 1; lane 2, mitochondria from patient no. 2; lane 3, mitochondria from controls. Identical amounts of protein (5 μg) were loaded.
Cell fusion and characterization of the resultant cells.
To demonstrate which part of the nuclear or mitochondrial genome was involved in the primary defect associated with complex I deficiency, patients’ fibroblasts were fused with ρ° cells fully depleted of mtDNA. The rationale for this experiment was based on the following hypotheses: (a) under appropriate conditions, fusion between cells would lead to the segregation of new cells (cybrid cells) in which patients’ mitochondria would be placed into a novel nuclear environment; (b) only patients’ cells with a nuclear defect could theoretically be transcomplemented by normal ρ°-cell nuclei; and (c) on the contrary, cybrid cells issued from patients’ cells harboring mtDNA defects would remain complex I deficient.
The growth capacity of the different cell lines in various media was assessed. The ρ° cells exhibited both auxotrophy for uridine and pyruvate (21) and resistance to BrdU, as they were obtained from the thymidine kinase–deficient (TK–) osteosarcoma 143B cell line (Figure 2). Unlike ρ° cells, patients’ fibroblasts exhibited BrdU sensitivity and were able to grow in DMEM lacking pyruvate and uridine. From these results, it was expected that none of the parental cells (ρ° cells and fibroblasts from patients or controls) could grow in a medium lacking pyruvate and uridine and containing BrdU. On the other hand, only cybrid cells that contained fibroblast mitochondria and were homozygous for the TK– nuclear marker of the ρ° cells were expected to survive in this medium. After fusion, selection of cybrids was carried out in the selective medium containing BrdU but lacking pyruvate and uridine (see Methods). After 15 days, 10 clones were collected and further analyzed. As expected, the different clones were able to grow in the selective medium, with growth kinetics similar to those of ρ° cells in a pyruvate- and uridine-supplemented medium (Figure 2).
Figure 2.

Growth kinetics of the cell types used in transnuclear complementation experiments. ρ° cells in DMEM + dialyzed FBS + BrdU (open squares); ρ° cells in DMEM + dialyzed FBS + BrdU + pyruvate + uridine (filled squares); fibroblasts from patient no. 1 in DMEM + dialyzed FBS + BrdU (open triangles); fibroblasts from patient no. 1 in DMEM + dialyzed FBS (filled circles); cybrid cells in DMEM + dialyzed FBS + BrdU (open circles). Concentrations used: dialyzed FBS, 10% (vol/vol); BrdU, 100 μg/mL; uridine, 50 μg/mL; pyruvate, 100 mg/mL. Control and fibroblasts from patient no. 2 essentially behaved the same as fibroblasts from patient no. 1 (not shown).
Morphological examination of parental (ρ° and fibroblasts) and cybrid cells was performed by electron microscopy. The ρ° cells contained enlarged spherical mitochondria with rare cristae and a poorly dense matrix (Figure 3b). This enlarged and rounded shape has already been observed in MRC5 ρ° fibroblasts (36). In contrast, both patients’ and control fibroblasts contained rod-shaped mitochondria with numerous cristae (Figure 3a, c, and d). Fifteen days after fusion, both types of mitochondria coexisted in fused cells, with a predominance of enlarged mitochondria from the ρ° parental cells (Figure 3e). After 2 additional weeks in culture, ρ° cell–type mitochondria disappeared from the cytoplasm, and rodlike mitochondria with cristae became largely predominant (Figure 3f). This is consistent with a previous observation (37) showing that after introduction of normal mitochondria into ρ° HeLa cells, all the ρ° mitochondria took normal shape within a few hours.
Figure 3.

Electron micrographs of the cultured cells. (a) Control fibroblasts. (b) ρ° cells. (c) Fibroblasts from patient no. 1. (d) Fibroblasts from patient no. 2. (e) BrdU-resistant cells obtained 15 days after fusion between ρ° cells and patient no. 2 fibroblasts. (f) The cybrid cell line shown in e, 4 weeks after fusion. Mitochondria are shown by arrowheads. Scale bar: 1 μm.
Finally, we examined both mitochondrial and nuclear genotypes of cybrid cells. The mitochondrial genotypes of the 143B cells and the 2 patients’ cells were characterized by amplifying and sequencing a PCR fragment ranging from mtDNA nucleotide 1 to 2000. Compared with the Cambridge sequence (38), the 3 resulting fragments were each unique, with specific sets of polymorphic changes. When aligned, the sequences of the mtDNA fragments obtained from the patients’ fibroblasts and the cybrid cells unambiguously revealed that the 2 types of cybrid cells contained only mtDNA from patients’ cells and no mtDNA of 143B cell origin (not shown). This was consistent with our finding of no PCR-detectable mtDNA in the ρ° cells, in agreement with previous observations (21). Nuclear genotypes of the parental cells (the ρ° cells and the 2 patients’ cells) were also characterized by assessing the size of a set of polymorphic markers present on different chromosomes. The patterns of these allelic markers differed between all parental cells. Only the allelic marker pattern found in the ρ° cells was evidenced in the cybrid cells (not shown). This clearly indicated that the nucleus in cybrid cells originated exclusively from ρ° cells.
Immunodetection of complex I and enzymatic measurements in cybrid cells.
Mitochondria-enriched preparations from patients’ cybrid cells were analyzed by immunoblotting. As a control, mitochondria prepared from ρ° cells and control fibroblasts were also investigated. Antibodies directed against the 23-, 24-, 30-, 49-, and 51-kDa subunits of complex I revealed that these subunits were present in comparable amounts in patients’ cybrid cells and in control fibroblasts (Figure 4, lanes 1, 4, and 6). Unexpectedly, these nuclear-encoded subunits were also found in appreciable amounts in ρ°-cell mitochondria. However, as expected, mitochondrial-encoded ND1 and COII subunits were absent in ρ°-cell mitochondria (Figure 4, lane 2). The presence of the 24-kDa and 51-kDa subunits in ρ°-cell mitochondria did not allow us to ascertain nuclear complementation of complex I in patient no. 1. In patient no. 2’s cybrid cells, the recovery of the ND1 subunit was a clear indication that complex I was present in the mitochondria (Figure 4, lane 6).
Figure 4.

Western blot analysis of mitochondrial proteins in transnuclear complemented cells. Experimental conditions were identical to those in Figure 1. Mitochondria were prepared from the following cell types: lane 1, control fibroblasts; lane 2, ρ° cells; lane 3, fibroblasts from patient no. 1; lane 4, patient no. 1 cybrid cells; lane 5, fibroblasts from patient no. 2; lane 6, patient no. 2 cybrid cells.
To ascertain whether cybrid cells contained fully assembled and active complex I, complex I activity and other respiratory chain complex activities were assayed in mitochondria-enriched preparations (Table 3). In the parental ρ° cells, activity of the 3 complexes containing mtDNA-encoded subunits (complexes I, III, and IV) was undetectable. In contrast, SDH and complex II activity was present in ρ° cells, in agreement with a previous report (39), and was found to be similar to that of the parental 143B cell line. These results were consistent with the fact that all the complex II subunits are nuclear encoded and that mitochondrial import of SDH subunits has been demonstrated in MRC5 ρ° fibroblasts (36). As shown previously (36, 39), citrate synthase activity was high in ρ° cells compared with the parental 143B cell line. Contrary to ρ° cells, control cybrid cells (obtained by fusion between ρ° cells and control fibroblasts) exhibited fully active respiratory complexes. In patients’ cybrid cells, activities of all respiratory chain enzymes were similar to those measured both in control cybrid cells and in the parental 143B cell line. In particular, the complex I activity, which was specifically and markedly decreased in the fibroblasts from both patients, appeared restored in cybrid cells. In addition, comparable ratios of enzyme activities were found in all cybrid cell types (Table 3). These results confirmed that fusion of the patients’ fibroblasts with ρ° cells concurred with the restoration of an active complex I.
Table 3.
Respiratory chain complex activities in ρ°, 143B parental, and cybrid cells

Transcriptional analysis and cDNA mutation screening.
As a first step toward characterizing the defects at the molecular level, we checked whether the genes coding for the major complex I subunits were actively transcribed. Steady-state levels of the transcripts were analyzed by Northern blot (Figure 5a), and then by RT-PCR (Figure 5b), because the nuclear transcripts were found in very limited amounts compared with mitochondrial ND transcripts in normal fibroblasts. Northern blot and RT-PCR experiments revealed similar steady-state levels of all the different transcripts in patients’ cells and normal cells. More specifically, this analysis failed to detect any reduction of the level of the 51- and 24-kDa transcripts in patient no. 1, or of the 51-, 49-, 30-, 24-, 23-, and 20-kDa and ND1 transcripts in patient no. 2, that may have correlated with the reduced amounts of corresponding proteins in the patients’ cells. Mutation screening was then performed by sequencing cDNAs coding for certain nuclear-encoded subunits. For both patients, the cDNAs coding for the 10-, 24- and 51-kDa subunits (these subunits constitute the flavoprotein fraction of the enzyme), the 18- and 23-kDa subunits, and the 7 mitochondrial ND genes were totally sequenced. The recently described (18–20) 5-bp duplication in the cDNA coding for the 18-kDa subunit, and the point mutations in the 23-kDa and 51-kDa subunits, were not found in either patient. We only detected a C→T transition in the 24-kDa cDNA from patient no. 1. This transition led to an Ala29→Val substitution in the presequence of the 24-kDa subunit and has been reported, at a homozygous state, as a susceptibility factor for Parkinson’s disease (40). Patient no. 1 was found to be heterozygous for this mutation, as were healthy individuals in the study of Hattori et al. (40).
Figure 5.

Transcription analysis of the genes coding for the complex I subunits in patients’ fibroblasts. (a) Northern blot of the ND mitochondrial transcripts and the 51- and 24-kDa nuclear transcripts. Twenty micrograms of total RNA were loaded in each lane. Subunit-specific cDNAs with a length ranging between 300 and 1,500 bp were radiolabeled and used as probes. Probing the 18S rRNA with an 18S rRNA gene fragment was used as a quantitative reference. Detection was performed by autoradiography. Lane 1, RNA from controls; lane 2, RNA from patient no. 1; lane 3, RNA from patient no. 2. Transcript names are indicated to the left. (b) RT-PCR detection of the nuclear-encoded transcripts. PCR experiments were performed under conditions that ensured that amplifications of 2 different concentrations of control RNAs gave unsaturated and proportional signals when samples were analyzed by electrophoresis in ethidium bromide–stained agarose gels. Lanes 1 and 2, control RNA (starting amounts: 24 ng and 48 ng, respectively); lane 3, patient no. 1 RNA (starting amount: 48 ng); lane 4, patient no. 2 RNA (starting amount: 48 ng).
Discussion
In this report, cell fusion between fibroblasts from 2 complex I–deficient patients presenting with FILA and ρ° cells were carried out to determine which of the 2 genomes — nDNA or mtDNA — was involved in these respiratory chain defects. Our data demonstrated that in both patients, the complex I defects arose from nuclear mutations. Biochemical investigations in tissues and cultured fibroblasts demonstrated severe and selective complex I dysfunctions as the probable causes of the rapid neurological deterioration and early death of both patients. Specific antibodies against nuclear- and mitochondrial-encoded subunits were used to analyze the subunit composition of complex I in patients’ mitochondria. Despite the lack of a comprehensive analysis (only 7 of the numerous subunits that compose complex I were tested), we were able to distinguish 2 different types of complex I misassembly. Patient no. 1 was deficient in 24-kDa and 51-kDa subunits, whereas in patient no. 2, all the investigated subunits were in very low amounts.
Cell fusions between ρ° cells and enucleated cells have already been used to study the impact of mtDNA mutations on the cellular energy production (21, 41, 42). This approach was also successfully used to show that nuclear complementation of fibroblasts with mtDNA depletion restored the mtDNA level, providing evidence for a nuclear-encoded factor involved in mtDNA depletion (43). Tiranti et al. (39) elegantly demonstrated the nDNA origin of cytochrome c oxidase deficiency in Leigh’s syndrome by generating 2 different transmitochondrial cybrid cell lines. The first was obtained by fusing ρ° cells with enucleated patient’s fibroblasts, and the second, by fusing ρ° transformant fibroblasts derived from the same patient with nDNA-less cytoplasts. Recently, analysis of cybrids obtained after fusion between platelets and ρ° cells suggested that the complex I defect in focal dystonia is not caused by a mtDNA mutation (44). Here, we undertook a straightforward approach by fusing whole cells, rather than a combination of nucleated and enucleated cells, to ascertain whether the structural and functional abnormalities of complex I in the 2 cases of FILA were under the control of the nuclear genome. Another approach involving fusion of whole cells was successfully used by Isobe et al. (45) to study the inheritance modes of cytochrome c oxidase and succinate dehydrogenase deficiencies.
In the present work, this approach raised a number of questions concerning the nature of the cybrids screened in the selective medium containing BrdU. The possibility that the cybrid cells may in fact be 143B cells was considered, as the metabolic selection imposed by the restrictive medium (without pyruvate and uridine) might have favored the segregation of 143B cells with a restored amount of mtDNA. This hypothesis was ruled out by the exclusive presence, in cybrid cells, of mtDNA originating from the patients’ fibroblasts. Moreover, the cybrids were unlikely to be spontaneous TK– patients’ fibroblasts, as the restoration of complex I activity in the cybrids also implied that these cells would have simultaneously acquired mutations correcting the complex I defect. The probability for such an event would seem to be very low. The origin of the nucleus in the cybrids was determined by a polymorphic marker analysis. This revealed that only the 143B206 ρ°-cell chromosomes were maintained in the cybrid cells. In addition, the absence of heterokaryons and the presence, after fusion, of small numbers of mitochondria with cristae suggested that cybrid cells may have resulted from partial mixing of cytoplasm from patients’ fibroblasts into ρ° cells and that nuclear fusion did not take place. This contrasts with the cybrids obtained by fusion between ρ° HeLa cells and fibroblasts, which contained a nuclear genome derived from both parental cells (45).
The effective transnuclear complementation of complex I deficiency indicated that in both patients, the complex I defects resulted from nuclear gene mutations and not from mtDNA mutations. However, it could be argued that the complex I defects may have arisen from a heteroplasmic mtDNA mutation and that, after fusion, only mitochondria with wild-type mtDNA prevailed in the cybrids. Because the 2 patients reported here showed a severe degree of complex I inactivation, the hypothesis of a disease-associated mtDNA mutation would presuppose a high percentage of mutated mtDNA. To test this hypothesis, we made cybrids with fibroblasts from a patient (different from patients nos. 1 and 2) harboring high levels (95%) of the T8993G mtDNA mutation (46). The same degree of heteroplasmy was found in these new cybrid clones (not shown), suggesting that fusion with ρ° cells did not promote segregation of wild-type mitochondria in our situation.
Sequencing of the ND and tRNA mtDNA genes failed to detect any mutations in the 2 patients compared with the consensus sequence (38) and the referenced polymorphic sites (17). Moreover, the A→G mutation in tRNALeu(UUR) at position 3302 (i.e., 5 bp upstream from the ND1 gene), related to severe complex I deficiency and abnormal mRNA processing (47), was not found in either patient. These findings gave additional support to an nDNA origin of the 2 reported complex I defects. Finally, analysis of the steady-state level of mRNAs coding for major mitochondrial- and nuclear-encoded subunits indicated that complex I deficiency did not correlate with an impaired transcription of any of the investigated genes.
In patient no. 1, the amount of the 24-kDa subunit, and to a lesser extent of the 51-kDa subunit, was reduced. Lowered level of the 24-kDa subunit (isolated or not) has already been described in some patients with complex I deficiency (5, 12, 48). In complex I, the 10-, 24-, and 51-kDa subunits closely interact to constitute the flavoprotein (FP) fraction of the enzyme. Therefore, the presence of a reduced amount of the 24- and 51-kDa subunits in patient no. 1’s mitochondria may be the consequence of a misassembly of the FP fraction. However, because no specific antibody against the 10-kDa subunit was available, the presence or absence of this subunit in the mitochondria from patient no. 1 could not be tested. At the molecular level, no mutation was found in the cDNAs coding for the 10- and the 51-kDa subunits. We could also exclude the pathogenicity of the Ala29→Val substitution found in the 24-kDa cDNA, because the heterozygous state found in the patient was also reported in healthy individuals (40). Loosening of the FP subunits might still be due to a mutation in another subunit, which tightly interacts with them. This hypothesis will be investigated by sequencing the cDNAs coding for other known complex I subunits.
In patient no. 2, immunoblots revealed a decreased level of all the investigated subunits. Generalized loss of complex I subunits in patients’ mitochondria has already been observed by immunoblotting (48) or by blue native 2-dimensional gel electrophoresis (10, 16, 47). However, the last three reports did not determine whether complex I was absent because of the absence of the majority of the nuclear-encoded subunits or because of the dissociation of a partially assembled complex I, a process that may occur during extraction and electrophoretic migration. In the case of patient no. 2, it may be hypothesized that extensive impairment of complex I assembly results from a mutation in a gene coding for a factor specifically required for assembly of this enzyme. Human genes whose products are involved in respiratory chain assembly or biogenesis of complex IV have recently been identified (50–52).
The transnuclear complementation strategy reported here helped to define the mendelian inheritance of the 2 investigated complex I defects. This is a valuable piece of information for genetic counseling in the 2 concerned families. Biochemical and immunochemical investigations also demonstrated that both a severe reduction of complex I activity and abnormal patterns of complex I subunit composition are specific features associated with these 2 lethal clinical phenotypes. Looking for such specific features by analyzing small mitochondrial samples from chorionic villi cells may allow prenatal diagnosis in these families.
Acknowledgments
We are extremely grateful to G. Attardi (California Institute of Technology, Pasadena, California) for the gift of 143B206 ρ° cells, and to M. Skehel and J. Walker (Medical Research Council, Cambridge, United Kingdom) for the gift of the anti–24-kDa bovine subunit antibody. We gratefully acknowledge M. Vidailhet (Hôpital d’Enfants, Vandoeuvre les Nancy, France) for sending us tissue samples from patient no. 2. We thank M.T. Zabot, I. Maire, and E. Boumendil-Amar (Laboratoire de Biochimie, Hôpital Debrousse, Lyon, France) for providing us with fibroblast cultures and expert technical assistance. This work was supported by grants from the Région Rhône-Alpes, the Ministère de l’Education Nationale, de l’Enseignement Supérieur et de la Recherche (ACC-SV), and the University J. Fourier. We thank A. Renaudin and J. Willison for careful reading of the manuscript.
References
- 1.Brown MD, Wallace DC. Molecular basis of mitochondrial DNA disease. J Bioenerg Biomembr. 1994;26:273–289. doi: 10.1007/BF00763099. [DOI] [PubMed] [Google Scholar]
- 2.Schon EA, Bonilla E, DiMauro S. Mitochondrial DNA mutations and pathogenesis. J Bioenerg Biomembr. 1997;29:131–149. doi: 10.1023/a:1022685929755. [DOI] [PubMed] [Google Scholar]
- 3.Zeviani M, Antozzi C. Mitochondrial disorders. Mol Hum Reprod. 1997;3:133–148. doi: 10.1093/molehr/3.2.133. [DOI] [PubMed] [Google Scholar]
- 4.Wallace DC, et al. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science. 1988;242:1427–1430. doi: 10.1126/science.3201231. [DOI] [PubMed] [Google Scholar]
- 5.Morgan-Hughes JA, Schapira AHV, Cooper JM, Clark JB. Molecular defects of NADH-ubiquinone oxidoreductase (complex I) in mitochondrial diseases. J Bioenerg Biomembr. 1988;20:365–382. doi: 10.1007/BF00769638. [DOI] [PubMed] [Google Scholar]
- 6.Robinson BH. Lacticacidemia. Biochim Biophys Acta. 1993;1182:231–244. doi: 10.1016/0925-4439(93)90064-8. [DOI] [PubMed] [Google Scholar]
- 7.Robinson BH. Human complex I deficiency: clinical spectrum and involvement of oxygen free radicals in the pathogenicity of the defect. Biochim Biophys Acta. 1998;1364:271–286. doi: 10.1016/s0005-2728(98)00033-4. [DOI] [PubMed] [Google Scholar]
- 8.Schapira AHV. Human complex I defects in neurodegenerative diseases. Biochim Biophys Acta. 1998;1364:261–270. doi: 10.1016/s0005-2728(98)00032-2. [DOI] [PubMed] [Google Scholar]
- 9.Pitkänen S, Feigenbaum A, Laframboise R, Robinson BH. NADH-coenzyme Q reductase (complex I) deficiency: heterogeneity in phenotype and biochemical findings. J Inherit Metab Dis. 1996;19:675–686. doi: 10.1007/BF01799845. [DOI] [PubMed] [Google Scholar]
- 10.Dionisi-Vici C, et al. New familial mitochondrial encephalopathy with macrocephaly, cardiomyopathy, and complex I deficiency. Ann Neurol. 1997;42:661–665. doi: 10.1002/ana.410420419. [DOI] [PubMed] [Google Scholar]
- 11.Moreadith RW, Cleeter MWJ, Ragan CI, Batshaw ML, Lehninger AL. Congenital deficiency of two polypeptide subunits of the iron-protein fragment of mitochondrial complex I. J Clin Invest. 1987;79:463–467. doi: 10.1172/JCI112834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schapira AHV, et al. Molecular basis of mitochondrial myopathies: polypeptide analysis in complex-I deficiency. Lancet. 1988;1:500–503. doi: 10.1016/s0140-6736(88)91296-2. [DOI] [PubMed] [Google Scholar]
- 13.Ichiki T, et al. Deficiency of subunits of complex I and mitochondrial encephalomyopathy. Ann Neurol. 1988;23:287–294. doi: 10.1002/ana.410230312. [DOI] [PubMed] [Google Scholar]
- 14.Robinson BH, et al. The use of skin fibroblast cultures in the detection of respiratory chain defects in patients with lacticacidemia. Pediatr Res. 1990;28:549–555. doi: 10.1203/00006450-199011000-00027. [DOI] [PubMed] [Google Scholar]
- 15.Slipetz DM, Goodyer PR, Rozen R. Congenital deficiency of a 20-kD subunit of mitochondrial complex I in fibroblasts. Am J Hum Genet. 1991;48:1121–1126. [PMC free article] [PubMed] [Google Scholar]
- 16.Bentlage H, et al. Human diseases with defects in oxidative phosphorylation. I. Decreased amounts of assembled oxidative phosphorylation complexes in mitochondrial encephalomyopathies. Eur J Biochem. 1995;227:909–915. doi: 10.1111/j.1432-1033.1995.tb20218.x. [DOI] [PubMed] [Google Scholar]
- 17.MITOMAP: a human mitochondrial genome database. Center for Molecular Medicine, Emory University, Atlanta, GA. http://www.gen.emory.edu/mitomap.html. Accessed 1999.
- 18.van den Heuvel L, et al. Demonstration of a new pathogenic mutation in human complex I deficiency: a 5-bp duplication in the nuclear gene encoding the 18-kD (AQDQ) subunit. Am J Hum Genet. 1998;62:262–268. doi: 10.1086/301716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loeffen J, et al. The first nuclear-encoded complex I mutation in a patient with Leigh syndrome. Am J Hum Genet. 1998;63:1598–1608. doi: 10.1086/302154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schuelke M, et al. Mutant NDUFV1 subunit of mitochondrial complex I causes leukodystrophy and myoclonic epilepsy. Nat Genet. 1999;21:260–261. doi: 10.1038/6772. [DOI] [PubMed] [Google Scholar]
- 21.King MP, Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science. 1989;246:500–503. doi: 10.1126/science.2814477. [DOI] [PubMed] [Google Scholar]
- 22.Rustin P, et al. Biochemical and molecular investigations in respiratory chain deficiencies. Clin Chim Acta. 1994;228:35–51. doi: 10.1016/0009-8981(94)90055-8. [DOI] [PubMed] [Google Scholar]
- 23.Mousson B, et al. An abnormal exercise test response revealing a respiratory chain complex III deficiency. Acta Neurol Scand. 1995;91:488–493. doi: 10.1111/j.1600-0404.1995.tb00451.x. [DOI] [PubMed] [Google Scholar]
- 24.Trounce IA, Kim YL, Jun AS, Wallace DC. Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Methods Enzymol. 1996;264:484–509. doi: 10.1016/s0076-6879(96)64044-0. [DOI] [PubMed] [Google Scholar]
- 25.Chevallet M, Procaccio V, Rabilloud T. A nonradioactive double detection method for the assignment of spots in two-dimensional blots. Anal Biochem. 1997;251:69–72. doi: 10.1006/abio.1997.2206. [DOI] [PubMed] [Google Scholar]
- 26.Procaccio V, et al. Mapping to 1q23 of the human gene (NDUFS2) encoding the 49-kD subunit of the mitochondrial respiratory complex I and immunodetection of the mature protein in mitochondria. Mamm Genome. 1998;9:482–484. doi: 10.1007/s003359900803. [DOI] [PubMed] [Google Scholar]
- 27.de Sury R, Martinez P, Procaccio V, Lunardi J, Issartel JP. Genomic structure of the human NDUFS8 gene coding for the iron-sulfur TYKY subunit of the mitochondrial NADH:ubiquinone oxidoreductase. Gene. 1998;215:1–10. doi: 10.1016/s0378-1119(98)00275-3. [DOI] [PubMed] [Google Scholar]
- 28.Pignot-Paintrand I. Orientation of human spermatozoa for electron microscopy: a fast, simple method. Microsc Res Tech. 1992;21:75–76. doi: 10.1002/jemt.1070210112. [DOI] [PubMed] [Google Scholar]
- 29.MitoPick: complex I database. Laboratoire de Bioénergétique Cellulaire et Pathologique, CEA Grenoble, France. http://www-dsv.cea.fr/MitoPick/Default.html. Accessed December 1997.
- 30.Dib C, et al. A comprehensive genetic map of the human genome based on 5,264 microsatellites. Nature. 1996;380:152–154. doi: 10.1038/380152a0. [DOI] [PubMed] [Google Scholar]
- 31.Zeviani M, et al. Maternally inherited myopathy and cardiomyopathy: association with mutation in mitochondrial DNA tRNALeu(UUR) Lancet. 1991;338:143–147. doi: 10.1016/0140-6736(91)90136-d. [DOI] [PubMed] [Google Scholar]
- 32.Moraes CT, et al. Two novel pathogenic mitochondrial DNA mutations affecting organelle number and protein synthesis. Is the tRNALeu(UUR) gene an etiologic hot spot? J Clin Invest. 1993;92:2906–2915. doi: 10.1172/JCI116913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tritschler HJ, et al. Mitochondrial myopathy of childhood associated with depletion of mitochondrial DNA. Neurology. 1992;42:209–217. doi: 10.1212/wnl.42.1.209. [DOI] [PubMed] [Google Scholar]
- 34.Mariotti C, et al. Early-onset encephalomyopathy associated with tissue-specific mitochondrial DNA depletion: a morphological, biochemical and molecular-genetic study. J Neurol. 1995;242:547–556. doi: 10.1007/BF00868806. [DOI] [PubMed] [Google Scholar]
- 35.Ducluzeau P-H, et al. Depletion of mitochondrial DNA associated with infantile cholestasis and progressive liver fibrosis. J Hepatol. 1999;30:149–155. doi: 10.1016/s0168-8278(99)80019-1. [DOI] [PubMed] [Google Scholar]
- 36.Marusich MF, et al. Expression of mtDNA and nDNA encoded respiratory chain proteins in chemically and genetically-derived ρ° human fibroblasts: a comparison of subunit proteins in normal fibroblasts treated with ethidium bromide and fibroblasts from a patient with mtDNA depletion syndrome. Biochim Biophys Acta. 1997;1362:145–159. doi: 10.1016/s0925-4439(97)00061-6. [DOI] [PubMed] [Google Scholar]
- 37.Hayashi JI, Takemitsu M, Goto YI, Nonaka I. Human mitochondria and mitochondrial genome function as a single dynamic cellular unit. J Cell Biol. 1994;125:43–50. doi: 10.1083/jcb.125.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anderson S, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 39.Tiranti V, et al. Nuclear DNA origin of cytochrome c oxidase deficiency in Leigh’s syndrome: genetic evidence based on patient’s-derived ρ° transformants. Hum Mol Genet. 1995;4:2017–2023. doi: 10.1093/hmg/4.11.2017. [DOI] [PubMed] [Google Scholar]
- 40.Hattori N, Yoshino H, Tanaka M, Suzuki H, Mizuno Y. Genotype in the 24-kDa subunit gene (NDUFV2) of mitochondrial complex I and susceptibility to Parkinson disease. Genomics. 1998;49:52–58. doi: 10.1006/geno.1997.5192. [DOI] [PubMed] [Google Scholar]
- 41.Trounce I, Neill S, Wallace DC. Cytoplasmic transfer of the mtDNA nt 8993 T→G (ATP6) point mutation associated with Leigh syndrome into mtDNA-less cells demonstrates cosegregation with a decrease in state III respiration and ADP/O ratio. Proc Natl Acad Sci USA. 1994;91:8334–8338. doi: 10.1073/pnas.91.18.8334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jun AS, Trounce IA, Brown MD, Shoffner JM, Wallace DC. Use of transmitochondrial cybrids to assign a complex I defect to the mitochondrial DNA-encoded NADH dehydrogenase subunit 6 gene mutation at nucleotide pair 14459 that causes Leber hereditary optic neuropathy and dystonia. Mol Cell Biol. 1996;16:771–777. doi: 10.1128/mcb.16.3.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bodnar AG, Cooper JM, Holt IJ, Leonard JV, Schapira AHV. Nuclear complementation restores mtDNA levels in cultured cells from a patient with mtDNA depletion. Am J Hum Genet. 1993;53:663–669. [PMC free article] [PubMed] [Google Scholar]
- 44.Tabrizi SJ, Cooper JM, Schapira AH. Mitochondrial DNA in focal dystonia: a cybrid analysis. Ann Neurol. 1998;44:258–261. doi: 10.1002/ana.410440218. [DOI] [PubMed] [Google Scholar]
- 45.Isobe K, et al. Identification of inheritance modes of mitochondrial diseases by introduction of pure nuclei from mtDNA-less HeLa cells to patient-derived fibroblasts. J Biol Chem. 1997;272:12606–12610. doi: 10.1074/jbc.272.19.12606. [DOI] [PubMed] [Google Scholar]
- 46.Ferlin T, et al. Segregation of the G8993 mutant mitochondrial DNA through generations and embryonic tissues in a family at risk of Leigh syndrome. J Pediatr. 1997;131:447–449. doi: 10.1016/s0022-3476(97)80074-1. [DOI] [PubMed] [Google Scholar]
- 47.Bindoff LA, et al. Abnormal RNA processing associated with a novel tRNA mutation in mitochondrial DNA. A potential disease mechanism. J Biol Chem. 1993;268:19559–19564. [PubMed] [Google Scholar]
- 48.Bentlage HACM, et al. Multiple deficiencies of mitochondrial DNA and nuclear-encoded subunits of respiratory NADH dehydrogenase detected with peptide- and subunit-specific antibodies in mitochondrial myopathies. Biochim Biophys Acta. 1995;1234:63–73. doi: 10.1016/0005-2736(94)00288-z. [DOI] [PubMed] [Google Scholar]
- 49.Bentlage HACM, et al. Lethal infantile mitochondrial disease with isolated complex I deficiency in fibroblasts but with combined complex I and IV deficiencies in muscle. Neurology. 1996;47:243–248. doi: 10.1212/wnl.47.1.243. [DOI] [PubMed] [Google Scholar]
- 50.Rötig A, et al. Sequence and structure of the human OXA1L gene and its upstream elements. Biochim Biophys Acta. 1997;1361:6–10. doi: 10.1016/s0925-4439(97)00031-8. [DOI] [PubMed] [Google Scholar]
- 51.Zhu Z, et al. SURF1, encoding a factor involved in the biogenesis of cytochrome c oxidase, is mutated in Leigh syndrome. Nat Genet. 1998;20:337–343. doi: 10.1038/3804. [DOI] [PubMed] [Google Scholar]
- 52.Tiranti V, et al. Mutations of SURF-1 in Leigh disease associated with cytochrome c oxidase deficiency. Am J Hum Genet. 1998;63:1609–1621. doi: 10.1086/302150. [DOI] [PMC free article] [PubMed] [Google Scholar]