

Abstract
Secretion of inflammatory products from neutrophils can be induced by a combination of signals from ligated integrins and receptors for soluble, physiological agonists such as TNF. Here we identify pyk2 in primary human neutrophils; localize it to focal adhesions and podosomes; and demonstrate its tyrosine phosphorylation, activation, and association with paxillin during stimulation of adherent cells by TNF. Tyrphostin A9 emerged as the most potent and selective of 51 tyrosine kinase inhibitors tested against the TNF-induced respiratory burst. Tyrphostin A9 inhibited TNF-induced tyrosine phosphorylation of pyk2 without blocking the cells’ bactericidal activity. Wortmannin, an inhibitor of phosphatidylinositol-3-kinase, potently blocked the TNF-induced respiratory burst and selectively inhibited tyrosine phosphorylation of pyk2. Thus, pyk2 appears to play an essential role in the ability of neutrophils to integrate signals from β2 integrins and TNF receptors.
Introduction
Secretion of microbicidal and histotoxic molecules by polymorphonuclear leukocytes (PMNs) stimulated by soluble, physiological agonists depends on the cells receiving 2 signals (1). Ligation of integrins informs the cells that they have made multiple contacts with extracellular matrix proteins in the tissues. The second signal is delivered through receptors for inflammatory cytokines, chemokines, eicosanoids, glyceryl ethers, formyl peptides, or activated complement. These responses not only underlie antimicrobial defense but contribute to tissue damage in such states as septic shock, respiratory distress syndrome, ischemia-reperfusion, and rheumatoid arthritis. Thus, understanding how PMNs integrate the components of the binary signals controlling their activation may hold a key to new anti-inflammatory therapies.
TNF is a powerful mediator of the innate immune response. Despite intense interest in TNF signaling, it remains unclear how TNF activates PMNs. Most studies of signal transduction through TNF receptors have dealt with gene expression, cell proliferation, or apoptosis, mostly in transformed cell lines. In primary PMNs, however, TNF elicits spreading, exocytosis, and a respiratory burst independently of transcription and translation (2). No role in these responses has been described for any of the proteins thus far known to be associated with the intracellular domains of TNF receptors.
In vitro, TNF-treated PMNs spread on matrix protein-coated surfaces (3) and tyrosine phosphorylate several focal adhesion proteins (4, 5). The latter include paxillin (4, 6) and the tyrosine kinases fgr (7), lyn (8), and syk (9). When spreading is advanced, the cells abruptly begin to release H2O2 and granule contents (10) at a maximal rate (2, 3).
Protein tyrosine phosphorylation is necessary for the respiratory burst of adherent PMNs (5). PMNs from mice rendered genetically deficient in either of 2 src-family tyrosine kinases, fgr or hck, responded to TNF normally, but PMNs from mice doubly deficient in fgr and hck neither spread nor secreted oxidants in response to TNF (11). However, further studies strongly suggested that activation of fgr is not essential for the integrin- and TNF-dependent respiratory burst in human PMNs. On the contrary, activation of fgr and lyn in human PMNs appears to be a consequence, rather than a cause, of the respiratory burst (12). As yet, there has been no identification of any specific tyrosine kinase whose activity is required for the cytokine-induced, adhesion-dependent respiratory burst of human PMNs.
Tyrosine phosphoproteins in TNF-stimulated, adherent human PMNs were localized to punctate structures on the adhering surface containing vinculin, a marker of focal adhesions (6). This observation directed our attention to tyrosine kinases associated with such structures. Among the tyrosine kinases localized to focal adhesions and involved in integrin signaling are the focal adhesion kinases, whose prototype is FAK (13). FAK is activated after β1 and β3 integrin stimulation in fibroblasts and platelets and binds β1 and β3 integrins, src, CSK, paxillin, GRAF, Cas, phosphatidylinositol 3-kinase (PI3K), and sos/Grb2 (reviewed in ref. 14). Human PMNs contain FAK, but its tyrosine phosphorylation is not altered by adhesion (6, 15) or TNF stimulation (6), although spontaneous spreading on laminin promotes it (15).
Recently, a second member of the FAK family has been identified, termed pyk2 (proline-rich tyrosine kinase) (16), CAKβ (17), and RAFTK (18). Pyk2 is more prominent than FAK in unseparated peripheral blood leukocytes. Although pyk2 and FAK are highly homologous, the amino acid sequences surrounding the conserved tyrosines are not identical, suggesting that pyk2’s binding partners, including the src kinases that activate it, may be different than FAK’s. Indeed, in cells possessing both kinases, one but not the other is activated by a given stimulus (19, 20). Pyk2 is activated by TNF in HL-60 leukemic cells (21) but has not been identified in primary PMNs.
The present study demonstrates pyk2 in primary human PMNs; localizes it to focal adhesions and podosomes; and demonstrates its tyrosine phosphorylation, activation, and association with paxillin during activation of the cells by TNF. Of 51 chemically distinct tyrosine kinase inhibitors tested, 1 was a particularly potent and selective inhibitor of the TNF-induced respiratory burst. This agent, tyrphostin A9, also inhibited the tyrosine phosphorylation of pyk2. Wortmannin, a PI3K inhibitor, also blocked pyk2 phosphorylation, while leaving most tyrosine phosphorylation intact. Thus, pyk2 appears to play an essential role in the ability of PMNs to integrate signals from β2 integrins and TNF receptors, and participates in a signal cascade of interest as a potential target for anti-inflammatory therapy.
Methods
PMNs and test agents.
Human PMNs were isolated from heparinized blood of normal donors and processed as described for immunoprecipitation (6), immunoblot (6), microscopy (1), H2O2 release (3), and bactericidal function (22). Pure recombinant human TNF was a gift of Genentech Inc. (South San Francisco, California, USA). PMA was from Sigma Chemical Co. (St. Louis, Missouri, USA). Tyrosine kinase inhibitors and wortmannin were obtained from Calbiochem-Novabiochem Corp. (San Diego, California, USA) or from Samuel Wright at Merck Research Laboratories (Rahway, New Jersey, USA), where they were assembled from commercial sources.
Immunoprecipitation, immunoblot, and immunocytochemistry.
Rabbit anti-pyk2 antibody was a gift of H. Avraham (Harvard Institute of Medicine, Boston, Massachusetts, USA) (18) and was also purchased from Upstate Biotechnology Inc. (Lake Placid, New York, USA). Goat anti-PYK2 was from Santa Cruz Biotechnology Inc. (Santa Cruz, California, USA). Rabbit anti-phosphotyrosine antibody, mouse anti-phosphotyrosine mAb (PY20-IgG2b), recombinant mouse anti-phosphotyrosine mAb coupled to horseradish peroxidase (RC20), and anti-paxillin (349-IgG1) mAb were from Transduction Laboratories (Lexington, Kentucky, USA). Anti-vinculin mAb (hVIN-1-IgG1) was from Sigma Chemical Co.
Immunoprecipitation and immunoblot were as reported (6), including pretreatment with 5 mM diisopropylfluorophosphate, except that for coimmunoprecipitation, cells were lysed in 200 μL of nondenaturing modified RIPA buffer (10 mM Tris-HCl [pH 7.5], 1% Triton X-100, 150 mM NaCl, 1 mM sodium vanadate, 0.1 mM sodium molybdate, 1 mM sodium pyrophosphate, 1 mM NaF, 1 mM phenylmethylsulfonyl fluoride, and 5 μg/mL each of pepstatin A, leupeptin, aprotinin, and chymostatin) on ice for 30 minutes. A Bio-Rad MRC600 laser scanning confocal microscope (Bio-Rad Microscience, Cambridge, Massachusetts, USA) with 2 photomultiplier tubes was used on an inverted Zeiss Axiovert microscope (Zeiss, Oberkochen, Germany) with a ×63 (NA 1.4) Zeiss Plan-apo objective for simultaneous detection of Cy2- and Cy3-conjugated antibodies. Images were processed with Metamorph software (Universal Image Corp., West Chester, Pennsylvania, USA).
Kinase assay.
After immunoprecipitation with goat or rabbit anti-pyk2 antibodies, immune complexes were further washed in kinase buffer (25 mM HEPES, 1 mM DTT, 10 mM MnCl2, 5 μM ATP [pH 7.4]) and then incubated for 30 minutes at room temperature in kinase buffer supplemented with [γ-32P]ATP (0.1 μCi, 10 Ci/mmol). Proteins were separated on SDS-PAGE gels, transferred electrophoretically to PVDF membrane, and autoradiographed.
Results
Identification and localization of pyk2 in human PMNs.
Pyk2 was detected by immunoblot in preparations containing more than 98% PMNs, but only when the cells were treated before cell lysis with diisopropylfluorophosphate. Other protease inhibitors (phenylmethylsulfonyl fluoride, pepstatin A, leupeptin, aprotinin, and chymostatin in combination) were unable to preserve pyk2 (Figure 1a). Thus, protease sensitivity may explain why pyk2 was not identified previously in primary PMNs.
Figure 1.

Identification of pyk2 in human PMNs and its localization to adhesion structures. (a) Pyk2 is evident only in PMNs treated with diisopropylfluorophosphate before lysis (first 2 lanes), in addition to the other protease inhibitors used in the second 2 lanes (see text). PMNs were plated on FBS-coated plates and treated with buffer alone (control; C) or TNF (250 ng/mL; T) for 60 minutes. Identical amounts of protein (100 μg) were loaded in each lane. (b) Indirect immunofluorescence. PMNs adherent to FBS-coated glass coverslips were stimulated with TNF (100 ng/mL) for 60 minutes, fixed, permeabilized, and stained with rabbit anti-pyk2 antibody (panel 1) or mouse anti-vinculin mAb (panel 2), followed by Cy3-conjugated donkey anti-rabbit (panels 1 and 3) or Cy2-conjugated donkey anti-mouse IgG (panels 2 and 4). In panel 3, primary antibody was omitted. In panel 4, an irrelevant mAb was used. The photomicrograph is focused at the plane of cell contact with the substratum. (c) Confocal microscopy of cells prepared as in b and stained with rabbit anti-pyk2 antibody (panel 5) or mouse anti-vinculin mAb (panel 6). Sum of four 0.32-μm-thick images taken from the plane of contact with the substratum.
In adherent PMNs, phosphotyrosine-containing proteins are predominantly localized in punctate podosomes (23) abutting the cell surface in contact with the subjacent matrix. The distribution of pyk2 in primary human PMNs was consistent with localization both to podosomes and to oblong structures at the cell periphery, typical of focal adhesions (Figure 1, b and c). Pyk2 colocalized with vinculin, a component of focal adhesions. Pyk2 in both sets of structures was confined to the cell surface in contact with the substratum (Figure 1b), as confirmed by confocal microscopy (Figure 1c).
TNF-dependent tyrosine phosphorylation of pyk2.
Stimulation of adherent PMNs with TNF causes a time-dependent increase in tyrosine phosphorylation of proteins with an apparent Mr of ∼120 kDa, compatible with pyk2 (5). When PMNs adherent to fibrinogen-coated or FBS-coated plates were stimulated with TNF for 60 minutes, immunoprecipitation with anti-pyk2 followed by immunoblot with anti-phosphotyrosine antibody demonstrated TNF-dependent tyrosine phosphorylation of pyk2 (Figure 2a). Similar amounts of pyk2 were precipitated in all samples (data not shown). No tyrosine phosphorylation of pyk2 was detected in suspended PMNs treated or not with TNF (data not shown). TNF-dependent tyrosine phosphorylation in adherent PMNs was shown to involve β2 integrins (6, 7). Because β2 integrins are the only known receptors for fibrinogen in PMNs (24), it appears that the dependence on β2 integrins extends to signaling for pyk2 phosphorylation.
Figure 2.
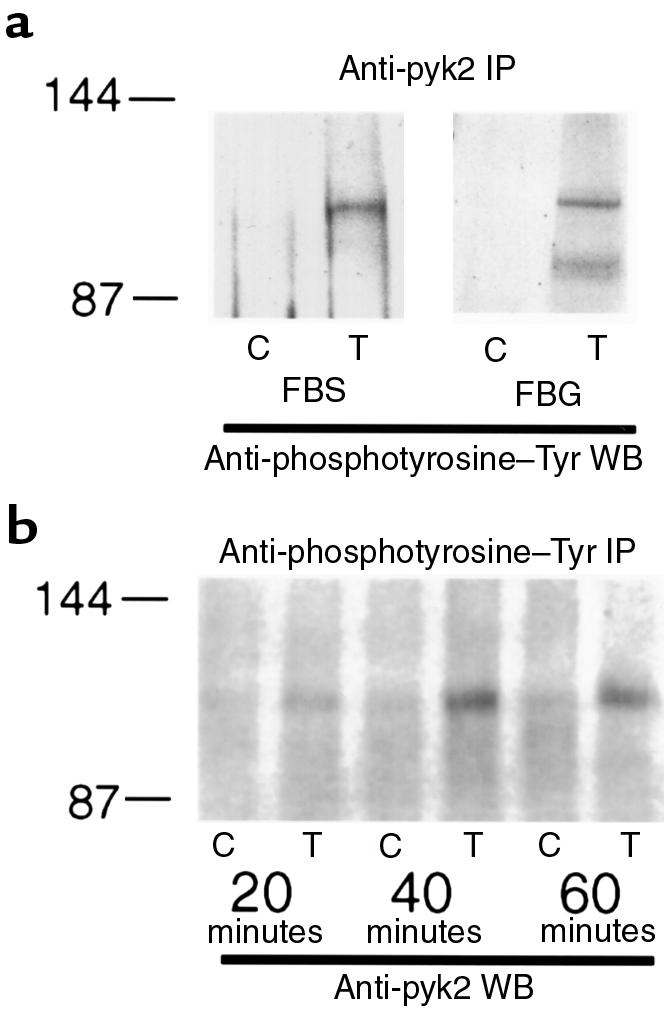
TNF-stimulated tyrosine phosphorylation of pyk2. (a) PMNs were plated on fetal bovine serum–coated (FBS) or fibrinogen-coated (FBG) plates and treated with buffer alone (control; C) or TNF (250 ng/mL; T) for 60 minutes (FBS) or 30 minutes (FBG). Cell lysates (200 μg) were immunoprecipitated (IP) with anti-pyk2 goat antibody. Proteins were separated by reducing SDS-PAGE, transferred to nitrocellulose, and Western blotted (WB) with horseradish peroxidase–conjugated (HRP-conjugated) anti-phosphotyrosine mAb followed by enhanced chemiluminescence (ECL) detection. (b) PMNs were plated on FBS-coated plates and treated with buffer alone (control; C) or TNF (250 ng/mL; T) for 20, 40, or 60 minutes as indicated. Cell lysates (500 μg) were immunoprecipitated (IP) with anti-phosphotyrosine mAb. Proteins were separated by reducing SDS-PAGE, transferred to nitrocellulose, and Western blotted (WB) with anti-pyk2 antibody followed by ECL detection. Molecular mass markers are indicated in kilodaltons.
Lysates of PMNs stimulated with TNF for various times were next immunoprecipitated with anti-phosphotyrosine antibody and immunoblotted with anti-pyk2. Tyrosine phosphorylation of pyk2 was detectable at 20 minutes, intense at 40 minutes, and still marked by 60 minutes, compatible with the time course of TNF-induced spreading (3) (Figure 2b).
Activation of pyk2 by TNF.
PMNs were lysed with nondenaturing buffer after a 60-minute incubation with or without TNF. Anti-pyk2 immunoprecipitates were washed, incubated in kinase buffer in the presence of [γ-32P]ATP, separated by SDS-PAGE, transferred to a PVDF membrane, and subjected to autoradiography (Figure 3). The 2 species to undergo the most prominent increase in incorporation of [γ-32P]ATP had Mr’s compatible with pyk2 and IgG heavy chain. Additional species undergoing TNF-triggered phosphorylation had apparent Mr’s of ∼150, 70, 45, and 34 kDa. Similar amounts of pyk2 were precipitated in the 2 samples (data not shown). These experiments showed that pyk2 becomes more enzymatically active when PMNs are treated with TNF and/or associates with an active kinase in response to TNF.
Figure 3.
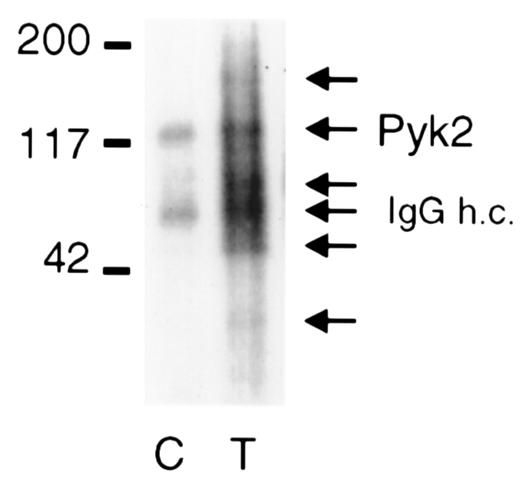
Activation of pyk2 by TNF. PMNs plated on FBS-coated dishes were treated with buffer alone (control; C) or TNF (250 ng/mL; T) for 60 minutes. PMNs were lysed in modified RIPA buffer. Lysates (200 μg) were immunoprecipitated with anti-pyk2 antibody, and the immunoprecipitates were subjected to in vitro kinase assay, separated on reducing SDS-PAGE, transferred to a nylon membrane, and autoradiographed. Prominent species are marked by arrows, including IgG heavy chain (h.c.). Molecular mass markers are indicated in kilodaltons.
TNF-induced association of pyk2 with paxillin.
A major substrate to become tyrosine- and serine- or threonine-phosphorylated in PMNs in response to TNF is the focal adhesion protein paxillin (6). Paxillin can bind FAK (25) or pyk2 (26–30) by an interaction that is phosphotyrosine independent (25). In the previous experiment,(Figure 3), a protein with an Mr of ∼70 kDa was immunoprecipitated with pyk2 from lysates of TNF-treated PMNs and phosphorylated in vitro, consistent with the Mr of paxillin. We therefore immunoprecipitated PMNs lysates with anti-pyk2 antibody and immunoblotted with anti-paxillin mAb. A small amount of paxillin was associated with pyk2 in control PMNs; the amount increased markedly upon stimulation by TNF (Figure 4a). The high apparent Mr of pyk2-associated paxillin suggested that both in basal and stimulated conditions, this fraction of paxillin was heavily phosphorylated, as previously documented for about 5% of paxillin in total lysates of TNF-treated PMNs (6). Results were the same when lysates were immunoprecipitated with anti-paxillin and immunoblotted with anti-pyk2 (Figure 4b).
Figure 4.

TNF-enhanced association of pyk2 with paxillin. (a) Coimmunoprecipitation of paxillin with pyk2. PMNs plated on FBS-coated dishes were treated with buffer alone (control; C) or TNF (250 ng/mL; T) for 60 minutes. PMNs were lysed in modified RIPA buffer. Lysates were immunoprecipitated (IP) with the indicated antibodies or with protein A-Sepharose beads alone, separated on reducing SDS-PAGE, transferred to nitrocellulose, and Western blotted (WB) with anti-paxillin mAb followed by anti-mouse HRP-conjugated antibody with ECL detection. Molecular mass markers are indicated in kilodaltons. (b) Coimmunoprecipitation of pyk2 with paxillin. PMNs plated on FBS-coated dishes were treated with buffer alone (control; C) or TNF (250 ng/mL; T) for 60 minutes and lysed in modified RIPA buffer. Lysates were immunoprecipitated with anti-paxillin mAb, separated on reducing SDS-PAGE, transferred to nitrocellulose, and Western blotted (WB) with anti-paxillin mAb or anti-pyk2 antibody followed by anti-mouse or anti-goat HRP-conjugated antibodies with ECL detection. Molecular mass markers are indicated in kilodaltons.
Screening of a panel of tyrosine kinase inhibitors.
Next, we tested a series of tyrosine kinase inhibitors for their ability to prevent the TNF-induced respiratory burst while sparing the respiratory burst triggered by the phorbol ester PMA. In dose-response fashion, we individually tested 51 structurally diverse compounds (Table 1) known to inhibit a variety of tyrosine kinases. Fourteen failed to exert more than 50% suppression of either the TNF- or PMA-induced respiratory burst up to the highest concentration of compound tested (7.7 μM). Another 10 were inhibitory but blocked the responses to TNF and to PMA equally, suggesting that they affected a shared pathway, were toxic, or interfered with the assay. Four were inhibitory for TNF response at higher concentrations than for PMA. Twenty-three inhibited the TNF response by more than 50% at or below 8 μM, and either did not inhibit the response to PMA or did so at a substantially higher concentration than required to inhibit the response to TNF. The 9 most selective compounds from the last group (i.e., most potent against the response triggered by TNF and least potent against the response triggered by PMA) were further tested by adding them to PMNs 30 minutes in advance of TNF or PMA, which improved the apparent potency of several. Of these, tyrphostin A9 was the compound with the lowest IC50 for TNF (40 nM) and the greatest ratio between the IC50 for PMA and that for TNF (106-fold) (Figure 5a). Lack of toxicity of tyrphostin A9 and selectivity of its actions were illustrated not only by its sparing of the response to PMA over the concentration range 30 nM–2.6 μM, but also by the lack of effect on the ability of PMNs to ingest and kill Salmonella typhimurium (Figure 5b).
Table 1.
Tyrosine kinase inhibitors tested for their effect on PMA- and TNF-stimulated H2O2 production.

Figure 5.

Tyrphostin A9 inhibits the TNF-induced respiratory burst of human PMNs and TNF-induced tyrosine phosphorylation of pyk2 but not their bactericidal activity. (a) Inhibition of TNF-stimulated H2O2 release. PMNs were plated in FBS-coated 96-well plates and preincubated for 30 minutes with the indicated concentrations of tyrphostin A9. Cells were then unstimulated (–) or stimulated (+) either with TNF (100 ng/mL; gray bars) or PMA (100 ng/mL; filled bars), and H2O2 release was recorded 60 minutes later as mean ± SEM of triplicates. (b) Lack of inhibition of bacterial killing. PMNs were preincubated (30 minutes at 4°C) with 0 (filled squares) or 5 μM (open squares) tyrphostin A9 and then infected with 10% autologous serum-opsonized Salmonella typhimurium. Cells were lysed at indicated times and CFUs were determined. Survival of bacteria without PMNs is shown with 0 (filled circles) or 5 μM (open circles) tyrphostin A9. Mean ± SEM of triplicates; most error bars fall within the symbols. (c) PMNs were plated on FBS-coated plates, preincubated for 30 minutes in the presence or absence of tyrphostin A9 (2 μM), and then stimulated with TNF (250 ng/mL) or left untreated for 60 minutes, as indicated. Cell lysates were immunoprecipitated (IP) with anti-pyk2 antibody. Total cell lysate and immunoprecipitates were separated by reducing SDS-PAGE, transferred to nitrocellulose, and Western blotted (WB) with anti-phosphotyrosine mAb (top) or anti-pyk2 antibody (bottom) followed by ECL detection. Molecular mass markers are indicated in kilodaltons.
Inhibition of pyk2 phosphorylation by tyrphostin A9.
A concentration of tyrphostin A9 that totally suppressed the TNF-induced respiratory burst while sparing the PMA-induced respiratory burst decreased the overall level of protein tyrosine phosphorylation stimulated by TNF in adherent cells. TNF-induced tyrosine phosphorylation of pyk2 was completely suppressed (Figure 5c).
Inhibition of pyk2 phosphorylation by wortmannin and PP2.
The most potent inhibitor of the TNF-induced respiratory burst identified in previous work was wortmannin (31), an antagonist of PI3K. We wanted to determine what relation wortmannin’s actions might have to the pathway involving pyk2. When added before or together with TNF, wortmannin inhibited H2O2 production with an IC50 of 0.8 ± 0.1 nM (mean ± SEM), whereas PMA-induced H2O2 release was unaffected (data not shown), demonstrating wortmannin’s lack of toxicity. Delayed addition of 10 nM wortmannin still resulted in complete inhibition of H2O2 production triggered by previously added TNF, provided that the drug was added during the lag period characteristic of the response to TNF (2). Once the lag period had expired, addition of wortmannin had markedly less or no effect on the ongoing secretion of H2O2 (Figure 6a). Thus, a wortmannin-sensitive process was essential for initiating the respiratory burst in response to TNF, but not for maintaining it.
Figure 6.

Wortmannin inhibits the TNF-induced respiratory burst and tyrosine phosphorylation of pyk2. (a) H2O2 release from PMNs measured 120 minutes after the addition of TNF (100 ng/mL; squares) or PMA (100 ng/mL; circles). Wortmannin (10 nM) was added to separate sets of cells at the indicated number of minutes after TNF or PMA. Results are mean ± SEM for triplicates in 1 experiment of 3; some error bars fall within the symbols. (b) Tyrosine phosphorylation. PMNs were plated on FBS-coated plates and treated for 60 minutes with buffer alone or TNF (100 ng/mL) in the presence or absence of wortmannin (Wort) at 10 nM or the src kinase inhibitor PP2 (2.5 μM) as indicated. Cell lysates were immunoprecipitated (IP) with anti-pyk2 antibody. Total cell lysate (top) and anti-pyk2 immunoprecipitates (bottom) were separated by reducing SDS-PAGE, transferred to nitrocellulose, and Western blotted (WB) with anti-phosphotyrosine mAb followed by ECL detection. Molecular mass markers are indicated in kilodaltons.
Next, we tested the effect of wortmannin of pyk2 phosphorylation, comparing it with the most selective src-family tyrosine kinase inhibitor commercially available — PP2 (32). Wortmannin (10 nM) had little effect on the overall level of protein tyrosine phosphorylation stimulated by TNF in adherent cells. Only 2 Mr classes of polypeptides (∼125 and ∼95 kDa, respectively) seemed to be affected (Figure 6b, top). Strikingly, however, tyrosine phosphorylation of pyk2 was completely abolished (Figure 6, bottom). In contrast, PP2 at much higher concentrations (2.5 μM) blocked almost all detectable tyrosine phosphorylation, including that of pyk2 (Figure 6b). Similar amounts of pyk2 were precipitated in each case (data not shown). These findings suggest that pyk2 participates in the signaling cascade leading to the respiratory burst in TNF-treated, adherent PMNs and that PI3K activity is required upstream of pyk2 activation. These results are also consistent with a role for src-family tyrosine kinases. However, attempts at reciprocal coimmunoprecipitation did not reveal a detergent-resistant association between pyk2 and fgr, hck, or syk (data not shown).
Inhibition of neutrophil spreading by wortmannin and tyrphostin A9.
Both wortmannin and tyrphostin A9 inhibited TNF-induced spreading of adherent PMNs, but in a different fashion (Figure 7). Cells pretreated with wortmannin (10 nM) stayed mostly round after TNF treatment, extending only 1 or 2 short pseudopodia. In contrast, tyrphostin A9-treated cells responded to TNF by extending filopodia several micrometers long and began to flatten. Nonetheless, their spreading was arrested compared with TNF-stimulated cells treated with vehicle alone. Thus, wortmannin and tyrphostin A9 both interfered with TNF-induced cytoskeletal reorganization, but wortmannin appeared to act earlier in the sequence than tyrphostin A9.
Figure 7.

Wortmannin and tyrphostin A9 inhibit TNF-induced neutrophil spreading. PMNs adherent to FBS-coated glass coverslips were left untreated (a) or stimulated with TNF (100 ng/mL) for 60 minutes (b–d), either alone (b) or in the presence of wortmannin (10 nM) (c) or tyrphostin A9 (2 μM) (d), and then fixed and photographed with phase-contrast microscopy.
Discussion
The present experiments demonstrate that pyk2 is present in primary human PMNs and that TNF triggers its tyrosine phosphorylation and activation in adherent cells; they also suggest that pyk2 plays an essential role in the TNF-triggered respiratory burst. The delayed and prolonged kinetic of pyk2’s tyrosine phosphorylation is consistent with a downstream role in TNF signaling, such as that leading to cytoskeletal reorganization. The localization of pyk2 primarily to podosomes and focal adhesions positions it for this role, and its TNF-induced association with paxillin may be one manifestation of its execution of this assignment.
Adhesion-controlled cell responsiveness to soluble agonists has become a major paradigm in cell biology and immunology to explain how matrix and soluble signals combine to regulate cellular behavior and fate. In PMNs, adhesion-dependent responses that are independent of transcription and translation constitute an important portion of what the cells contribute to the survival of the organism as well as to its pathology. Experiments with blocking antibodies and/or genetic deficiency states have indicated that β2 and β3 integrins play a major role in the adhesion-dependent responses of PMNs (1, 33). It remains unknown, however, how integrins signal in this setting and how their signals are integrated with those elicited by soluble agonists. Both PI3K action and protein tyrosine phosphorylation are essential for physiological agonists to trigger secretion from adherent human PMNs (5, 9, 31, 34). Pyk2 is the only specific tyrosine kinase in PMNs shown to depend on PI3K for its activation. Thus, a key action of PI3K may be to engineer the activation of pyk2.
The observation that low-dose wortmannin interfered with the initial stages of TNF-induced cell spreading and blocked the TNF-induced tyrosine phosphorylation of pyk2 suggested that PI3K acted early in the TNF signaling pathway and upstream of pyk2. However, wortmannin also blocked the TNF-induced respiratory burst when added at the end of the lag period at a time when cells were fully spread. This suggested that PI3K activity was also required after cell spreading had advanced past the relatively late point at which tyrphostin A9 could interrupt it. The latter observation suggests an additional role for PI3K downstream of pyk2.
Soluble, inflammatory stimuli of PMNs increase the affinity and number of β2 integrins expressed at the cell surface. This process is thought to be mediated in large part by activation of PI3K (35). This could account for the action of PI3K upstream of pyk2, insofar as integrin activation is required to activate pyk2. PIP2 and PIP3 produced by PI3K can activate the serine protein kinase Akt (36). Akt can phosphorylate and activate protein kinase C and the phox47 component of the respiratory burst oxidase (37). Syk associates with both CD18 and fgr in activated, adherent PMNs (38), and activation of syk in response to formylated peptide appears to be required for cell spreading and paxillin phosphorylation (9). Binding of pyk2 to phosphorylated paxillin could help localize pyk2 to podosomes and focal adhesions. We speculate that activated syk and/or src kinases phosphorylate pyk2, and that phosphorylated pyk2 provides binding sites in podosomes and focal adhesions for additional molecules of PI3K. The ensuing tyrosine phosphorylation of PI3K would lead to its activation, membrane association, and further augmentation of integrin activation. This scenario could help explain why the long lag period in the TNF-stimulated respiratory burst has features of a positive-feedback system, terminating abruptly with the release of H2O2 at the maximal rate.
Defining the role of an enzyme within primary human neutrophils is more difficult than in many other types of cells. Neutrophils are not amenable to transfection. They cannot be loaded with presumptive dominant-negative peptides by osmotic shock or electroporation if the goal is to follow the cells’ response to physiological agonists over subsequent hours. Leukemic cell lines such as HL-60 can respond to PMA but do not manifest a TNF-stimulated respiratory burst after differentiation. Primary mouse neutrophils are genetically manipulatable through the germ line, but their TNF-induced respiratory burst is about 20-fold smaller than in human neutrophils (11) and, in our laboratory, has been detectable in only a small proportion of experiments; responses to a formylated peptide have been completely lacking (data not shown).
As an alternative to genetic intervention, we took a pharmacologic approach, surveying 51 tyrosine kinase inhibitors. We sought compounds at least equal in potency to those already shown to be inhibitory of the integrin-dependent, TNF-induced respiratory burst (6) and inactive against the response triggered by phorbol ester, which is integrin independent (1). Tyrphostin A9 was previously identified as a relatively selective inhibitor of the tyrosine kinase activity of the PDGF receptor, with an IC50 (40 nM) similar to that recorded in the present experiments (39). The potency of tyrphostin A9 on PMNs matches that of K252a, the most potent tyrosine kinase inhibitor of the TNF-induced respiratory burst yet described (IC50 = 30 nM), and exceeds by far the potency of others previously studied (genistein, IC50 = 10 μM; ST638, IC50 = 30 μM; piceatannol, IC50 = 1–10 μM) (5, 9). Tyrphostin A9’s inhibition was relatively selective, sparing not only the PMA-induced respiratory burst but also the ability of the PMNs to ingest and kill bacteria. This illustrates the potential to exert an anti-inflammatory action without blocking intracellular bactericidal function.
Identification of the steps proximal and distal to pyk2 in the TNF- and adhesion-dependent responses of human PMNs may reveal relatively myeloid-specific, enzymatic targets for anti-inflammatory therapy.
Acknowledgments
We thank H. Avraham for the gift of anti-pyk2 antibody; S. Wright and M. Shiloh for advice and review of the manuscript; S. Wright, R. Newbold, P. Detmers, V. Bansal, and S. Vaidya for help assembling reagents; and Genentech Inc. for recombinant TNF. This study was supported by National Institutes of Health (NIH) grant CAAI45218 and by NIH Medical Science Training Program grant GM07739 (to G.J. Lyon).
References
- 1.Nathan CF, et al. Cytokine-induced respiratory burst of human neutrophils: dependence on extracellular matrix proteins and CD11/CD18 integrins. J Cell Biol. 1989;109:1341–1349. doi: 10.1083/jcb.109.3.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nathan CF. Neutrophil activation on biological surfaces. Massive secretion of hydrogen peroxide in response to products of macrophages and lymphocytes. J Clin Invest. 1987;80:1550–1560. doi: 10.1172/JCI113241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nathan C, Sanchez E. Tumor necrosis factor and CD11/CD18 (β2) integrins act synergistically to lower cAMP in human neutrophils. J Cell Biol. 1990;111:2171–2181. doi: 10.1083/jcb.111.5.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Graham IL, Anderson DC, Holers VM, Brown EJ. Complement receptor 3 (CR3, Mac-1, integrin alpha M beta 2, CD11b/CD18) is required for tyrosine phosphorylation of paxillin in adherent and nonadherent neutrophils. J Cell Biol. 1994;127:1139–1147. doi: 10.1083/jcb.127.4.1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fuortes M, Jin WW, Nathan C. Adhesion-dependent protein tyrosine phosphorylation in neutrophils treated with tumor necrosis factor. J Cell Biol. 1993;120:777–784. doi: 10.1083/jcb.120.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fuortes M, Jin WW, Nathan C. Beta 2 integrin–dependent tyrosine phosphorylation of paxillin in human neutrophils treated with tumor necrosis factor. J Cell Biol. 1994;127:1477–1483. doi: 10.1083/jcb.127.5.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berton G, Fumagalli L, Laudanna C, Sorio C. Beta 2 integrin–dependent protein tyrosine phosphorylation and activation of the FGR protein tyrosine kinase in human neutrophils. J Cell Biol. 1994;126:1111–1121. doi: 10.1083/jcb.126.4.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yan SR, Fumagalli L, Berton G. Activation of SRC family kinases in human neutrophils. Evidence that p58C-FGR and p53/56LYN redistributed to a Triton X-100–insoluble cytoskeletal fraction, also enriched in the caveolar protein caveolin, display an enhanced kinase activity. FEBS Lett. 1996;380:198–203. doi: 10.1016/0014-5793(96)00029-4. [DOI] [PubMed] [Google Scholar]
- 9.Fernandez R, Suchard SJ. Syk activation is required for spreading and H2O2 release in adherent human neutrophils. J Immunol. 1998;160:5154–5162. [PubMed] [Google Scholar]
- 10.Berton G, Laudanna C, Sorio C, Rossi F. Generation of signals activating neutrophil functions by leukocyte integrins: LFA-1 and gp150/95, but not CR3, are able to stimulate the respiratory burst of human neutrophils. J Cell Biol. 1992;116:1007–1017. doi: 10.1083/jcb.116.4.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lowell CA, Fumagalli L, Berton G. Deficiency of Src family kinases p59/61hck and p58c-fgr results in defective adhesion-dependent neutrophil functions. J Cell Biol. 1996;133:895–910. doi: 10.1083/jcb.133.4.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yan SR, Berton G. Regulation of src family tyrosine kinase activities in adherent human neutrophils. J Biol Chem. 1996;271:23464–23471. doi: 10.1074/jbc.271.38.23464. [DOI] [PubMed] [Google Scholar]
- 13.Schaller MD, et al. pp125FAK a structurally distinctive protein-tyrosine kinase associated with focal adhesions. Proc Natl Acad Sci USA. 1992;89:5192–5196. doi: 10.1073/pnas.89.11.5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hanks SK, Polte TR. Signaling through focal adhesion kinase. Bioessays. 1997;19:137–145. doi: 10.1002/bies.950190208. [DOI] [PubMed] [Google Scholar]
- 15.Fernandez R, Boxer LA, Suchard SJ. Beta2 integrins are not required for tyrosine phosphorylation of paxillin in human neutrophils. J Immunol. 1997;159:5568–5575. [PubMed] [Google Scholar]
- 16.Lev S, et al. Protein tyrosine kinase pyk2 involved in Ca(2+)-induced regulation of ion channel and MAP kinase functions. Nature. 1995;376:737–745. doi: 10.1038/376737a0. [DOI] [PubMed] [Google Scholar]
- 17.Sasaki H, et al. Cloning and characterization of cell adhesion kinase beta, a novel protein-tyrosine kinase of the focal adhesion kinase subfamily. J Biol Chem. 1995;270:21206–21219. doi: 10.1074/jbc.270.36.21206. [DOI] [PubMed] [Google Scholar]
- 18.Avraham S, et al. Identification and characterization of a novel related adhesion focal tyrosine kinase (RAFTK) from megakaryocytes and brain. J Biol Chem. 1995;270:27742–27751. doi: 10.1074/jbc.270.46.27742. [DOI] [PubMed] [Google Scholar]
- 19.Schaller MD, Sasaki T. Differential signaling by the focal adhesion kinase and cell adhesion kinase beta. J Biol Chem. 1997;272:25319–25325. doi: 10.1074/jbc.272.40.25319. [DOI] [PubMed] [Google Scholar]
- 20.Zheng CH, et al. Differential regulation of Pyk2 and focal adhesion kinase (FAK). The C-terminal domain of FAK confers response to cell adhesion. J Biol Chem. 1998;273:2384–2389. doi: 10.1074/jbc.273.4.2384. [DOI] [PubMed] [Google Scholar]
- 21.Tokiwa G, Dikic I, Lev S, Schlessinger J. Activation of Pyk2 by stress signals and coupling with JNK signaling pathway. Science. 1996;273:792–794. doi: 10.1126/science.273.5276.792. [DOI] [PubMed] [Google Scholar]
- 22.Shiloh MU, Ruan J, Nathan C. Evaluation of bacterial survival and phagocyte function with a fluorescence-based microplate assay. Infec Immun. 1997;65:3193–3198. doi: 10.1128/iai.65.8.3193-3198.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones SL, Brown EJ. FcgammaRII-mediated adhesion and phagocytosis induce L-plastin phosphorylation in human neutrophils. J Biol Chem. 1996;271:14623–14630. doi: 10.1074/jbc.271.24.14623. [DOI] [PubMed] [Google Scholar]
- 24.Loike JD, et al. CD11c/CD18 on neutrophils recognizes a domain at the N terminus of the A alpha chain of fibrinogen. Proc Natl Acad Sci USA. 1991;88:1044–1048. doi: 10.1073/pnas.88.3.1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hildebrand JD, Schaller MD, Parsons JT. Paxillin, a tyrosine phosphorylated focal adhesion-associated protein binds to the carboxyl terminal domain of focal adhesion kinase. Mol Biol Cell. 1995;6:637–647. doi: 10.1091/mbc.6.6.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Salgia R, et al. The related adhesion focal tyrosine kinase forms a complex with paxillin in hematopoietic cells. J Biol Chem. 1996;271:31222–31226. doi: 10.1074/jbc.271.49.31222. [DOI] [PubMed] [Google Scholar]
- 27.Hiregowdara D, Avraham H, Fu YG, London R, Avraham S. Tyrosine phosphorylation of the related adhesion focal tyrosine kinase in megakaryocytes upon stem cell factor and phorbol myristate acetate stimulation and its association with paxillin. J Biol Chem. 1997;272:10804–10810. doi: 10.1074/jbc.272.16.10804. [DOI] [PubMed] [Google Scholar]
- 28.Li X, Earp HS. Paxillin is tyrosine phosphorylated by and preferentially associates with the calcium-dependent tyrosine kinase in rat liver epithelial cells. J Biol Chem. 1997;272:14341–14348. doi: 10.1074/jbc.272.22.14341. [DOI] [PubMed] [Google Scholar]
- 29.Ganju RK, et al. RAFTK, a novel member of the focal adhesion kinase family, is phosphorylated and associates with signaling molecules upon activation of mature T lymphocytes. J Exp Med. 1997;185:1055–1063. doi: 10.1084/jem.185.6.1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ostergaard HL, Lou O, Arendt CW, Berg NN. Paxillin phosphorylation and association with Lck and Pyk2 in anti-Cd3– or anti-Cd45–stimulated T cells. J Biol Chem. 1998;273:5692–5696. doi: 10.1074/jbc.273.10.5692. [DOI] [PubMed] [Google Scholar]
- 31.Laudanna C, Rossi F, Berton G. Effect of inhibitors of distinct signalling pathways on neutrophil O2- generation in response to tumor necrosis factor-alpha, and antibodies against CD18 and CD11a: evidence for a common and unique pattern of sensitivity to wortmannin and protein tyrosine kinase inhibitors. Biochem Biophys Res Commun. 1993;190:935–940. doi: 10.1006/bbrc.1993.1139. [DOI] [PubMed] [Google Scholar]
- 32.Hanke JH, et al. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J Biol Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- 33.Lindberg FP, et al. Decreased resistance to bacterial infection and granulocyte defects in IAP-deficient mice. Science. 1996;274:795–798. doi: 10.1126/science.274.5288.795. [DOI] [PubMed] [Google Scholar]
- 34.Ding J, Vlahos CJ, Liu R, Brown RF, Badwey JA. Antagonists of phosphatidylinositol 3-kinase block activation of several novel protein kinases in neutrophils. J Biol Chem. 1995;270:11684–11691. doi: 10.1074/jbc.270.19.11684. [DOI] [PubMed] [Google Scholar]
- 35.Jones SL, Knaus UG, Bokoch GM, Brown EJ. Two signaling mechanisms for activation of alphaM beta2 avidity in polymorphonuclear neutrophils. J Biol Chem. 1998;273:10556–10566. doi: 10.1074/jbc.273.17.10556. [DOI] [PubMed] [Google Scholar]
- 36.Klippel A, Kavanaugh WM, Pot D, Williams LT. A specific product of phosphatidylinositol 3-kinase directly activates the protein kinase Akt through its pleckstrin homology domain. Mol Cell Biol. 1997;17:338–344. doi: 10.1128/mcb.17.1.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Didichenko SA, et al. Constitutive activation of protein kinase B and phosphorylation of p47phox by a membrane-targeted phosphoinositide 3-kinase. Curr Biol. 1996;6:1271–1278. doi: 10.1016/s0960-9822(02)70713-6. [DOI] [PubMed] [Google Scholar]
- 38.Yan SR, Huang M, Berton G. Signaling by adhesion in human neutrophils: activation of the p72syk tyrosine kinase and formation of protein complexes containing p72syk and Src family kinases in neutrophils spreading over fibrinogen. J Immunol. 1997;158:1902–1910. [PubMed] [Google Scholar]
- 39.Bilder GE, et al. Tyrphostins inhibit PDGF-induced DNA synthesis and associated early events in smooth muscle cells. Am J Physiol. 1991;260:C721–C730. doi: 10.1152/ajpcell.1991.260.4.C721. [DOI] [PubMed] [Google Scholar]