

Abstract
Ischemia and reperfusion activate cardiac myocyte apoptosis, which may be an important feature in the progression of ischemic heart disease. The relative contributions of ischemia and reperfusion to apoptotic signal transduction have not been established. We report here that severe chronic hypoxia alone does not cause apoptosis of cardiac myocytes in culture. When rapidly contracting cardiac myocytes were exposed to chronic hypoxia, apoptosis occurred only when there was a decrease in extracellular pH ([pH]o). Apoptosis did not occur when [pH]o was neutralized. Addition of acidic medium from hypoxic cultures or exogenous lactic acid stimulated apoptosis in aerobic myocytes. Hypoxia-acidosis–mediated cell death was independent of p53: equivalent apoptosis occurred in cardiac myocytes isolated from wild-type and p53 knockout mice, and hypoxia caused no detectable change in p53 abundance or p53-dependent transcription. Reoxygenation of hypoxic cardiac myocytes induced apoptosis in 25–30% of the cells and was also independent of p53 by the same criteria. Finally, equivalent levels of apoptosis, as demonstrated by DNA fragmentation, were induced by ischemia-reperfusion, but not by ischemia alone, of Langendorff-perfused hearts from wild-type and p53 knockout mice. We conclude that acidosis, reoxygenation, and reperfusion, but not hypoxia (or ischemia) alone, are strong stimuli for programmed cell death that is substantially independent of p53.
J. Clin. Invest. 104:239–252 (1999).
Introduction
Cardiac myocyte cell death by apoptosis accompanies heart disease of both ischemic and nonischemic origin (reviewed in refs. 1–3). It has been demonstrated in the myocardium from failing human hearts (4–7), in patients with arrhythmogenic right ventricular dysplasia (8), and in association with myocardial infarction, both within the infarcted area itself and in the surrounding viable tissue (5, 9–12). In animal models, increased apoptosis accompanies pacing-induced dilated cardiomyopathy (13, 14), pressure-overload hypertrophy (15, 16), hypertension (17), hibernating myocardium (18), and both phases of ischemia and reperfusion (3). The precise initiating stimuli, signaling pathways, and mechanisms of apoptosis are not understood.
A number of factors may contribute to ischemia-mediated cell death (19–21). Hypoxia causes inhibition of oxidative phosphorylation and a switch to glycolytic metabolism, resulting in decreased levels of high-energy phosphates, increased lactic acid production, and lower intracellular pH ([pH]i) (22–24). If ATP is rapidly depleted, necrosis will occur because of passive loss of transmembrane ion gradients, followed by cell swelling and loss of membrane integrity (19–21, 25). Unlike necrosis, apoptosis is an active, energy-consuming process that must be executed in the presence of sufficient cellular ATP (26), and one in which membrane integrity is preserved until very late stages. It is widely accepted that apoptosis accounts for some of the cell death observed after myocardial infarction, although the relative contributions of necrosis and apoptosis are unclear (27, 28). Initiating signals for apoptosis of ischemic heart cells may be related to cellular energy levels, osmotic/ionic changes, altered gene expression, altered Ca2+ handling, oxidative stress, and accumulation of waste metabolites that are normally cleared from the interstitial space.
Recently, it has been suggested that proton pumps and pH regulation may play a role in apoptosis signaling (29–32). Ischemic cardiac myocytes generate excess H+ through increased anaerobic metabolism, net hydrolysis of ATP, and CO2 retention (33). These protons are extruded from the myoplasm to the interstitial space by the combined action of 3 major ion-specific membrane transporters, including the Na+/H+ exchanger, the Na+/HCO3– cotransporter, and the vacuolar proton ATPase (30, 34, 35). Increased activity of the Na+/H+ exchanger can cause Ca2+ overload because the elevated intracellular Na+ is subsequently exchanged for Ca2+ via the Na+/Ca2+ exchanger (36). Inhibition of Na+/H+ exchange has been shown to protect against ischemic injury, possibly by preventing this increase in Ca2+ (37, 38). Conversely, inhibition of the vacuolar ATPase promotes apoptosis, in part by shifting the proton load toward the Na+/H+ transporter and thus increasing Ca2+ uptake, and in part by reducing the myocyte capacity to control [pH]i (29–31). Acidosis has been shown to correlate with apoptosis in a number of other systems (39–41), but its role, if any, in ischemic cardiac myocyte apoptosis is not understood.
There are a number of important and controversial questions about the mechanism of cardiac myocyte apoptosis during ischemia (32). One concerns the initiating signals for cell death. Is hypoxia alone sufficient to induce apoptosis, or does it require a combination of hypoxia and acidosis or accumulation of other extracellular waste metabolites? Another question relates to the role of p53. Increased activity of p53 protein has been reported to accompany apoptosis in response to volume overload and mechanical stretch (42). In these models, increased levels of angiotensin II induced expression of p53, promoting changes in the balance between Bcl-2 and Bax in favor of apoptosis (14, 42). Hypoxia has been shown to induce p53 in transformed cells (43) and both p53 and Fas in cardiac myocytes (44, 45). p53 mRNA transcript levels can also be induced in cardiac myocytes by inhibition of vacuolar ATPase with bafilomycin A (31). However, these studies did not establish a direct cause-and-effect relationship between enhanced p53 expression and hypoxia-mediated apoptosis of cardiac myocytes. Ischemia has been shown to induce apoptosis equally well in wild-type and p53-deficient mice (46).
The goal of the present study was to evaluate the separate effects of hypoxia, pH, and reoxygenation on apoptosis in actively contracting cardiac myocytes. We provide evidence that severe chronic hypoxia is not sufficient to cause apoptosis; however, hypoxia associated with acidosis or reoxygenation caused extensive apoptosis that was independent of changes in the activity of p53. The results with isolated myocytes in vitro were reproduced in an adult mouse model of ischemia and reperfusion.
Methods
Reagents.
Antibodies to p53, p21, Bax, Bcl-2, and actin were from Santa Cruz Biotechnology Inc. (Santa Cruz, California, USA), and anti-Bak was from LXR Biotechnology (Richmond, California, USA). Anti–sarcomeric myosin antibody (MF-20) was obtained from the Developmental Studies Hybridoma Bank at the University of Iowa (Ames, Iowa, USA). HOECHST 33342 and propidium iodide (PI) dyes were purchased from Calbiochem-Novabiochem Corp. (San Diego, California, USA). Ad-p53 and AdDN-p53 were obtained from F. Graham (Microbix Inc., Ontario, Ontario, Canada). Plasmids p21-Luc and p21-M–Luc, containing the wild-type and deleted p21 promoters, respectively, were provided by W. El-Deiry (Howard Hughes Medical Institute, Philadelphia, Pennsylvania, USA). Other plasmids have been described previously (47). All other reagents were purchased from Sigma Chemical Co. (St. Louis, Missouri, USA).
Cell culture.
All procedures involving animals were performed in accordance with University of Miami and SRI International guidelines for the care and use of animals. Methods for primary culture of neonatal rat cardiac myocytes have been described previously (48, 49). In brief, enriched cultures of myocyte and nonmyocyte cells were obtained from 1- to 2-day-old rats by stepwise trypsin dissociation, and were plated at a density of 4 × 106 cells/60-mm dish or on 2-well glass dishes (Nalge Nunc International, Naperville, Illinois, USA) at a density of 4 × 105 cells/cm2, in MEM supplemented with 5% FCS, penicillin, and streptomycin (MEM + 5% FCS). After 3–5 days, cells were rinsed 3 times in MEM and transferred to a defined serum-free DMEM/M-199 (4:1) medium supplemented with transferrin, vitamin B12, and insulin. The final cultures contained more than 95% cardiac myocytes, contracting at greater than 200 beats per minute (bpm). Bromodeoxyuridine (BrdU; 0.1 mM) was included in the medium for the first 3 days after plating to inhibit fibroblast growth. In some experiments, nonmyocytes from the same cultures (∼95% fibroblasts, with small percentages of smooth muscle and endothelial cells) were used. These cells were used at passages 2 or 3, maintained in MEM + 5% serum as described previously (49), and transferred to serum-free DMEM/M-199 (4:1) before experiments.
Hypoxia and reoxygenation.
Cultures were placed in serum-free DMEM/M-199 (4:1) containing 3.8 g/L glucose and transferrin, insulin, and vitamin B12, 24 hours before exposure to hypoxia. Details of our methods for exposing cells to hypoxia by incubation in an environmental chamber have been described previously (48–50). Oxygen inside the chamber was continuously monitored with an oxygen electrode (Controls Katharobic, Philadelphia, Pennsylvania, USA), and contractility was monitored by edge detection as described previously (48, 49). The chamber oxygen concentration was maintained at greater than 10 mmHg. For replacement of the culture medium under hypoxia, fresh medium was incubated under hypoxia at room temperature for 24 hours; the medium was brought to 37°C; and the pO2 was measured before adding to the dishes. Cultures exposed only to hypoxia (without reoxygenation) were lysed under hypoxia using ice-cold deoxygenated buffers. For reoxygenation, plates were removed from the chamber and reoxygenated by replacing the medium with oxygenated medium and incubating under 21% O2 (air/5% CO2). Cells were harvested and lysed for apoptosis, Western blots, or biochemical assays as described later here.
Quantitative analysis of apoptotic nuclei.
Cells were examined for morphological evidence of apoptosis or necrosis after staining with the fluorescent DNA-binding dyes HOECHST 33342 and PI as described previously (51). Treated and control cell monolayers grown on uncoated Nunc 2-well coverslip dishes were rinsed with PBS, stained with 5 μg/mL HOECHST 33342 and 5 μg/mL PI for 15 minutes, and viewed ×400 on a Zeiss IM fluorescence microscope (Carl Zeiss Inc., Thornwood, New York, USA). Cells were scored as apoptotic if they exhibited unequivocal nuclear chromatin condensation and/or fragmentation; PI-stained cells with normal nuclear morphology were scored as necrotic. At the later time points (72 hours of hypoxia-acidosis), cells with condensed nuclei that costained with PI were also scored as apoptotic. In some experiments, apoptotic nuclei were localized within cardiac myocytes by staining with an mAb against sarcomeric myosin (MF-20). Cells were fixed in ice-cold methanol, rinsed, and stained with the anti-myosin antibody and HOECHST 33342, followed by an FITC-tagged anti-mouse IgG secondary antibody. Cells were imaged and photographed on a Zeiss IM inverted-phase fluorescence microscope using a mounted Contax 35-mm camera (Yashica, Tokyo, Japan) and ASA 400 Kodak color transparency film (Eastman Kodak Co., Rochester, New York, USA). To quantitate apoptosis, an average of 400 nuclei from random fields were analyzed, and apoptotic cell counts were expressed as a percentage of the total number of nuclei counted.
Analysis of DNA fragmentation.
Cells were lysed for 5 hours at 37°C in a buffer containing 100 mM NaCl, 10 mM Tris-Cl (pH 8.0), 5 mM EDTA, 0.5% SDS, and 1 μg/mL proteinase K; proteins were precipitated with 0.8 M NaCl; and DNA was extracted with phenol/chloroform and precipitated with an equal volume of isopropanol. The resulting washed pellet was resuspended in Tris-EDTA buffer and treated with 100 μg/mL of DNase-free RNase for 30 minutes at 37°C. The DNA content was quantitated by spectrophotometry at 260/280 nm. Samples (5 μg) were subjected to electrophoresis in 2% agarose gels and were imaged by ethidium bromide staining and digital photography. In some cases, the extent of DNA fragmentation was quantified by densitometry of subchromosomal DNA fragments on digitized images using Adobe Photoshop 4.0 (Adobe Systems Inc., Mountain View, California, USA) for Macintosh.
Western blot analysis.
For detection of p53, Bax, Bak, Bcl2, and actin proteins, cells were harvested in ice-cold lysis buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1% Triton X-100, and 50 mM NaF) with freshly added 1 mM Na3VO4, 0.5 mM DTT, 1 mM PMSF, 10 μg/mL leupeptin, and 2 μg/mL aprotinin. Cells were triturated with a pipette tip 10 times, and the resulting lysates were centrifuged at 14,000 g for 10 minutes to remove cell debris. Protein content was determined using a Pierce BCA kit (Pierce Chemical Co., Rockford, Illinois, USA). Equal amounts of protein (10–200 μg) were fractionated on 12% or 15% SDS-polyacrylamide gels and were electroblotted to nitrocellulose (Bio-Rad Laboratories Inc., Hercules, California, USA). Blots were stained with Ponceau red to monitor the transfer of proteins. Membranes were blocked for 1 hour at room temperature with 5% nonfat milk in TBS (25 mM Tris, 137 mM NaCl, and 2.7 mM KCl) containing 0.05% Tween-20, and were incubated with specific antibodies for 2–4 hours in the same buffer. After washing, the blots were incubated for 1 hour with 1:7,500 dilution of horseradish peroxidase–conjugated (HRP-conjugated) anti-rabbit IgG or HRP-conjugated donkey anti-goat IgG, and were viewed using an enhanced chemiluminescence detection system (Pierce Chemical Co.).
Northern blots.
Northern blot procedures were exactly as described previously (47, 52). Full-length rat p21 and p53 probes were generated by RT-PCR on rat template mRNA using primers derived from the published sequences (53, 54).
Electrophoretic gel mobility shift.
Nuclear extracts were prepared from confluent plates as described previously (55). Sequences of the oligonucleotide probes (sense strands) were as follows: p53 wild-type, TACAGAACATGTCTAAGCATGCTGGGG; p53 mutant, TACAGAATCTGTCTAAGC ATGCTGGGG; HRE competitor, AAAGAGAGGCGGGGCTGGCTGGG (47). Gel-purified double-stranded oligonucleotides were end labeled with [32P]ATP using T4 polynucleotide kinase (Promega Biotech, Promega Corp., Madison, Wisconsin, USA) and [γ-32P]ATP (Du Pont NEN Research Products, Boston, Massachusetts, USA). Equal amounts of radioactive probe (1.5 × 104 to 2.5 × 104 cpm) were added to binding reactions that contained 8 μg of nuclear extract protein in 20 μL of a buffer containing 4 mM Tris (pH 7.8), 12 mM HEPES (pH 7.9), 60 mM KCl, 30 mM NaCl, 0.1 mM EDTA, and 1 μg poly(dI-dC) (Amersham Pharmacia Biotech, Piscataway, New Jersey, USA). Reactions were incubated for 20 minutes at 22°C before separating on nondenaturing 5% polyacrylamide gels at 4°C. Competitor DNAs were added in 200-fold excess immediately before the radioactive probe. Proteins were determined with a Pierce BCA kit.
Transient expression.
Cardiac myocytes were transfected the day after isolation using calcium phosphate as described previously (47, 49). Plates (60 mm) were transfected with 9 μg of the test plasmid (p21-Luc, p21-M–Luc, pα-MHC–HRE-Luc, and Rous sarcoma virus-Luc [RSV-Luc]) and 1 μg of pTK-RN (Promega Corp.) as the internal control to correct for variations in transfection efficiency. For adenoviral infections, transfected cells were infected with virus at 10 plaque-forming units (PFU) per cell the day after transfection. Plates were exposed to hypoxia or air after an additional 2–3 days. Adenoviruses infected 100% of the cells under these conditions, as determined by X-gal staining after infection with Ad–β-gal, and wild-type and mutant p53 viruses generated equivalent levels of protein expression by Western blots (data not shown; see Figure 7). Cultures were harvested after treatments and lysed for luciferase assays. Equal amounts of protein were assayed for expression of luciferase using the Promega dual-luciferase reporter assay system according to the manufacturer’s protocol. Protein was assayed using a Bio-Rad assay kit.
Figure 7.

Hypoxia and reoxygenation do not affect expression of p53, Bak, or Bax. Cultures of cardiac myocytes were exposed to hypoxia for 24 hours and then to reoxygenation as described in Figure 6. Proteins were extracted and analyzed by Western blots as described in Methods.
p53–/– knockout mice.
Mice heterozygous for a disruption in the p53 gene locus (56) were a kind gift from R. Kitsis (Albert Einstein College of Medicine, Bronx, New York, USA), and were backcrossed into C57BL/6 wild-type mice. Offspring of heterozygous pairs were genotyped by PCR analysis of tail DNA by PCR with primers X6.5 (5′-ACAGCGTGGTACCTTAT-3′) and X7 (5′-TATACTCAGAGCCGGCCT-3′) to amplify the endogenous allele, and primers neo 18.5 (5′-TCCTCGTGCTTTACGGTATC-3′) and X7 to amplify the disrupted allele. Homozygous offspring were bred to produce F1 litters of p53–/– pups, which were used to generate neonatal mouse cardiac myocyte cultures exactly as already described here for the neonatal rat. Wild-type C57BL/6 litters were used as controls.
Langendorff perfusions.
Mouse hearts were perfused by the Langendorff method essentially as described previously (57). Briefly, C57BL/6 or p53 knockouts, as described here (25–35 g body weight), were anesthetized with pentobarbital (60 mg/kg intraperitoneally). The chests were opened, and the hearts were rapidly excised and placed in iced Krebs-Henseleit buffer (KHB) (4°C). Thymic and fatty tissues were carefully trimmed to reveal the ascending aorta, which was then cannulated with a blunted 20-gauge needle. During transfer to the Langendorff apparatus, the cannula was perfused to avoid air entry. The heart was attached to the apparatus and retrograde perfused at constant pressure (80 mmHg) with a flow rate of 2–4 mL/min. The time from excision of the heart to commencement of perfusion did not exceed 5 minutes. The perfusion buffer consists of a phosphate-free KHB as follows (mM): 118 NaCl, 24 KCl, 2.5 CaCl2, 1.2 MgSO4, 0.5 EDTA, 25 NaHCO3, 0.5 pyruvate, and 10 glucose. The KHB buffer was equilibrated at 37°C with 5% CO2/95% O2 (pH 7.4), and the Langendorff apparatus was housed in a temperature-controlled (37°C) and humidified incubator. Any hearts in which coronary flow was excessive (>5 mL/min; indicative of an aortic tear), that contracted at more than 300 bpm, or that maintained persistent arrhythmias after stabilization were excluded from further study. Before experimentation, all hearts were exposed to an initial 30-minute stabilization period. For controls, hearts were perfused for 3 additional hours; ischemia consisted of no flow for the periods indicated; ischemia-reperfusion entailed a 20-minute no-flow with reperfusion for the indicated times. At the end of each experiment, hearts were frozen at –95°C.
Isolation of DNA from perfused hearts.
The left ventricle was removed from frozen hearts, sliced into approximately 2-mm squares, and digested for 16 hours at 37°C in a lysis buffer containing 10% SDS, 10 mM Tris-HCl (pH 7.8), 1 mM EDTA, and 200 μg/mL proteinase K. The DNA was purified by twice extracting with phenol/chloroform and precipitating with isopropanol. The DNA pellet was rinsed with 70% ethanol and resuspended in Tris-EDTA buffer. DNA samples were subjected to electrophoresis as described earlier here.
Biochemical assays.
ATP and glucose were measured as described previously (48, 49), and pH was measured directly in the culture medium using an Orion meter with a micro Ross electrode (Orion Research Inc., New South Wales, Australia).
Statistical analysis.
Results are expressed as mean ± SEM. Differences between means were evaluated by 2-tailed Student’s t test. ANOVA was carried out using InStat 2.0 (GraphPad Software for Science Inc., San Diego, California, USA) for Macintosh.
Results
Correlation of hypoxia-mediated apoptosis with change in extracellular pH.
We reported previously that rapidly contracting cardiac myocytes remained fully viable and contractile during culture under severe hypoxia for up to 6 days (48). Glycolysis was induced approximately 10-fold within 1 hour, and there was no evidence of major cell loss. The intracellular ATP of hypoxic myocytes dropped to about 70% of control plates, and a slightly reduced contractility correlated with lower intracellular cAMP in the hypoxic cultures. These results contrast with more recent reports of significant cardiac myocyte cell loss by apoptosis after 48–72 hours of exposure to an equivalent degree of hypoxia (44, 45). To investigate this apparent discrepancy, we exposed cultures of myocytes to 2 different hypoxic regimens and measured the levels of apoptosis by DNA ladders and HOECHST staining. In the first set, the cultures were maintained continuously under hypoxia, and the medium was replaced twice daily with fresh hypoxic medium to prevent the buildup of waste metabolites (48). In the second set, the cultures were maintained in parallel, but the medium was not replaced. The cultures were also monitored for ATP, glucose, and extracellular pH ([pH]o) as described previously (48, 58) and in Methods. The results are shown in Figure 1, a and b. There was no evidence of DNA fragmentation under the first set of conditions (Figure 1a), but cultures subjected to hypoxia without medium changes showed significant DNA laddering after 48 hours and extensive laddering after 72 hours (Figure 1b). In agreement with our previous observations, when the medium was replaced twice daily, ATP levels dropped to about 75% of aerobic control levels after 24 hours and remained stable thereafter; [pH]o did not change, and glucose levels remained high (Figure 1c). Under these conditions, the myocytes continued to contract for the duration of the experiment, as reported previously, and there was no significant loss of total DNA or protein (data not shown; ref. 48). When the medium was not replaced, intracellular ATP levels were sustained up to 48 hours but dropped dramatically at 72 hours, coincident with the loss of most of the cells by apoptosis. Glucose declined progressively over the 72-hour period but was not depleted, and [pH]o declined steadily to a final value of about 6.0 (Figure 1c). Under these conditions, contractions ceased before 48 hours (data not shown).
Figure 1.

Contributions of waste metabolic buildup to apoptosis induced by chronic hypoxia. (a and b) Parallel cultures of cardiac myocytes were exposed to hypoxia as described in Methods. In a, the medium was replaced with fresh hypoxic medium every 12 hours; in b, there was no medium replacement. Cultures were harvested at the indicated times and processed for DNA fragmentation. (c) Intracellular ATP, medium glucose, and [pH]o were measured in parallel cultures as described in Methods; results are means of 3 separate experiments. Open circles are results from cultures without medium replacement; closed circles, with medium replacement. (d–f) Typical fields of myocytes stained with HOECHST 33342 and anti-myosin antibody as described in Methods. (g) Quantitations of HOECHST-stained condensed nuclei, also described in Methods. At 24 and 48 hours, less than 2% of cells were PI positive (scored as necrotic) under any condition; at 72 hours, hypoxia-acidic cultures had more PI-positive cells, and these were scored as apoptotic if they contained condensed nuclei. Costaining populations were not distinguished from PI-excluding cells at this stage. Results are representative of at least 3 experiments.
Cardiac myocytes were grown on glass coverslips and were fixed and double stained with HOECHST 33342 and anti-myosin antibody (Figure 1, d–f). In cultures grown aerobically, abundant myofilaments with clear cross-striations were evident with the myosin stain (Figure 1d, right panel). As indicated by the white arrows, most of the nuclei were oval, and there was sparse evidence of condensation or internal fragmentation. In this field, 29 nuclei were scored normal and 1 was condensed; 25 nuclei were localized within cells that were myosin positive. At 72 hours, the unfed hypoxic cultures (Figure 1e) still stained strongly with myosin antibody, but there was clear deterioration of the myofilaments, and cross-striations were no longer clearly visible. In the field shown, 22 nuclei were scored condensed (examples are indicated by the arrows), and 14 were normal; only 2 nuclei in this field were localized to myosin-negative cells. In contrast, cells that received medium replacement still exhibited myofilaments with intact cross-striations after 72 hours of hypoxia (Figure 1f, right panel, arrow at far right), and most of the nuclei were normal. In this field, 27 nuclei were scored normal and 3 condensed; 2 nuclei were localized to nonmyocytes. White arrows indicate normal cardiac myocyte nuclei; the pink arrow indicates a condensed nucleus.
These data were quantitated as shown in Figure 1g. The bar graphs indicate the percentage of apoptotic nuclei scored for each condition. Control aerobic cultures contained 5–7% apoptotic cells, similar to previous reports (51). This increased to 44% after 48 hours of hypoxia with metabolite buildup, and to 60% after 72 hours of hypoxia. In hypoxic cultures without metabolite buildup, the apoptotic index was significantly lower (11 ± 1.5% at 72 hours). This slight increase in apoptosis over aerobic myocytes could be due to transitory acidosis under these conditions or to other factors associated with hypoxic incubation. Except for the 72-hour hypoxia-acidosis condition, necrotic cells (PI-positive noncondensed nuclei) were less than 5% of the total cells counted (see Methods; data not shown).
These results suggest either that proapoptotic factors accumulate in the medium during hypoxia or that vital components are depleted. To test these possibilities, medium from 48-hour hypoxic cultures that were just beginning to show signs of DNA laddering (Figure 2, top) was transferred to fresh cardiac myocytes. These cells were incubated for 24 hours under either hypoxic or aerobic conditions. Apoptosis was monitored by DNA fragmentation. Control plates received medium from hypoxic cells that underwent medium replacement (+ med). A typical experiment is shown in Figure 2. In this case, significant apoptosis was apparent in both aerobic and hypoxic cultures 24 hours after exposure to the spent medium. Glucose and ATP levels were maintained in all cultures (data not shown). The [pH]o in the aerobic plates remained stable at 7.0, whereas the [pH]o under hypoxia dropped to 6.1. More DNA fragmentation appeared in the sample from the 24-hour hypoxic plate, correlating with the lower [pH]o. In control plates (Figure 2, bottom right), there was only slight DNA laddering at 24 hours, and the [pH]o remained high in these cultures.
Figure 2.

Induction of apoptosis by conditioned medium. Cultures were grown under hypoxia with or without medium change. After 48 hours, the medium was removed and the cells were analyzed for DNA fragmentation (top). The spent medium was centrifuged at 800 g for 5 minutes, to pellet cells and debris, and was added directly to a second set of plates. These plates were incubated in air or under hypoxia as indicated. After 24 hours, these cells were also harvested and analyzed for DNA fragmentation. Controls show untreated cells harvested at the time of medium change. [pH]o was measured in all cases immediately before harvesting the cells. Control samples shown in the last 2 lanes of the bottom left panel did not receive spent medium. Results are representative of 3 separate experiments.
The induction of apoptosis of fresh cardiac myocytes by spent medium could be due to the accumulation or depletion of factors in this medium. Obvious candidates for proapoptotic factors are the protons extruded from the hypoxic cells. To determine whether this was the case, the pH of the spent medium from 48-hour hypoxic cultures was readjusted to 7.6 with NaOH and HEPES before adding back to fresh cardiac myocytes. Apoptosis was again monitored in the recipient cells as described in Figure 2. These results are shown in Figure 3a. As before, acidic spent medium from hypoxic myocytes again caused extensive apoptosis after 24 hours (Figure 3a, bottom, lanes 4 and 5). In contrast, minimal DNA fragmentation was detected in the control cells (normal medium, lanes 2 and 3) and in samples from cells exposed to the same spent medium after pH neutralization (lanes 6 and 7). This suggests that an acidic pH is important for the induction of nuclear fragmentation by spent medium from hypoxic cultures. It also suggests that acidic [pH]o may directly induce apoptosis. To test this hypothesis directly, parallel cardiac myocyte cultures were again exposed to hypoxia as described in Figure 1. In the first set, the medium was not replaced and became acidic (conditions were the same as described in Figure 1b). In the second set of cultures, the medium was not replaced, but [pH]o was maintained higher than 7.0 by adding predetermined amounts of HEPES and NaOH at 12-hour intervals. The results are shown in Figure 3b. In the absence of additional buffer, [pH]o dropped to 5.7 at 48 hours, and there was extensive apoptosis at both 48 hours and 72 hours. In the cultures with neutralized medium, DNA fragmentation was significantly reduced.
Figure 3.

Neutralized medium prevents apoptosis. Conditioned medium was generated as described in Figure 2. (a, top) DNA ladders from the cardiac myocytes used to generate the spent medium. (a, bottom) Spent medium was added directly to fresh plates of cardiac myocytes (middle 2 lanes), or it was neutralized to pH 7.4 with HEPES (20 mM final concentration) and NaOH and then added to a second set of fresh cardiac myocytes. Both sets of plates were incubated under hypoxia for 24 hours and analyzed for DNA fragmentation. Control plates were incubated under aerobic or hypoxic conditions in parallel. (b) Parallel sets of cardiac myocytes exposed to hypoxia without medium change. In the first set (b, left), the acid was allowed to accumulate exactly as described in Figure 1b; in the second set (b, right) aliquots of 250 mM HEPES and 250 mM NaOH were added every 12 hours to maintain a [pH]o of approximately 7.1. Measurements of DNA fragmentation and determinations of percent condensed nuclei (c) were as described in Methods. In all cases, the medium pH was measured immediately before the cultures were harvested. (d) Typical field of myocytes stained with HOECHST 33342 and anti-myosin antibody as described in Figure 1d. Results in a, b, and c are from typical experiments; error bars in c are SEM (n = 3).
These results were confirmed by quantitation of HOECHST-stained condensed nuclei (Figure 3c). Cardiac myocytes were cultured for 72 hours under hypoxia without medium change, with or without the addition of sufficient buffer and alkali to neutralize the pH every 12 hours. These cells were fixed and double stained with HOECHST 33342 and MF-20 (Figure 1). Examples are shown in Figure 3d. Hypoxic, pH-neutralized myocytes exhibited some degree of myofilament deterioration, but cross-striations were still apparent, and most of the nuclei were normal (compare with Figure 1e). In the field shown, 25 nuclei were scored normal and 3 were apoptotic; 1 nucleus was localized to a nonmyocyte.
Induction of apoptosis by extracellular lactic acid.
The results described here demonstrate that low [pH]o is critical for the induction of apoptosis by hypoxia in this model. They do not show that acidosis can activate apoptosis independently of other factors that may accumulate or disappear from the extracellular medium during exposure to hypoxia. To test the effect of acidosis directly, we added lactic acid to fresh cardiac myocyte cultures at similar concentrations to those seen in conditioned medium after 72 hours of hypoxia (48), and adjusted the pH to 6.8 with phosphoric acid. These cultures were exposed to aerobic or hypoxic conditions as before. As shown in Figure 4, the addition of acid induced DNA laddering in both aerobic and hypoxic cardiac myocytes. The fragmentation induced by exogenous acid was not as pronounced as that caused by 24 hours of conditioned medium (Figure 2). This could be due to a number of factors, including the site of generation of the acid or additional proapoptotic components in the conditioned medium. However, there was more DNA fragmentation in the acidified aerobic cultures than in either the aerobic or hypoxic controls, even though the hypoxia control cultures achieved the same final pH. These results confirm that extracellular acidosis can induce significant apoptosis independently of hypoxia in cardiac myocytes.
Figure 4.
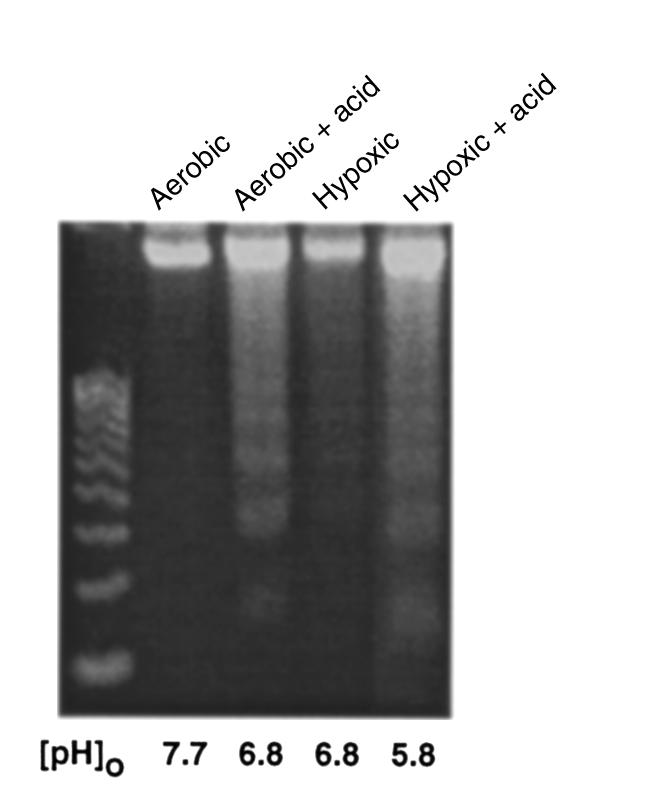
Exogenous lactic acid induces apoptosis. Lactic acid was added to culture medium to a final concentration of 16.5 mM, and the pH was adjusted to 6.8 by adding phosphoric acid. The medium was added to fresh cultures of cardiac myocytes, and the cultures were incubated under air or hypoxia as indicated. DNA fragmentation was determined as described in Methods. The initial pH in both aerobic and hypoxic cultures was 6.8; the aerobic cultures maintained this pH, but the pH of hypoxic cultures decreased further to a final pH of 5.8.
Hypoxia-acidosis–mediated apoptosis is independent of changes in p53.
The results described here demonstrate that extracellular acidosis, and possibly other metabolites, induces apoptosis of cardiac myocytes independently of hypoxia. Because p53 has been implicated in numerous pathways of apoptosis in other systems, as well as in cardiac myocytes subjected to stretch, angiotensin II, and hypoxia (42–44), we asked whether p53 was involved in initiating the signaling pathways mediated by hypoxia-acidosis. Four assays for p53 activity are shown in Figure 5, a–d. Figure 5a shows typical Western blots of proteins from cardiac myocytes and mouse embryo fibroblasts (MEFs) subjected to increasing periods of hypoxia (as in Figure 1, a and b). The p53 levels in the myocytes were very low at all times and did not change after short- or long-term exposure to hypoxia, with or without metabolic buildup. Infection with a p53 adenovirus caused a large accumulation of p53 (Figure 5a, third panel). As a positive control, p53 was measured in MEFs exposed to hypoxia (Figure 5a, bottom panel). In agreement with a previous report (43), p53 protein was elevated approximately 4-fold in these cells after 12 hours.
Figure 5.

Hypoxia-acidosis–mediated apoptosis is independent of p53. (a) Cardiac myocytes were exposed to hypoxia as described in Methods. Plates were harvested at the indicated times, and proteins were extracted. For the 36-hour time points in a, the culture medium was changed every 12 hours (no metabolite buildup) or there was no medium change (metabolite buildup). In the bottom panel, proteins were extracted from MEFs as described in Methods. Western blots and probes are described in Methods. (b) Nuclear extracts were prepared from aerobic and (unfed) hypoxic cultures and from cultures infected for 48 hours with an adenoviral vector expressing p53 as described in Methods. Competition lanes include self (p53) and an oligonucleotide with the hypoxia-inducible factor-1 consensus binding site (HRE) (73). All probes and procedures are described in Methods. Equal amounts of protein (8 μg) were loaded in each lane. (c) Cardiac myocytes were exposed to hypoxia (without medium change) for the indicated times; protein was extracted and analyzed by Western blot using anti-Bax, anti-Bak, and anti-actin antibodies, described in Methods. (d) Cardiac myocytes were transfected with the indicated plasmid using calcium phosphate as described in Methods. Transfected cultures were incubated under aerobic or hypoxic conditions for 24, 48, or 72 hours (without medium change) as indicated. For viral infections, transfected cultures were inoculated with wild-type p53 at 5 PFU/cell plus 5 PFU of a virus expressing LacZ (Ad-p53), or with 5 PFU/cell of wild-type p53 plus 5 PFU of dominant-negative p53 (Ad-p53DN; contains a Cys-135→Ser mutation; ref. 74) as described in Methods. (e) Cardiac myocytes were isolated from wild-type or p53 knockout neonatal mice as described in Methods. The myocytes were cultured and exposed to hypoxia under the same conditions described in Figure 1b.
Changes in the activity of p53 may occur independently of net synthesis, e.g., through phosphorylation or nuclear translocation (59, 60). To determine whether there were changes in the activity of nuclear p53, DNA binding was measured by electrophoretic mobility shifts using a p53 binding site oligonucleotide and nuclear extracts from aerobic and hypoxic cardiac myocytes (as described in Methods). Previous reports have demonstrated the weak but specific nature of p53 binding by this assay (42, 43). The arrow in Figure 5b indicates the p53-specific binding complex. This band was competed by excess cold probe (lane 5), but not by excess of a non–p53 binding site probe (HRE, lane 6), and it was not present when the labeled probe contained a p53 binding site mutation (lanes 7–10). There was no change in the specific p53 binding complex between aerobic and hypoxic nuclear extracts. In agreement with a previous report (44), infection with a p53 adenovirus caused an increase of p53 complex binding (lane 4).
Changes in the expression of p53 target genes Bax and Bcl2 have been implicated in apoptosis caused by pressure overload, angiotensin II, and stretch (42). Increased expression of Bcl2 and Fas proteins and no change of Bax were reported in ischemic myocardial tissue (61). Western blot assays were used to determine whether chronic hypoxia caused changes of Bax, Bcl2, or Bak. As shown in Figure 5c, exposure of myocytes to hypoxia for 24 or 48 hours caused no apparent changes in Bak or Bax; Bcl-2 protein levels were too low to detect, consistent with a previous report (62).
To assay for changes in function of p53, we measured the effects of chronic hypoxia on the expression of a transfected p21 promoter (43, 44). As shown in Figure 5d, there was no significant effect of hypoxia on the expression of luciferase from either wild-type or mutant p21 promoters (in p21-M, the p53 binding sites of the wild-type p21 promoter are deleted). The slight trend toward increased p21 promoter activity after 48 hours of hypoxia (1.2 ± 0.3–fold), although differing from the trends of the α-MHC and RSV promoters at this time point, was not significant when compared with the p21 aerobic control or the p21-M promoter at 48 hours of hypoxia (0.9 ± 0.15). As a positive control, p21-Luc–transfected cells were infected with a p53 adenovirus (p53-Ad); in this case, expression of the p21 promoter was induced by about 3-fold. When the p21-Luc–transfected cultures were infected with equal amounts (5 PFU/cell) of Ad-p53 and an adenoviral vector containing a dominant-negative mutant p53 (Ad-p53DN), the induction was quenched (Figure 5d, lane 10). Expression of the hypoxia-responsive vector pα-MHC–HRE-Luc (lanes 11 and 12) confirmed that the culture conditions used here were sufficient to activate hypoxia-regulated genes, whereas there was no effect on the expression of the RSV promoter (47, 63).
Finally, we detected no change in endogenous p53 or p21 transcripts by Northern blots of RNA from cardiac myocytes exposed to hypoxia or hypoxia-acidosis for 6, 12, 24, or 36 hours, compared with aerobic controls (data not shown).
These results indicate that p53 activity in cardiac myocytes does not change during the exposure of these cells to chronic hypoxia and suggest that the ensuing apoptosis is independent of changes in p53 activity. To confirm this, cardiac myocytes were isolated from wild-type and p53 knockout mice and were subjected to chronic hypoxia-acidosis (without medium change) as described in Figure 1b. As shown in Figure 5e, although the levels of apoptosis were lower than previously seen with rat cardiac myocytes, there was clear genomic DNA fragmentation in both wild-type and knockout mouse cells; the arrows in the figure indicate fragments of about 200-bp increments, highly characteristic of apoptotic genomic DNA (2). No fragmentation was detected in the control (aerobic) samples. There appeared to be slightly more DNA loaded in the knockout lanes, and more intense fragmentation; densitometric scanning indicated approximately equivalent ratios of fragmented/intact DNA in the 2 samples (n = 2; data not shown). The reason for lower levels of apoptosis in mouse versus rat cardiac myocytes is probably related to the slightly decreased contractility and slower rate of acid production in the mouse cultures (data not shown). In other experiments, rat myocytes treated with Ad-p53DN, as described in Figure 5d, displayed identical DNA fragmentation as the controls after exposure to chronic hypoxia-acidosis (data not shown). Together, these results strongly suggest that hypoxia-acidosis–mediated apoptosis of cardiac myocytes is independent of p53 activity.
Reoxygenation of hypoxic cardiac myocytes induces apoptosis.
The results reported here show that chronic hypoxia alone does not cause apoptosis of cardiac myocytes. Our previous results indicated that chronic hypoxia caused loss of glutathione and mediated a stress response that included the induction of c-Jun NH2-terminal kinase (JNK) activity when the cultures were reoxygenated (64). These conditions mimic the changes in oxygen tension that occur during ischemia and reperfusion. We therefore asked whether reoxygenation of hypoxic cultures also induced apoptosis. These results are shown in Figure 6, a and b. There was no evidence of apoptosis after 24 hours of hypoxia, in agreement with the results in Figure 1. However, DNA laddering was apparent 8–12 hours after reoxygenation. Maximal DNA fragmentation occurred 16–24 hours after reoxygenation and then declined to baseline levels. Figure 6b shows representative fields of HOECHST-stained nuclei. Arrows in the bottom left panel indicate examples of condensed nuclei that were scored positive. In this field, 12 nuclei were scored condensed and 15 were normal. Figure 6c shows the quantitation of condensed nuclei under the different conditions, determined as described in Figure 1; approximately 30% of the cells became apoptotic at the peak time of 16–24 hours after reoxygenation.
Figure 6.

Induction of apoptosis by hypoxia-reoxygenation. Cultures of cardiac myocytes were exposed to hypoxia for 24 hours and then reoxygenated by replacing the medium with fresh aerobic medium and incubating in air (21% O2/5% CO2). Plates were harvested at the indicated times and analyzed for DNA fragmentation (a) or nuclear condensation (b and c) as described in Methods. HOECHST staining and the quantitation of condensed nuclei were as described in Figure 1. Typical fields of HOECHST-stained cardiac myocytes (b) were photographed at ×400. The 48-hour hypoxic incubation (c) was subjected to medium replacement and did not become acidic.
Reoxygenation-mediated apoptosis is independent of changes in p53.
To determine the role of p53 or its associated genes in reoxygenation-mediated apoptosis, we measured protein levels of p53, Bak, Bax, and Bcl-2 at intervals during exposure of cells to hypoxia-reoxygenation. These results are shown in Figure 7. As with hypoxia alone, there were no major changes in the levels of p53, Bak, or Bax after hypoxia-reoxygenation. Bcl-2 was not detectable. The slight increase in p53 seen at the 24-hour hypoxia time point was not reproduced in 2 other experiments in which there was a slight decrease and no change (data not shown).
To confirm these results, we asked whether hypoxia-reoxygenation induced DNA fragmentation of cardiac myocytes in the absence of p53. Myocytes were isolated from p53-null homozygous knockout mice and their wild-type littermates and were subjected to hypoxia-reoxygenation (Figure 6). The results are shown in Figure 8. DNA internucleosomal cleavage peaked earlier and was at least equivalent in cardiac myocytes from p53–/– versus p53+/+ animals. This confirms that apoptosis mediated by either hypoxia-reoxygenation or hypoxia-acidosis does not require p53.
Figure 8.

Induction of apoptosis in cardiac myocytes from p53–/– knockout mice. Cardiac myocytes were isolated from 1- to 2-day-old wild-type (a) or p53-nullizygous (b) mice as described in Methods. Cell culture and treatments were exactly as described in Figure 6.
Ischemia with reperfusion causes equivalent apoptosis in wild-type and p53 knockout mouse hearts.
As one approach to determine whether the results obtained from the in vitro model reflected pathological changes in the intact organ, we measured DNA fragmentation from mouse hearts subjected to ischemia and reperfusion. Adult hearts from wild-type and p53 knockout mice were perfused by the Langendorff technique as described in Methods. Previous studies demonstrated that DNA fragmentation induced by 24 hours of permanent occlusion of the coronary artery (ischemia alone) occurred at equivalent levels in wild-type and p53 knockout mice (46). Apoptosis induced by ischemia and reperfusion of Langendorff-perfused mouse hearts has not been reported previously. Figure 9 compares nucleosomal ladders from the 2 groups. There was clear genomic DNA fragmentation in both wild-type and p53 knockout cells subjected to 20 minutes of ischemia and 3 hours of reperfusion (Figure 9, panels 2 and 4, respectively). DNA fragmentation in the knockout hearts was at least equivalent to that in the wild-type. No fragmentation was detected in the aerobic samples (controls, 3-hour aerobic perfusion; Figure 9, all panels) or in samples from hearts subjected to ischemia alone for 20 minutes to 3 hours (Figure 9, panels 1 and 3); a faint smear of degraded DNA was sometimes evident in samples subjected to extended ischemia, but no distinct ladders were seen. In the ischemia-reperfused samples (Figure 9, panel 2), DNA ladders appeared only after more than 2 hours of reperfusion, and the degree of fragmentation was maximal after 3–4 hours of reperfusion. Apoptosis in the ischemia-reperfused hearts of both wild-type and p53 knockout mice was confirmed by TUNEL assay, using an ApopTag kit (Intergen Co., Purchase, New York, USA) in which TUNEL-positive cells colocalized with cardiac myocytes (data not shown). These results show that ischemia with reperfusion, but not ischemia alone, induces equivalent DNA fragmentation in Langendorff-perfused hearts from wild-type and p53–/– knockout mice.
Figure 9.

Nucleosomal fragmentation in genomic DNA from ischemia-reperfused mouse hearts. Hearts were perfused on a Langendorff apparatus as described in Methods. Left ventricle DNA was isolated from wild-type and p53–/– knockout mice subjected to ischemia alone or to ischemia and reperfusion. Controls were perfused with aerobic perfusion buffer (95% O2/5% CO2) for 3 hours. Nucleosomal ladders are apparent from the ischemia-reperfused hearts of both genotypes.
Hypoxia, acidosis, and reoxygenation do not induce DNA fragmentation in nonmuscle fibroblasts.
Finally, in results not shown, confluent cultures of nonmuscle fibroblasts were exposed to the same conditions of hypoxia, acidosis, and reoxygenation as described earlier here for cardiac myocytes, and were analyzed for DNA fragmentation. There was no evidence of DNA fragmentation in these cells under any condition, even under the most extreme conditions of hypoxia with acidosis. This indicates that the responses described here are at least partially specific for cardiac myocytes; it also suggests that the apoptosis observed and quantitated in this report is probably exclusively from cardiac myocytes.
Discussion
Our results demonstrate that chronic severe hypoxia does not cause apoptosis of cardiac myocytes unless it is accompanied by acidosis or reoxygenation. Exposure of cultures to 72 hours of continuous hypoxia with twice-daily medium replacement resulted in levels of apoptosis that were only slightly higher than aerobic controls, as determined by DNA laddering or nuclear condensation. This is consistent with a previous report from this laboratory wherein we exposed cardiac myocytes to 6 days of continuous hypoxia and observed no significant cell loss (48). Extensive apoptosis was apparent when cultures were exposed to hypoxia for 48–72 hours with no medium change. Under these conditions, the [pH]o decreased progressively, usually to less than 6.0 at 72 hours. At this time, contractions failed, myofilaments deteriorated, most of the remaining nuclei were condensed, and there was very pronounced DNA fragmentation with little evidence of high–molecular-weight DNA, indicating that most of the cells were either dead or dying.
Four direct lines of evidence support an essential if not singular role for [pH]o in the activation of apoptosis by hypoxia. First, acidic spent medium induced apoptosis of fresh myocytes with or without additional hypoxia. Second, neutralized spent medium was no longer proapoptotic, even with additional hypoxia. Third, apoptosis was prevented in hypoxic cultures by neutralizing extracellular acid as it was produced. Fourth, acidification of the medium with lactic and phosphoric acids was sufficient to induce myocyte apoptosis under either aerobic or hypoxic conditions. These results suggest that the direct stimulus for apoptosis of cardiac myocytes during hypoxia is the extracellular accumulation of protons.
Excess protons are expelled from the intracellular compartment of the cardiac myocytes through the actions of 3 main transporters: the Na+/H+ exchanger, the Na+/HCO3– cotransporter, and the vacuolar proton ATPase (30, 34, 35). Each of these exchangers contributes to the maintenance of neutral [pH]i by extruding excess protons under conditions of hypoxia or ischemia. The activity of each transporter is regulated, at least in part, by the myoplasmic membrane pH gradient, determined by [pH]i and [pH]o (34). A decrease of [pH]o below [pH]i requires that protons are transported against the proton gradient; as this gradient increases, transporter activities decrease and the [pH]i falls correspondingly. If the [pH]o falls low enough, proton extrusion stops; this has been reported to occur at a [pH]o of about 6.0 for Na+/H+ exchange in chick cardiac myocytes (34). Therefore, when extracellular protons are not cleared, decreased [pH]o will be paralleled by a corresponding, or even greater, drop of [pH]i. This is likely to be the situation under conditions of hypoxia or ischemia with pronounced acidosis, and the signal for apoptosis under these conditions probably involves both intracellular and extracellular acidosis. It should be noted that apoptosis was initiated by hypoxia-acidosis in our studies well before there was significant loss of ATP; therefore, failure of ATP-sensitive channels, including the vacuolar proton ATPase or ATP-sensitive K+ channel, is probably not a part of this mechanism (29, 30, 65). Also, the effect is probably not mediated by secondary accumulation of Ca2+, because more Na+/H+ exchange and consequent Ca2+ accumulation will occur under conditions of extracellular H+ clearance when there is no significant apoptosis (Figure 1a).
Our results also indicate that apoptosis resulting from hypoxia-acidosis was independent of changes in the tumor-suppressor protein p53. Five independent lines of evidence favor a p53-independent pathway for hypoxia-acidosis–activated apoptosis in this model. First, there was no change of p53 protein at any time during the exposure of myocytes to hypoxia. Second, there was no change of p53 binding activity in nuclear extracts from cardiac myocytes subjected to 24 or 48 hours of hypoxia with developing acidosis. Third, there was no change in the expression of a p53-dependent p21 promoter in cardiac myocytes subjected to hypoxia-acidosis. Fourth, there was no change in expression of p53-dependent genes or proteins. Finally, and more importantly, equivalent genomic DNA fragmentation occurred in hypoxic-acidotic myocytes isolated from the hearts of wild-type or homozygous p53–/– knockout mice.
The proapoptotic potential of p53 has been demonstrated in numerous systems (reviewed in refs. 59, 60, 66), including cardiac myocytes responding to stress (42), hypoxia-mediated apoptosis of transformed cells (67), and DNA-damaging agents in multiple cell types (68). Numerous pathways of apoptosis have also been described that are independent of p53 (59, 69). The activity of p53 is highly dependent on dose, cell type, and cell context. For example, p53 overexpression does not by itself induce apoptosis in smooth muscle (70, 71); overexpression of p53-dependent p21 protects skeletal myocytes from apoptosis (72); and γ-radiation activation of p53 may mediate apoptosis or cell-cycle withdrawal, depending on the cell type (68). A previous study reported that p53 was induced in neonatal cardiac myocytes in response to hypoxia (44). Although we cannot account for the apparent discrepancies between this report and our observations, they may be related to differences in the models and the pleiotropic and conditional properties of p53. The previous study suggested that hypoxia-mediated changes in p53 correlated with increased apoptosis, although it did not demonstrate that p53 was required for apoptosis.
Also, to our knowledge, we present the first definitive evidence that reoxygenation is a direct and distinct stimulus for apoptosis of cardiac myocytes. Nucleosomal fragmentation was evident 8–12 hours after reoxygenation and peaked at 16–24 hours in different experiments. Quantitation of condensed nuclei by HOECHST staining indicated that approximately 30% of myocytes were apoptotic after 1 cycle of hypoxia-reoxygenation. This clearly demonstrates that reoxygenation of hypoxic cardiac myocytes is extremely stressful, and it supports our previous report of an intense induction of JNK activity under the same conditions (64). Apoptosis caused by hypoxia-reoxygenation was also independent of p53 by the same criteria described earlier here for hypoxia-acidosis–mediated apoptosis.
We found no evidence for modulation of the expression of the Bcl-2 family members Bak and Bax in hypoxia-acidosis– or hypoxia-reoxygenation–mediated apoptosis. Unchanged levels of Bax protein have also been reported in ischemic myocardial tissue (61). Although this does not exclude a role for posttranslational modification of these proteins, the results support our conclusion that neither of the apoptosis pathways described here is dependent on p53.
Although it is not possible to extrapolate all of these results to conditions in vivo, it is noteworthy that hearts from p53 knockout mice show the same degree of apoptosis as wild-type hearts subjected to chronic ischemia (46) or to ischemia and reperfusion (Figure 9). Therefore, in the mouse heart, p53 is clearly not required for apoptosis in response to either ischemia or reperfusion. This does not mean that apoptosis in response to other stresses is independent of p53, or that p53 will not contribute to ischemia-mediated apoptosis under some circumstances, because ischemia is a complex stress. Our results suggest that hypoxia per se is not a signal for apoptosis, because cardiac myocytes can remain completely viable for extended times under severe hypoxia, provided they have sufficient glycolytic substrate and waste metabolite removal. There may be direct molecular parallels between the in vitro models of hypoxia, acidosis, and reoxygenation described here and ischemia in the intact adult heart. Myocardial tissue subjected to severe chronic ischemia by coronary artery occlusion is likely to be subjected to varying degrees of both hypoxic stress and acidosis, whereas ischemia-reperfused tissue experiences hypoxia-reoxygenation. All of these conditions can cause apoptosis in a p53-null cellular environment.
Acknowledgments
This work was supported by the National Institutes of Health (HL-44578 to K. Webster; HL-49891 to N.H. Bishopric), by the Cigarette and Tobacco Surtax of the State of California through the Tobacco-Related Disease Research Program of the University of California (1RT-402 to K.Webster), by an Established Investigator Award from the American Heart Association (to N.H. Bishopric), and, in part, by a grant from the Miami Heart Foundation. We acknowledge the excellent contributions of P. Andreka in generating and characterizing the p53 knockout mice, and we thank M. Javadpour and N. Maulik for their helpful suggestions in setting up Langendorff-perfused mouse hearts.
Footnotes
A preliminary report of this work has appeared in abstract form (1998. Circulation. 98:I743).
Nanette H. Bishopric and Daryl J. Discher contributed equally to this work.
References
- 1.Haunstetter A, Izumo S. Apoptosis: basic mechanisms and implications for cardiovascular disease. Circ Res. 1998;82:1111–1129. doi: 10.1161/01.res.82.11.1111. [DOI] [PubMed] [Google Scholar]
- 2.MacLellan WR, Schneider MD. Death by design: programmed cell death in cardiovascular biology and disease. Circ Res. 1997;81:137–144. doi: 10.1161/01.res.81.2.137. [DOI] [PubMed] [Google Scholar]
- 3.Fliss H, Gattinger D. Apoptosis in ischemic and reperfused rat myocardium. Circ Res. 1996;79:949–956. doi: 10.1161/01.res.79.5.949. [DOI] [PubMed] [Google Scholar]
- 4.Olivetti G, et al. Apoptosis in the failing human heart. N Engl J Med. 1997;336:1131–1141. doi: 10.1056/NEJM199704173361603. [DOI] [PubMed] [Google Scholar]
- 5.Olivetti G, et al. Acute myocardial infarction in humans is associated with activation of programmed myocyte cell death in the surviving portion of the heart. J Mol Cell Cardiol. 1994;28:2005–2016. doi: 10.1006/jmcc.1996.0193. [DOI] [PubMed] [Google Scholar]
- 6.Kajstura J, et al. Myocyte proliferation in end-stage cardiac failure in humans. Proc Natl Acad Sci USA. 1998;95:8801–8805. doi: 10.1073/pnas.95.15.8801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Narula J, et al. Apoptosis in myocytes in end-stage heart failure. N Engl J Med. 1996;335:1182–1189. doi: 10.1056/NEJM199610173351603. [DOI] [PubMed] [Google Scholar]
- 8.Mallat Z, et al. Evidence of apoptosis in arrhythmogenic right ventricular dysplasia. N Engl J Med. 1996;335:1190–1196. doi: 10.1056/NEJM199610173351604. [DOI] [PubMed] [Google Scholar]
- 9.Misao J, et al. Expression of bcl-2 protein, an inhibitor of apoptosis, and Bax, an accelerator of apoptosis, in ventricular myocytes of human hearts with myocardial infarction. Circulation. 1996;94:1506–1512. doi: 10.1161/01.cir.94.7.1506. [DOI] [PubMed] [Google Scholar]
- 10.Itoh G, et al. DNA fragmentation of human infarcted myocardial cells demonstrated by the nick end labeling method and DNA agarose gel electrophoresis. Am J Pathol. 1995;146:1325–1331. [PMC free article] [PubMed] [Google Scholar]
- 11.Saraste A, et al. Apoptosis in human acute myocardial infarction. Circulation. 1997;95:320–323. doi: 10.1161/01.cir.95.2.320. [DOI] [PubMed] [Google Scholar]
- 12.Cheng W, et al. Programmed myocyte cell death affects the viable myocardium after infarction in rats. Exp Cell Res. 1996;226:316–327. doi: 10.1006/excr.1996.0232. [DOI] [PubMed] [Google Scholar]
- 13.Kajstura J, et al. The cellular basis of pacing-induced dilated cardiomyopathy: myocyte cell loss and myocyte cellular reactive hypertrophy. Circulation. 1995;92:2306–2317. doi: 10.1161/01.cir.92.8.2306. [DOI] [PubMed] [Google Scholar]
- 14.Leri A, et al. Pacing-induced heart failure in dogs enhances the expression of p53 and p53-dependent genes in ventricular myocytes. Circulation. 1998;97:194–203. doi: 10.1161/01.cir.97.2.194. [DOI] [PubMed] [Google Scholar]
- 15.Gottlieb RA, Burleson KO, Kloner RA, Babior BM, Engler RL. Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J Clin Invest. 1994;94:1612–1628. doi: 10.1172/JCI117504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Teiger E, et al. Apoptosis in pressure overload-induced heart hypertrophy in the rat. J Clin Invest. 1996;97:2891–2897. doi: 10.1172/JCI118747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hamet P, et al. The time window of apoptosis: a new component in the therapeutic strategy for cardiovascular remodeling. J Hypertens Suppl. 1996;14:S65–S70. [PubMed] [Google Scholar]
- 18.Chen C, et al. Myocardial cell death and apoptosis in hibernating myocardium. J Am Coll Cardiol. 1997;30:1407–1412. doi: 10.1016/s0735-1097(97)00309-4. [DOI] [PubMed] [Google Scholar]
- 19.Buja LM, Eigenbrodt ML, Eigenbrodt EH. Apoptosis and necrosis: basic types and mechanisms of cell death. Arch Pathol Lab Med. 1993;117:1208–1214. [PubMed] [Google Scholar]
- 20.Majno G, Joris S. Apoptosis, oncosis, and necrosis: an overview of cell death. Am J Pathol. 1995;146:3–15. [PMC free article] [PubMed] [Google Scholar]
- 21.Reimer KA, Ideker RE. Myocardial ischemia and infarction: anatomic and biochemical substrates for ischemic cell death and ventricular arrhythmias. Hum Pathol. 1987;18:462–475. doi: 10.1016/s0046-8177(87)80031-x. [DOI] [PubMed] [Google Scholar]
- 22.Allen DG, Orchard CH. Myocardial contractile function during ischemia and hypoxia. Circ Res. 1987;60:153–168. doi: 10.1161/01.res.60.2.153. [DOI] [PubMed] [Google Scholar]
- 23.Allen DG, Lee JA, Smith GL. The consequences of simulated ischaemia on intracellular Ca2+ and tension in isolated ferret ventricular muscle. J Physiol. 1989;410:297–323. doi: 10.1113/jphysiol.1989.sp017534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neely JR, Grotyohann LW. Role of glycolytic products in damage to ischemic myocardium. Circ Res. 1984;55:816–824. doi: 10.1161/01.res.55.6.816. [DOI] [PubMed] [Google Scholar]
- 25.Hochachka PW, Buck LT, Doll CJ, Land SC. Unifying theory of hypoxia tolerance: molecular/metabolic defense and rescue mechanisms for surviving oxygen lack. Proc Natl Acad Sci USA. 1996;93:9493–9498. doi: 10.1073/pnas.93.18.9493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buja LM, Entman ML. Modes of myocardial cell injury and cell death in ischemic heart disease. Circulation. 1998;98:1355–1357. doi: 10.1161/01.cir.98.14.1355. [DOI] [PubMed] [Google Scholar]
- 27.Kajstura J, et al. Apoptotic and necrotic myocyte cell deaths are independent contributing variables of infarct size in rats. Lab Invest. 1996;74:86–107. [PubMed] [Google Scholar]
- 28.Ohno M, et al. “Apoptotic” myocytes in infarct area in rabbit hearts may be oncotic myocytes with DNA fragmentation: analysis by immunogold electron microscopy combined with in situ nick end-labeling. Circulation. 1998;98:1422–1430. doi: 10.1161/01.cir.98.14.1422. [DOI] [PubMed] [Google Scholar]
- 29.Gottlieb RA, Gruol DL, Zhu JY, Engler RL. Preconditioning in rabbit cardiomyocytes. J Clin Invest. 1996;97:2391–2398. doi: 10.1172/JCI118683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karwatowska-Prokopczuk E, Nordberg JA, Li HL, Engler RL, Gottlieb RA. Effect of vacuolar proton ATPase on pHi, Ca2+, and apoptosis in neonatal cardiac myocytes during metabolic inhibition/recovery. Circ Res. 1998;82:1139–1144. doi: 10.1161/01.res.82.11.1139. [DOI] [PubMed] [Google Scholar]
- 31.Long X, et al. Enhanced expression of p53 and apoptosis induced by blockade of the vacuolar proton ATPase in cardiomyocytes. J Clin Invest. 1998;101:1453–1461. doi: 10.1172/JCI345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anversa P, Kajstura J. Myocyte cell death in the diseased heart. Circ Res. 1998;82:1231–1233. doi: 10.1161/01.res.82.11.1231. [DOI] [PubMed] [Google Scholar]
- 33.Dennis SC, Gevers W, Opie LH. Protons in ischemia: where do they come from; where do they go? J Mol Cell Cardiol. 1991;23:1077–1086. doi: 10.1016/0022-2828(91)91642-5. [DOI] [PubMed] [Google Scholar]
- 34.Lazdunski M, Frelin C, Vigne P. The sodium/hydrogen exchange system in cardiac cells: its biochemical and pharmacological properties and its role in regulating internal concentrations of sodium and internal pH. J Mol Cell Cardiol. 1985;17:1029–1042. doi: 10.1016/s0022-2828(85)80119-x. [DOI] [PubMed] [Google Scholar]
- 35.Lagadic-Gossmann D, Buckler KJ, Vaughan-Jones RD. Role of bicarbonate in pH recovery from intracellular acidosis in the guinea-pig ventricular myocyte. J Physiol. 1992;458:361–384. doi: 10.1113/jphysiol.1992.sp019422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pierce GN, Czubryt MP. The contribution of ionic imbalance to ischemia/reperfusion-induced injury. J Mol Cell Cardiol. 1995;27:53–63. doi: 10.1016/s0022-2828(08)80007-7. [DOI] [PubMed] [Google Scholar]
- 37.Shimada Y, Hearse DJ, Avkiran M. Impact of extracellular buffer composition on cardioprotective efficacy of Na+/H+ exchange inhibitors. Am J Physiol. 1996;270:H692–H700. doi: 10.1152/ajpheart.1996.270.2.H692. [DOI] [PubMed] [Google Scholar]
- 38.Bond JM, Chacon E, Herman B, Lemasters JJ. Intracellular pH and Ca2+ homeostasis in the pH paradox of reperfusion injury to rat neonatal cardiac myocytes. Am J Physiol. 1993;265:C129–C137. doi: 10.1152/ajpcell.1993.265.1.C129. [DOI] [PubMed] [Google Scholar]
- 39.Li J, Eastman A. Apoptosis in an interleukin-2–dependent cytotoxic T lymphocyte cell line is associated with intracellular acidification. J Biol Chem. 1995;270:3203–3211. doi: 10.1074/jbc.270.7.3203. [DOI] [PubMed] [Google Scholar]
- 40.Gottlieb RA, et al. Cell acidification in apoptosis: granulocyte colony-stimulating factor delays programmed cell death in neutrophils by up-regulating the vacuolar H+ ATPase. Proc Natl Acad Sci USA. 1995;92:5965–5968. doi: 10.1073/pnas.92.13.5965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perez-Sala D, Collado-Escobar D, Mollinedo F. Intracellular alkalinization suppresses lovastatin-induced apoptosis in HL-60 cells through the inactivation of a pH-dependent endonuclease. J Biol Chem. 1998;270:6235–6242. doi: 10.1074/jbc.270.11.6235. [DOI] [PubMed] [Google Scholar]
- 42.Leri A, et al. Stretch-mediated release of angiotensin II induces myocyte apoptosis by activating p53 that enhances the local renin-angiotensin system and decreases the Bcl-2-to-Bax protein ratio. J Clin Invest. 1998;101:1326–1342. doi: 10.1172/JCI316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Graeber TG, et al. Hypoxia induces accumulation of p53 protein but activation of a G1-phase checkpoint by low-oxygen is independent of p53 status. Mol Cell Biol. 1994;14:6264–6277. doi: 10.1128/mcb.14.9.6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Long X, et al. p53 and the hypoxia-induced apoptosis of cultured neonatal rat cardiac myocytes. J Clin Invest. 1997;99:2635–2643. doi: 10.1172/JCI119452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tanaka M, et al. Hypoxia induces apoptosis with enhanced expression of Fas antigen messenger RNA in cultured neonatal rat cardiomyocytes. Circ Res. 1994;75:426–433. doi: 10.1161/01.res.75.3.426. [DOI] [PubMed] [Google Scholar]
- 46.Bialik S, et al. Myocyte apoptosis during acute myocardial infarction in the mouse localizes to hypoxic regions but occurs independently of p53. J Clin Invest. 1997;100:1363–1372. doi: 10.1172/JCI119656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Discher DJ, Bishopric NH, Wu X, Peterson CA, Webster KA. Hypoxia regulates b-enolase and pyruvate kinase-M promoters by modulating Sp1/Sp3 binding to a conserved GC element. J Biol Chem. 1998;273:26087–26093. doi: 10.1074/jbc.273.40.26087. [DOI] [PubMed] [Google Scholar]
- 48.Webster KA, Bishopric NH. Molecular regulation of cardiac myocyte adaptations to chronic hypoxia. J Mol Cell Cardiol. 1992;24:741–751. doi: 10.1016/0022-2828(92)93388-z. [DOI] [PubMed] [Google Scholar]
- 49.Webster KA, Discher D, Bishopric NH. Induction and nuclear accumulation of fos and jun proto-oncogenes in hypoxia cardiac myocytes. J Biol Chem. 1993;268:16852–16859. [PubMed] [Google Scholar]
- 50.Bishopric NH, Simpson PC, Ordahl CP. Induction of the skeletal actin gene in alpha1-adrenoceptor mediated hypertrophy of rat cardiac myocytes. J Clin Invest. 1987;80:1194–1199. doi: 10.1172/JCI113179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bishopric NH, Zeng G, Sato B, Webster KA. Adenovirus E1A inhibits cardiac myocyte-specific gene expression through its amino terminus. J Biol Chem. 1997;272:20584–20594. doi: 10.1074/jbc.272.33.20584. [DOI] [PubMed] [Google Scholar]
- 52.Webster KA, Muscat GEO, Kedes L. Adenovirus E1A products suppress myogenic differentiation and inhibit transcription from muscle-specific promoters. Nature. 1988;332:553–561. doi: 10.1038/332553a0. [DOI] [PubMed] [Google Scholar]
- 53.Soussi T, de Fromentel CC, Breugnot C, May E. Nucleotide sequence of a cDNA encoding the rat p53 nuclear oncoprotein. Nucleic Acids Res. 1988;16:11383. doi: 10.1093/nar/16.23.11384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Belinski SA, Middleton SK, Picksley SM, Hahn FF, Niku K. Analysis of the K-ras and p53 pathways in x-ray–induced lung tumors in the rat. Radiat Res. 1996;145:449–456. [PubMed] [Google Scholar]
- 55.Wu X, Bishopric NH, Discher DJ, Murphy BJ, Webster KA. Physical and functional sensitivity of zinc finger transcription factors to redox change. Mol Cell Biol. 1996;16:1035–1046. doi: 10.1128/mcb.16.3.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jacks T, et al. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 57.Summeray MS, Yellon DM. Characterisation and validation of a murine model of global ischaemia-reperfusion injury. Mol Cell Biochem. 1998;186:61–68. [PubMed] [Google Scholar]
- 58.Hoppe-Seyler F, Butz K. Activation of human papillomavirus type 18 E6-E7 oncogene expression by transcription factor Sp1. Nucleic Acids Res. 1992;20:6701–6706. doi: 10.1093/nar/20.24.6701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bellamy COC. p53 and apoptosis. Br Med Bull. 1997;53:522–538. doi: 10.1093/oxfordjournals.bmb.a011628. [DOI] [PubMed] [Google Scholar]
- 60.Canman CE, Kastan MB. Role of p53 in apoptosis. Adv Pharmacol. 1997;41:429–460. doi: 10.1016/s1054-3589(08)61068-6. [DOI] [PubMed] [Google Scholar]
- 61.Kajstura J, Cheng W, Reiss K, Clark WA. Apoptotic and necrotic myocyte cell death are independent contributing variables of infarct size. Lab Invest. 1996;74:86–107. [PubMed] [Google Scholar]
- 62.Wang L, Ma W, Markovich R, Chen J, Wang PH. Regulation of cardiomyocyte apoptotic signaling by insulin-like growth factor. Circ Res. 1998;83:516–522. doi: 10.1161/01.res.83.5.516. [DOI] [PubMed] [Google Scholar]
- 63.Prentice H, et al. Regulated expression of a foreign gene targeted to the ischemic myocardium. Cardiovasc Res. 1997;35:567–574. doi: 10.1016/s0008-6363(97)00158-2. [DOI] [PubMed] [Google Scholar]
- 64.Laderoute KR, Webster KA. Hypoxia/reoxygenation stimulates jun kinase activity through redox signaling in cardiac myocytes. Circ Res. 1997;80:336–344. doi: 10.1161/01.res.80.3.336. [DOI] [PubMed] [Google Scholar]
- 65.Forgac M. Structure and function of vacuolar class of ATP-driven proton pumps. Physiol Rev. 1989;69:765–796. doi: 10.1152/physrev.1989.69.3.765. [DOI] [PubMed] [Google Scholar]
- 66.Ko LJ, Prives C. p53: puzzle and paradigm. Genes Dev. 1996;10:1054–1072. doi: 10.1101/gad.10.9.1054. [DOI] [PubMed] [Google Scholar]
- 67.Graeber TG, et al. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumors. Nature. 1996;379:88–91. doi: 10.1038/379088a0. [DOI] [PubMed] [Google Scholar]
- 68.Agarwal ML, Taylor WR, Chernov MV, Chernova OB, Stark GR. The p53 network. J Biol Chem. 1998;273:1–4. doi: 10.1074/jbc.273.1.1. [DOI] [PubMed] [Google Scholar]
- 69.Steller H. Mechanisms and genes of cellular suicide. Science. 1995;267:1445–1449. doi: 10.1126/science.7878463. [DOI] [PubMed] [Google Scholar]
- 70.Bennett MR, Littlewood TD, Schwartz SM, Weissberg PL. Increased sensitivity of human vascular smooth muscle cells from atherosclerotic plaques to p53-mediated apoptosis. Circ Res. 1997;81:591–599. doi: 10.1161/01.res.81.4.591. [DOI] [PubMed] [Google Scholar]
- 71.Bennet MR, Evan GI, Schwartz SM. Apoptosis of rat vascular smooth muscle cells is regulated by p53-dependent and -independent pathways. Circ Res. 1995;77:266–273. doi: 10.1161/01.res.77.2.266. [DOI] [PubMed] [Google Scholar]
- 72.Wang J, Guo K, Wills KN, Walsh K. Rb functions to inhibit apoptosis during myocyte differentiation. Cancer Res. 1997;57:351–354. [PubMed] [Google Scholar]
- 73.Hu J, Discher DJ, Bishopric NH, Webster KA. Hypoxia regulates expression of the endothelin-1 gene through a proximal hypoxia-inducible factor-1 binding site on the antisense strand. Biochem Biophys Res Commun. 1998;245:894–899. doi: 10.1006/bbrc.1998.8543. [DOI] [PubMed] [Google Scholar]
- 74.Bacchetti S, Hraham FL. Inhibition of cell proliferation by an adenovirus vector expressing the human wild type p53 protein. Int J Oncol. 1993;3:781–788. doi: 10.3892/ijo.3.5.781. [DOI] [PubMed] [Google Scholar]