

Significance
The photosynthetic cyanobacteria are promising candidates for the sustainable production of a plethora of plant secondary metabolites, which are difficult to produce and purify in other systems. Many secondary metabolites are beneficial to human health. For instance, the phenylpropanoids, which are derived from p-coumaric acid, have anticancer, antiviral, and anti-inflammatory properties. Here, we constructed a strain of cyanobacterium Synechocystis 6803 that heterologously expressed a foreign gene encoding a tyrosine ammonia lyase, which converts tyrosine into p-coumaric acid and lacked a native laccase that degrades p-coumaric acid. The strain secreted ∼82.6 mg/L p-coumaric acid, which was readily extracted and purified from the culture medium. We thus show that cyanobacteria may indeed be used to sustainably produce plant secondary metabolites.
Abstract
p-Coumaric acid is the precursor of phenylpropanoids, which are plant secondary metabolites that are beneficial to human health. Tyrosine ammonia lyase catalyzes the production of p-coumaric acid from tyrosine. Because of their photosynthetic ability and biosynthetic versatility, cyanobacteria are promising candidates for the production of certain plant metabolites, including phenylpropanoids. Here, we produced p-coumaric acid in a strain of transgenic cyanobacterium Synechocystis sp. Pasteur Culture Collection 6803 (hereafter Synechocystis 6803). Whereas a strain of Synechocystis 6803 genetically engineered to express sam8, a tyrosine ammonia lyase gene from the actinomycete Saccharothrix espanaensis, accumulated little or no p-coumaric acid, a strain that both expressed sam8 and lacked slr1573, a native hypothetical gene shown here to encode a laccase that oxidizes polyphenols, produced ∼82.6 mg/L p-coumaric acid, which was readily purified from the growth medium.
Phenylpropanoids, including coumaric acid, caffeic acid, and resveratrol, represent the largest group of secondary metabolites produced by plants. Secondary metabolites are mainly produced to protect the plant against stresses, such as infections, wounding, and UV irradiation (1). Many plant-derived phenolic compounds, such as flavonoids, coumarins, stilbenoids, and lignin, are derivatives of phenylpropanoids and play important roles in plant growth and development (2). Phenylalanine or tyrosine are the precursors of the general phenylpropanoid pathway. Phenylpropanoid biosynthesis begins with the deamination of tyrosine to p-coumaric acid by tyrosine ammonia lyase (TAL) (Fig. 1), or the deamination of phenylalanine to trans-cinnamic acid. Both of these steps are regulated by developmental and environmental stimuli (3).
Fig. 1.

Partial phenylpropanoid biosynthetic pathway. Tyrosine is the starting compound for phenylpropanoid biosynthesis. p-Coumaric acid, an intermediate of other phenylpropanoids, is formed by TAL. p-Coumaric acid is further converted into 4-coumarcyl-CoA in the presence of 4CL. Three malonyl-CoA molecules are added to 4-coumaroyl-CoA by an STS enzyme to form trans-resveratrol. C4H, cinnamate-4-hydroxylase; PAL, phenylalanine ammonia lyase.
Because of their anticancer, antioxidant, antiviral, and anti-inflammatory properties, phenylpropanoid compounds are the focus of intensive research (4–9). However, the phenylpropanoid compounds currently used in medicinal and cosmetic applications are mainly isolated and purified from plant extracts or from cultivated plant cells with relatively low yield, because it is technically challenging and expensive to chemically synthesize these compounds (10). Therefore, there is much interest in developing novel biosynthesis techniques to produce phenylpropanoids in an efficient and cost-effective manner.
Microorganisms are promising candidates for the large scale production of phenylpropanoids. The basic strategy for engineering microbial production is to transform microorganisms with a specific set of genes encoding biosynthetic enzymes. For instance, a recombinant Saccharomyces cerevisiae (yeast) strain heterologously expressing coumaroyl-CoA ligase (4CL) from Populus trichocarpa Torr. & Gray × Populus deltoids Marsh (hybrid poplar) and stilbene synthase (STS) from Vitis vinifera (grapevine) produced 1.45 µg/L trans-resveratrol when the culture medium was supplemented with the resveratrol precursor p-coumaric acid (11). Furthermore, a metabolically engineered Escherichia coli strain carrying a 4CL gene from Lithospermum erythrorhizon (gromwell) and a STS gene from Arachis hypogaea (groundnut) produced 171 mg/L resveratrol (12) upon supplementation with p-coumaric acid. In addition, a transgenic E. coli strain overexpressing an endogenous 4-hydroxyphenlacetate 3-hydroxylase gene synthesized 50 mg/L caffeic acid (13).
Because the production of many plant secondary metabolites requires chloroplast and membrane functions (e.g., photosynthesis and cellular reduction), systems that rely on yeast and E. coli are limited. Cyanobacteria have not been used extensively to produce proteins in industrial and pharmaceutical settings. However, this system offers potential advantages over other bioproduction systems. Cyanobacteria may be used to generate high levels of protein and, because of their photosynthetic activity, may be ideal for synthesizing plant secondary metabolites. Furthermore, these autotrophic organisms can be cultured sustainably on low-cost growth media.
We previously expressed Arabidopsis p-coumarate-3-hydroxylase (C3H) in Synechocystis sp. Pasteur Culture Collection 6803 (hereafter Synechocystis 6803) and showed that this protein catalyzed the conversion of exogenously supplied p-coumaric acid into caffeic acid (14). In this study, we report for the first time to our knowledge the successful production of p-coumaric acid by a strain of Synechocystis 6803 heterologously expressing a TAL gene from Saccharothrix espanaensis and lacking a native putative gene encoding a laccase (Slr1573). The transgenic Synechocystis 6803 strain produced ∼82.6 mg/L p-coumaric acid, most of which was readily extracted and purified from the growth medium.
Results
Integration of sam8 into the Synechocystis 6803 Genome and Expression Analysis.
A construct targeting sam8 to a neutral site of the Synechocystis 6803 genome was assembled and introduced into the cyanobacterium as described in Experimental Procedures. To verify that the gene was stably integrated into the genome, genomic DNA extracted from the sam8 transformant was subjected to PCR analysis using three pairs of primers that bind to different regions of the sam8 locus (Fig. 2B). Genomic DNA from the wild-type strain was used as a control. PCRs using genomic DNA from the sam8 transformant as template with primer pairs a+c, b+d, and a+d are expected to generate amplification products of specific sizes (2.3 kb, 3.9 kb, and 4.9 kb, respectively). For PCR using the wild-type genomic DNA as template, only primer pair a+d is expected to produce an amplification product (1.2 kb), and the product is expected to be smaller than that obtained using sam8 genomic DNA as template (4.9 kb). As shown in Fig. 2C, we inserted sam8 into each copy of the genome in the transformant (i.e., sam8 was completely segregated).
Fig. 2.
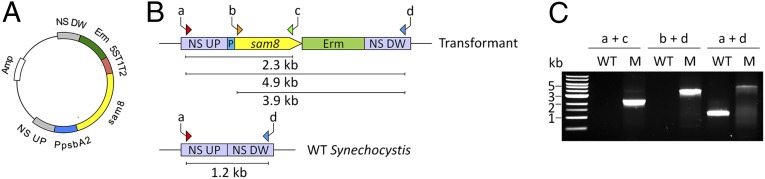
Plasmid construction and transformation of Synechocystis 6803 with sam8. (A) sam8 was cloned into the pBluescript SK(+) plasmid. The psbA2 promoter (PpsbA2), erythromycin-resistance cassette (Erm), upstream (NS UP) and downstream (NS DW) regions of the neutral site, and the E. coli 5S t1t2 terminator were cloned into the plasmid as well. Amp: ampicillin-resistance cassette. (B) Simplified scheme of wild-type (empty vector) and sam8 transformant Synechocystis 6803. Primers used for PCR screening are indicated (arrows labeled a-d) and the expected sizes of PCR products are shown as lines beneath each map. (C) PCR screen to test for the insertion of sam8 in the mutant. WT, wild type; T, sam8 transformant. The positions of the primers are indicated in B.
To determine whether the sam8 transformant is able to express the integrated sam8 gene, we analyzed the transcription of sam8 by reverse transcriptase (RT)-PCR. Total RNA was extracted from the transformant culture, and first-strand cDNA was prepared using random primers. The same procedure was used to obtain the wild-type Synechocystis 6803 cDNA, which was used as a negative control. Whereas sam8-specific primers amplified a fragment of the expected size from cDNA isolated from the sam8 transformant, they did not yield a product when combined with the wild-type cDNA or with samples that lacked RT or contained water instead of template (Fig. 3A).
Fig. 3.

Expression analysis of the sam8 transformant. (A) RT-PCR analysis of the sam8 Synechocystis 6803transformant. (Upper) PCR analysis using primers that bind to sam8. (Upper) PCR analysis using primers that bind to cDNA from 16S rRNA, which was used as a loading control. Lanes: M, DNA ladder; dH2O, negative control for PCR using water as template; WT+RT: RT-PCR using RNA from wild-type Synechocystis 6803 as template for the reverse transcription; WT-RT: as for WT+RT but with no RT in the reaction; sam8+RT: RT-PCR using RNA from the Synechocystis 6803 mutant transformed with sam8 as template for the reverse transcription; sam8-RT: as for sam8+RT, but with no RT in the reaction. (B) SDS/PAGE of the sam8 Synechocystis 6803 transformant. Total cell extract of wild-type Synechocystis 6803 and the sam8 transformant were fractionated by SDS/PAGE and stained with Coomassie blue. Lanes: WT, crude extracts from wild-type Synechocystis 6803; sam8, crude extracts from the sam8-expressing Synechocystis 6803 transformant. TAL is labeled. (C) Western blot analysis of the sam8 Synechocystis 6803 transformant. The proteins in B were blotted onto a nitrocellulose membrane and probed with anti-TAL antibody.
Total cell extract from the sam8 strain and wild-type Synechocystis 6803 was analyzed for TAL expression using SDS/PAGE (Fig. 3B). A 55-kDa band was present in total cell extract from the sam8 strain but not in that from the wild type. Immunoblot analysis was performed using anti-TAL antibody (Fig. 3C), and TAL was detected in the sam8 strain but not in the wild type. This confirmed that TAL was expressed in the Synechocystis 6803 transformant.
LC/MS Analysis of p-Coumaric Acid in the Synechocystis 6803 Strain Expressing sam8.
To test whether the Synechocystis 6803 strain expressing TAL is able to produce p-coumaric acid, the culture was initially grown to an optical density at 730 nm (hereafter OD730) of ∼0.7 (∼6 ×107 cells/mL), concentrated to ∼6 ×108 cells per milliliter in fresh medium, and grown continuously for 7 d. Samples of culture medium were collected every day, extracted with ethyl acetate, and analyzed using high-performance liquid chromatography (HPLC) and liquid chromatography/mass spectrometry (LC/MS).
No peak in the HPLC chromatogram (Fig. 4A) matched the retention time and spectrum of the p-coumaric acid standard. Rather, we observed a peak at 24 min with an m/z value of 488.8, which is much larger than that of p-coumaric acid (m/z 162.9). The identity of the peak was not investigated, but we predict that it is the polymerization compound of 4-vinylphenol, which is the decarboxylation product of p-coumaric acid.
Fig. 4.

LC/MS analysis of p-coumaric acid production. Synechocystis 6803 cells were incubated in BG-11 medium at 30 °C. The culture medium was extracted with ethyl acetate and subjected to LC/MS analysis. (A) HPLC profile of medium extract from the stable sam8 transformant. (B) HPLC profile of medium extract from Synechocystis 6803 transformed by triparental mating with sam8. (C) HPLC profile of medium extract from the sam8∆slr1573 strain. (D) HPLC profile of medium extract from the wild-type Synechocystis 6803 strain. (E) p-Coumaric acid standard (molecular mass: 164 kDa). (F) The mass spectrum of the peak at 18.2 min (p-coumaric acid) in C.
This initial result suggests that the heterogeneous expression level of TAL was not sufficient to produce p-coumaric acid or that the p-coumaric acid produced was modified or degraded inside the cells.
Triparental Mating for sam8 Overexpression in Synechocystis 6803.
To increase the expression level of sam8, we expressed this gene in Synechocystis 6803 using pSL1211, a high-level expression vector, through triparental mating. Transformed pSL1211-sam8 was extracted from Synechocystis 6803 transformant, and no rearrangements were detected using restriction enzyme digestion analysis.
The sam8 exconjugant was cultured under selective medium (Experimental Procedures), and culture medium was extracted daily with organic solvent for a week and analyzed by LC/MS. We detected a small p-coumaric acid peak in the sample at day 4 (Fig. 4B; 18.2 min). In addition, we identified a peak at 15.3 min as 4-vinylphenol, the decarboxylation product of p-coumaric acid. A large unidentified molecule with an m/z value of 488.8 was also present in this HPLC chromatogram.
Enzymatic Analysis of Slr1573.
The low level or lack of p-coumaric acid in the transgenic strain prompted us to investigate whether Synechocystis 6803 contains a bacterial laccase that degrades phenolic compounds such as p-coumaric acid. To this end, we searched the Synechocystis 6803 genome for sequences with homology to the Bacillus subtilis laccase CotA (15) gene and identified Slr1573 as a candidate laccase protein in Synechocystis 6803. The amino acid sequence of Slr1573 exhibits 31% similarity with that of CotA (Fig. S1). To test whether Slr1573 possesses laccase activity, we expressed slr1573 in E. coli strain BL21 (Fig. 5A). Total cell extracts were prepared from culture at 0, 1, 2, 3, and 4 h after isopropyl-β-thiogalactoside (IPTG) induction. The BL21 strain that lacked plasmid was similarly treated and used as a control. Cell extracts containing about 20 µg of protein were analyzed by SDS/PAGE (Fig. 5B). By 3 h after induction, a 32-kDa protein started to accumulate in the strain expressing slr1573. The size of the band matches the calculated molecular mass of His-laccase (i.e., ∼33 kDa). We then purified the Slr1573 protein by affinity chromatography (Fig. 5B, lane P1) and verified the protein by MS (Fig. S2). The N-terminal His-tag of the affinity-purified Slr1573 was removed using a Thrombin Cleavage Capture Kit, which also further purified the protein (Fig. 5B, lane P2). The overall purification achieved about 107.7-fold of enrichment, and the yield was about 27.7% (Table S1).
Fig. 5.
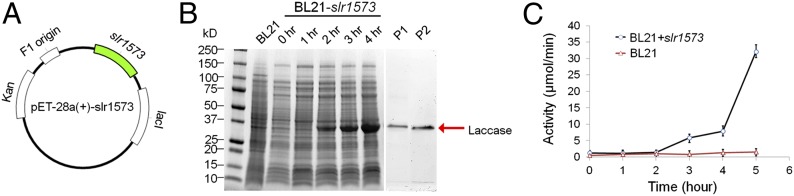
Functional analysis of slr1573 in E. coli. (A) Plasmid construction for laccase expression in E. coli. The slr1573 gene was inserted into pET-28(+) at the multiple cloning sites between BamHI and SacI. (B) Laccase expression in E. coli. Putative laccase proteins are indicated by an arrow. Samples were collected at 0 h, 1 h, 2 h, 3 h, and 4 h after IPTG induction. P1: proteins in cell extract were purified by affinity chromatography; P2: proteins from P1 were treated by thrombin to remove the associated His tags. Calculated molecular mass of His-laccase: 32.66 kDa. (C) Laccase activity of cell extracts from E. coli harboring slr1573. Error bars represent 1 SD for triplicate assays.
We next monitored the laccase activity of the expressed Slr1573 protein in cells using 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulphonic acid as substrate in a spectrophotometric method (16). As shown in Fig. 5C, the laccase activity of the BL21 strain remained low (<2 µmol/min) during the entire assay period. In contrast, the enzymatic activity increased at 3 h and peaked at 5 h (∼35 µmol/min) for the strain expressing Slr1573. After affinity chromatography and thrombin cleavage, the enzymatic activity of the purified Slr1573 was determined to be about 334 U/mg (Table S1). This confirms that Slr1573 possesses strong laccase activity.
Deletion of slr1573 from the Synechocystis 6803 Genome.
We next examined whether inactivation of Slr1573, which exhibits strong laccase activity, would increase p-coumaric acid production in the stable sam8 transformant. We constructed a slr1573-deletion plasmid (Fig. 6A) and transformed it into the stable sam8 transformant to yield sam8 ∆slr1573. The slr1573 gene was replaced by a spectinomycin-resistance cassette. To verify that slr1573 was successfully knocked out, we conducted a PCR analysis using the following three primer pairs (Fig. 6B): e + g (primer pair 1); f + h (primer pair 2); and e + h (primer pair 3). Whereas primer pairs 1 and 2 only amplify fragments from the mutant genomic DNA, primer pair 3 amplifies both mutant and wild-type genomic DNA. All amplification products were of the predicted size (Fig. 6C). Primers e + h only amplified the mutated slr1573 allele (the larger band) from the sam8 ∆slr1573 DNA and not the wild-type allele (smaller band). PCR analysis confirmed that all slr1573 copies in the sam8 ∆slr1573 genome were replaced by the deletion cassette.
Fig. 6.
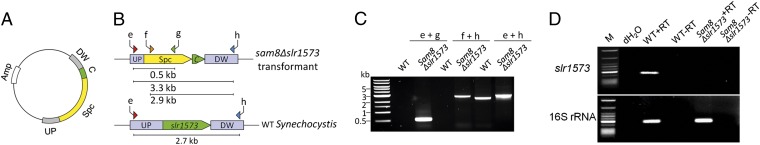
Inactivation of slr1573 in the Synechocystis 6803 sam8 transformant. (A) Plasmid construction for slr1573 knockout. The spectinomycin-resistance cassette was flanked by upstream (UP) and downstream (DW) regions of slr1573 and cloned into the pGEM-T plasmid. C: short C-terminal region of slr1573, which has no gene function; Amp: ampicillin-resistance cassette. (B) Simplified scheme of the wild-type Synechocystis 6803 and sam8-expressing Synechocystis 6803 mutant lacking slr1573. Primers for PCR screening are indicated and the expected sizes of PCR products are shown as bars beneath each diagram. (C) PCR amplification to evaluate slr1573 knockout in the sam8 Δslr1573 strain. Lane WT: wild type; lane sam8Δslr1573: the sam8 Δslr1573 strain. (D) RT-PCR analysis of the sam8 Δslr1573 transformant using primers that bind to cDNA generated from slr1573 (Upper) or 16S rRNA as a loading control (Lower). Lanes: M, DNA ladder; dH2O, negative PCR control using water as template; WT+RT: RT-PCR using RNA from wild-type Synechocystis 6803 as template for the reverse transcription; WT-RT: as for WT+RT, but with no RT in the reaction; sam8 Δslr1573+RT: RT-PCR using RNA from the Synechocystis 6803 sam8-expressing strain that lacked slr1573 as template for the reverse transcription; sam8 Δslr1573-RT: as for sam8 Δslr1573+RT, but with no RT in the reaction.
Removal of slr1573 from the genome was verified by RT-PCR (Fig. 6D). Primers were designed to bind specifically to slr1573. We only detected the slr1573 transcript when total RNA isolated from wild-type Synechocystis 6803 was analyzed in the presence of RT (lane WT+RT). No amplification product was detected when using sam8 ∆slr1573 RNA (lane sam8Δslr1573+RT) or in samples lacking RT or containing water instead of template.
The correct integration and complete segregation of sam8 and deletion of slr1573 were further confirmed by Southern blot analyses (Fig. S3).
Production of p-Coumaric Acid by the Synechocystis 6803 sam8 Δslr1573 Strain.
To analyze the biosynthesis of p-coumaric acid by the Synechocystis 6803 sam8 Δslr1573 strain, cells were grown at 30 °C in Blue–Green (BG)-11 liquid medium to high density for 7 d, and aliquots of culture medium were taken daily and extracted for LC/MS analysis.
Fig. 4C shows the HPLC profiles of the solvent extract of the medium aliquot taken on the fourth day. The peak at 18.2 min was confirmed to be p-coumaric acid by its absorption and mass spectra (Fig. 4F) in comparison with the standards. The titer of p-coumaric acid is about 82.6 mg/L (Fig. 7). Small amounts of 4-vinylphenol were also found at 15.3 min, but this is dramatically reduced by the deletion of slr1573. Thus, the sam8 Δslr1573 strain synthesizes and secretes the plant secondary metabolite p-coumaric acid.
Fig. 7.

Comparison of p-coumaric acid productivity between Synechocystis 6803 sam8 transformants. sam8: stable sam8 transformant; Tp-sam8: Synechocystis 6803 stably transformed with sam8 by triparental mating; sam8Δslr1573: the sam8 Δslr1573 strain. Error bars represent 1 SD for triplicate assays.
Discussion
In this study, we sought to produce p-coumaric acid in Synechocystis 6803. We first expressed sam8, which encodes a TAL, under the control of a native psbA2 promoter. However, this strain did not produce detectable amounts of p-coumaric acid (Figs. 4A and 7). This was somewhat surprising, because C3H, a plant cytochrome P450 enzyme that converts p-coumaric acid into caffeic acid, was previously successfully expressed in Synechocystis 6803 after codon optimization (14). sam8 is derived from the prokaryote S. espanaensis, and all codons exhibit high use frequency in Synechocystis 6803. In addition, previous studies (17) showed that sam8 is transcribed at high levels when heterologously expressed in E. coli and that the encoded TAL enzyme is functional. Therefore, no codon optimization was performed for the present study. Interestingly, we detected a compound with a large m/z value in the culture medium of the sam8 strain but not in that of the wild type. This suggests that the enzyme encoded by sam8 is active in the transgenic cells and that the larger compound might be a derivative of p-coumaric acid (or of another compound synthesized by the heterologously expressed TAL).
After our initial attempt to express sam8 failed, we used two different approaches to produce p-coumaric acid in Synechocystis 6803. The first approach was to increase the copy number of sam8 inside the cell by introducing the gene in a self-replicating plasmid into Synechocystis 6803 through conjugation (i.e., triparental mating), in which a cargo plasmid DNA is transferred from one bacterium (usually E. coli) to another bacterium (Synechocystis 6803 in this case) through cell-to-cell contact (18). To transform Synechocystis 6803 by conjugation, no helper plasmid is needed, because Synechocystis 6803 lacks restriction enzymes (19–21). There were about 29 copies of cargo plasmid per transformant cell, which is equivalent to about 3.3 copies per chromosome (22). Therefore, this transformation approach produces a higher copy number of transgene than does the genome-integration approach. However, this strategy was minimally effective, because only trace amounts of p-coumaric acid were detected in the strain by LC/MS (3.04 mg/L; Fig. 7).
The second approach was to search for and then eliminate genes encoding laccases, which break down p-coumaric acid. Laccases are present in many plants, fungi, and microorganisms and are multicopper-containing proteins belonging to the blue copper oxidase family. These enzymes catalyze the oxidation of a variety of phenolic substrates, including diphenols, polyphenols, diamines, and aromatic amines (23). In this reaction, one electron is removed from the substrate, and oxygen is the terminal electron acceptor. After losing an electron, the substrate becomes a free radical and is unstable. It can be further modified by nonenzymatic reactions, such as hydration, decarboxylation, polymerization, and disproportionation (24, 25). We hypothesized that the sam8-expressing transgenic strain is capable of producing p-coumaric acid but that the p-coumaric acid is oxidized into free radicals and further converted into other derivatives in the presence of endogenous laccase. The compound observed in our LC/MS analysis (the peak at 24 min in Fig. 4A) is likely derived from p-coumaric acid. Based on its m/z value (488.8) and its relation to p-coumaric acid, it is likely that this compound is poly-4-vinylphenol, the polymerization compound of 4-vinylphenol, which is generated by decarboxylation of p-coumaric acid.
We identified a hypothetical gene (slr1573) by a Blastp search of the Synechocystis 6803 genome database using the laccase gene sequence cotA from Bacillus subtilis (15) as the query sequence. We then confirmed that this gene encodes a laccase by conducting an enzymatic assay (Fig. 5) and deleted the gene from the strain expressing sam8. Using this approach, we were able to increase p-coumaric acid production by more than 25-fold compared with the sam8 strain generated by triparental mating (82.6 mg/L; Fig. 7), while decreasing the amount of the putative compound 4-vinylphenol. This further supports our previous hypothesis that the native laccase in Synechocystis 6803 modified p-coumaric acid in vivo, which was then converted into a related compound (Fig. 4A), most likely 4-vinylphenol.
Cyanobacteria have been described as sunlight-driven cell factories that can be harnessed to sustainably produce numerous products, such as biofuels, foods, nutraceuticals, and other biomaterials (26). Several favorable characteristics make cyanobacteria highly suitable for the production of p-coumaric acid, caffeic acid, and other related compounds found in plants. As organisms capable of integrating foreign DNA by double homologous recombination, cyanobacteria are ideal candidates for a synthetic biological approach. They use carbon dioxide from the air and sunlight as their carbon and energy sources, respectively. Some cyanobacteria are natural nitrogen fixers and do not require fixed nitrogen for growth. They produce large quantities of chemical reductants, which are required for the biosynthesis of plant secondary metabolites. More importantly, as demonstrated by the successful functional expression of C3H, which is not functional in E. coli (14), cyanobacteria are able to express plant hydroxylases, which are required for the biosynthesis of phenylpropanoids, a class of plant secondary metabolites. The work reported here represents an important step toward the sustainable production of a plethora of plant secondary metabolites using cyanobacteria.
Experimental Procedures
E. coli Strains and Growth Conditions.
E. coli strains XL1-Blue (Stratagene) and TOP10 (Invitrogen) were used for DNA cloning and plasmid construction. E. coli strain BL21 (DE3) (Invitrogen) was used to overexpress Slr1573. E. coli strain HB101 (American Type Culture Collection) was used in a triparental mating to introduce foreign genes into Synechocystis 6803. Recombinant proteins were overexpressed in BL21 in 2x yeast extract tryptone medium at 30 °C to avoid the formation of inclusion bodies. IPTG (Sigma-Aldrich) was added to the culture (optical density at 600 nm, ∼0.4) to a final concentration of 0.5 mM to induce gene expression. All other E. coli strains were grown at 37 °C with shaking (250 rpm) in LB medium containing the appropriate antibiotics. The following antibiotic concentrations were used: ampicillin (Sigma-Aldrich), 100 µg/mL in both liquid medium and on agar plates; erythromycin (Sigma-Aldrich), 10 µg/mL in liquid medium and 100 µg/mL in agar plates; gentamicin (Sigma-Aldrich), 15 µg/mL in liquid medium and 5 µg/mL in agar plates; and spectinomycin (Sigma-Aldrich), 25 µg/mL in liquid medium and 50 µg/mL in agar plates.
Plasmid Construction.
To construct the sam8 expression plasmid, the 1.5-kb Saccharothrix espanaensis sam8 gene was amplified from the pMB9 plasmid using primers 5′-TCAGAATTCATGACGCAGGTCGTGGAACGTCAGGCT-3′ and 5′-TCACTGCAGTCATCCGAAATCCTTCCCGTCTGCCTCGT-3′ and inserted into pBluescript II SK+ (Stratagene) between the EcoRI and PstI sites. A psbA2 promoter fragment was amplified from the Synechocystis 6803 genome and inserted into the EcoRI –SalI sites of pBluescript II SK+. The sequences of the two primers were 5′-TCAGTCGACGGTATATGGATCATAATTGTATGC-3′ and 5′-TCAGAATTCTTGGTTATAATTCCTTATGTATTTG-3′. The E. coli rrnB (5S rRNA) T1 and T2 terminator (GenBank accession no. U02439) was subcloned into the plasmid between the PstI and SmalI sites. Fragments 1,000 bp upstream and 900 bp downstream of a neutral site (near slr0646 in the Synechocystis 6803 genome) were inserted into the plasmid between KpnI and SalI and SpeI and SacII, respectively. The following primers were used: 5′-TCAGGTACCTTCGATCGTTAGCGCCAA-3′ and 5′-TCAGTCGACATGGGGATCAGCGCTAAAT-3′ for the upstream fragment; and 5′-TCAGCATGCAAGCCCATTTACGTCGTGTT-3′ and 5′-TCACCGCGGCCAACGGTTCCAGGTGACTAT-3′ for the downstream fragment. An erythromycin-resistance cassette was cloned between the SmaI and BamHI sites.
To construct a plasmid for slr1573 expression in E. coli, slr1573 was PCR-amplified from the wild-type Synechocystis 6803 genome using primers 5′-GTACGGATCCATGGGGGACGATCTCTGGGG-3′ and 5′-GTACGAGCTCTCAGTAACTGACAATGCCAG-3′. The PCR fragment was digested with BamHI and SacI and ligated into the pET-28a(+) vector, which had been digested with the same restriction enzymes. The resulting plasmid, pET-28a(+)-slr1573, was transformed into BL21 E. coli-competent cells.
A plasmid was constructed to inactivate slr1573 in the Synechocystis 6803 genome. The 0.8-kb slr1573 gene, together with the 1.2-kb upstream region and 1.2-kb downstream region, was amplified from Synechocystis 6803 genomic DNA using primers 5′-ATGGTCCTCGGAATTTTGCTCG-3′ and 5′-TTGCTCCACCAGCAGAGTTAC-3′ and ligated into the pGEM-T vector (Promega). Using primers 5′-ATCAGGATCCGAATTCCCGGGGATCCGGT-3′ and 5′-TAGTGAATTCGTGATTGATTGAGCAAGCTT-3′, the 2-kb spectinomycin-resistance gene was amplified from php45Ω (27), digested with MfeI, and ligated into the pGEM-T-slr1573 plasmid, which had been digested with MfeI (removing most of slr1573). The resulting plasmid was verified by sequencing and transformed into the Synechocystis 6803 strain containing stably integrated sam8. Transformants were selected for erythromycin and spectinomycin resistance on BG-11 plates. To inactivate every copy of slr1573 in the genome, the transformant was subcultured for four to five generations on agar plates containing increasing concentrations of spectinomycin (from 50 to 150 µg/mL).
Synechocystis 6803 Culture Conditions and Transformation.
Synechocystis 6803 was grown in BG-11 liquid medium (28) at 30 °C under 50 µmol of photons per square meter per second light intensity. When the OD730 reached 0.5–0.7, the cell cultures were centrifuged at 3,500 × g for 10 min and resuspended in fresh BG-11 liquid medium to 8.7 × 108 cells/mL. Then, 2 mg of plasmid DNA was mixed with 400 µL of cells and incubated at 30 °C for 6 h. Cells were then spread on a sterile Nuclepore Track-Etch Membrane (Whatman) on top of a BG-11 agar plate. After a 24-h incubation at 30 °C, the membrane was transferred to fresh BG-11 plates containing the appropriate antibiotics (15 µg/mL) and incubated at 30 °C for about 2 wk, until colonies appeared. Single colonies were isolated on a new BG-11 plate containing antibiotics (50 µg/mL) and inoculated in liquid medium for analysis.
Triparental Mating for sam8 Overexpression in Synechocystis 6803.
sam8 was inserted into pSL1211, a high-level expression vector (22), between XhoI and HindIII to generate the cargo plasmid pSL1211-sam8. For the triparental mating, three strains were mixed: (i) Synechocystis 6803 wild-type strain; (ii) E. coli HB101 strain containing pRL443 plasmid (21); and (iii) E. coli HB101 strain transformed with pSL1211-sam8. The mixed cultures were mated on a nitrocellulose membrane on a BG-11 agar plate at 30 °C for 12 h under 50 µmol of photons per square meter per second light intensity. The membrane was then transferred to a BG-11 plate containing 15 µg/mL gentamicin sulfate. Exconjugants that appeared on the membrane were cultured in BG-11 liquid medium supplemented with 5 µg/mL gentamicin sulfate.
To determine the identity and stability of cargo plasmid transformed into Synechocystis 6803, the total DNA from exconjugants was extracted and transformed into E. coli XL1-Blue–competent cells. Transformant colonies were selected for gentamicin resistance on LB solid medium. Plasmids were extracted from E. coli transformants and verified by restriction digest.
RT-PCR.
Total RNA was isolated from 100-mL Synechocystis 6803 cultures at an OD730 of ∼0 0.5, as previously described (29). RNase-free DNase (Qiagen) was used to remove contaminating genomic DNA. The reverse transcription reaction was carried out using SuperScript III enzyme (Invitrogen) and random primers. The resulting cDNA molecules were amplified by PCR using the following gene-specific primers: 5′-AAGGTTTCCTCAGGCTGGTT-3′ and 5′-CGCGTCGATGTGAACTCT-3′ for amplification of the 16S rRNA gene, which was used as a loading control; 5′-GCTCAACGGGGCGAACTC-3′ and 5′-GGCGTCCAGCGGCGGGCAGAT-3′ for amplification of sam8; and 5′-CCCTATCTCACCTGTGCTTTGTTG-3′ and 5′-GCTGGGGGATGGGTACTGTCTGG-3′ for amplification of slr1573. PCR products were analyzed by 0.8% agarose gel electrophoresis.
Extraction of p-Coumaric Acid from Culture Media.
Synechocystis 6803 cultures were grown in BG-11 medium at 30 °C under normal light (50 µmol of photons per square meter per second) until an OD730 of ∼0.7. Cells were concentrated by centrifugation at 3,500 × g for 10 min and resuspended in BG-11 medium at 1/10 of the original volume. Then, 5 mM glucose was added to the culture, and samples were collected for about 7 d. For extraction, 2 mL of culture was centrifuged at 13,000 rpm for 2 min to remove the cells. The supernatant (medium) was transferred to a new tube, the pH was adjusted to 5.0 with 1 N HCl, and the sample was stored at −20 °C overnight. The medium was extracted twice with an equal volume of ethyl acetate, and the combined extract was dried under continuous nitrogen gas flow. The residue was resuspended in 80% (vol/vol) methanol and used for HPLC and LC/MS analyses.
Synechocystis 6803 Cell Extract Preparation, SDS/PAGE, and Western Blot.
Synechocystis 6803 cultures were grown at 30 °C in BG-11 medium with the addition of glucose and appropriate antibiotics until OD730 reached 0.6–0.8. Cells were washed with and resuspended in thylakoid buffer [20 mM Mes/NaOH (pH 6.4), 5 mM MgCl2, 5 mM CaCl2, 20% glycerol (vol/vol), 1 mM freshly made phenylmethylsulfonyl fluoride, and 5 mM benzamidine] to a final volume of 0.6 mL. The cell suspension was mixed with 0.6-mL glass beads (diameter 0.1 mm; BioSpec Products) prewetted with thylakoid buffer and shaken in a MiniBeadBeater (BioSpec Products) three times for 30 s at 2-min intervals on ice. After centrifugation at 3,000 × g for 10 min to remove unbroken cells and cell debris, the cell suspension was collected as total cell extract.
Protein concentrations were determined using the Coomassie (Bradford) Protein Assay Kit (Thermo Fisher Scientific). SDS/PAGE was performed using 12% resolving gel and 5.5% stacking gel. Cell extracts were incubated with sample loading buffer [60 mM Tris⋅HCl (pH 6.8), 2% (wt/vol) SDS, 5% (vol/vol) 2-mercaptoethanol, 25% (vol/vol) glycerol, and 0.1% bromophenol blue] at 37 °C for 20 min before loading onto the gel. A total of 20 µg of protein was loaded per lane. After electrophoresis, the gels were stained with Coomassie brilliant blue R250. For immunoblot analysis, proteins on SDS/PAGE were transferred onto a nitrocellulose membrane using the Bio-R Trans-Blot system (Bio-Rad), probed with anti-TAL primary antibody (a kind gift from Qiang Wang, Chinese Academy of Sciences, Wuhan, China) and alkaline phosphatase-conjugated secondary antibody (Sigma-Aldrich). The specific protein on the blots was visualized using the 5-bromo-4-chloro-3-indolyl phosphate/nitroblue tetrazolium Kit (Invitrogen).
HPLC and LC/MS Analysis of p-Coumaric Acid Production.
HPLC was carried out by injecting 10 µL of sample into a Thermo HPLC UV6000 system (Thermo Fisher Scientific) with a GRACE Prevail C18 column (4.6 mm × 250 mm; 5 µm) (Alltech Associates). p-Coumaric acid was eluted with 0.5% phosphoric acid (A) and 100% methanol (B) at a flow rate of 1 mL/min. The solvent ratios (A/B) used were as follows: 95/5 from 0 to 2 min, 60/40 from 2 to 4 min, a gradient from 60/40 to 20/80 from 4 to 20 min, 10/90 from 20 to 22 min, and back to 95/5 from 22 to 26 min. The p-coumaric acid peak was identified by comparison with the retention time and UV/Vis spectrum of standards (Sigma-Aldrich) and verified by MS. LC/MS was carried out using an Agilent LC/MSD Trap VL mass spectrophotometer with a Grace Prevail C18 column (4.6 mm × 250 mm; 5 µm). The elution method was the same as that used for HPLC analysis. Electrospray ionization interface was used for mass fragmentation. The negative ion value of the p-coumaric acid standard is m/z 163.1.
Supplementary Material
Acknowledgments
We thank Dr. Peter Wolk for the pRL443 plasmid, Dr. Himadri Pakrasi for the pSL1211 plasmid, Dr. Andreas Bechthold for sam8, and Dr. Qiang Wang for anti-TAL antibody. This work was supported by National Science Foundation Grant MCB1120153, Shandong Province Taishan Scholar Foundation Grant tshw20091014, and Arkansas P3 Center Pilot Seed Grant P3-203.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1323725111/-/DCSupplemental.
References
- 1.Douglas CJ. Phenylpropanoid metabolism and lignin biosynthesis: From weeds to trees. Trends Plant Sci. 1996;1(6):171–178. [Google Scholar]
- 2.Korkina LG. Phenylpropanoids as naturally occurring antioxidants: From plant defense to human health. Cell Mol Biol (Noisy-le-grand) 2007;53(1):15–25. [PubMed] [Google Scholar]
- 3.Dixon RA, Paiva NL. Stress-induced phenylpropanoid metabolism. Plant Cell. 1995;7(7):1085–1097. doi: 10.1105/tpc.7.7.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee KH. Novel antitumor agents from higher plants. Med Res Rev. 1999;19(6):569–596. doi: 10.1002/(sici)1098-1128(199911)19:6<569::aid-med7>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 5.Duthie G, Crozier A. Plant-derived phenolic antioxidants. Curr Opin Lipidol. 2000;11(1):43–47. doi: 10.1097/00041433-200002000-00007. [DOI] [PubMed] [Google Scholar]
- 6.Kris-Etherton PM, et al. Bioactive compounds in foods: Their role in the prevention of cardiovascular disease and cancer. Am J Med. 2002;113(9) Suppl 9B:71S–88S. doi: 10.1016/s0002-9343(01)00995-0. [DOI] [PubMed] [Google Scholar]
- 7.Gescher A. Polyphenolic phytochemicals versus non-steroidal anti-inflammatory drugs: Which are better cancer chemopreventive agents? J Chemother. 2004;16(Suppl 4):3–6. doi: 10.1179/joc.2004.16.Supplement-1.3. [DOI] [PubMed] [Google Scholar]
- 8.Karakaya S. Bioavailability of phenolic compounds. Crit Rev Food Sci Nutr. 2004;44(6):453–464. doi: 10.1080/10408690490886683. [DOI] [PubMed] [Google Scholar]
- 9.de la Lastra CA, Villegas I. Resveratrol as an anti-inflammatory and anti-aging agent: Mechanisms and clinical implications. Mol Nutr Food Res. 2005;49(5):405–430. doi: 10.1002/mnfr.200500022. [DOI] [PubMed] [Google Scholar]
- 10.Fowler ZL, Koffas MA. Biosynthesis and biotechnological production of flavanones: Current state and perspectives. Appl Microbiol Biotechnol. 2009;83(5):799–808. doi: 10.1007/s00253-009-2039-z. [DOI] [PubMed] [Google Scholar]
- 11.Becker JV, et al. Metabolic engineering of Saccharomyces cerevisiae for the synthesis of the wine-related antioxidant resveratrol. FEMS Yeast Res. 2003;4(1):79–85. doi: 10.1016/S1567-1356(03)00157-0. [DOI] [PubMed] [Google Scholar]
- 12.Katsuyama Y, Funa N, Miyahisa I, Horinouchi S. Synthesis of unnatural flavonoids and stilbenes by exploiting the plant biosynthetic pathway in Escherichia coli. Chem Biol. 2007;14(6):613–621. doi: 10.1016/j.chembiol.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 13.Lin Y, Yan Y. Biosynthesis of caffeic acid in Escherichia coli using its endogenous hydroxylase complex. Microb Cell Fact. 2012;11:42. doi: 10.1186/1475-2859-11-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xue Y, Zhang Y, Grace S, He Q. Functional expression of an Arabidopsis p450 enzyme, p-coumarate-3-hydroxylase, in the cyanobacterium Synechocystis PCC 6803 for the biosynthesis of caffeic acid. J Appl Phycol. 2014;26(1):219–226. [Google Scholar]
- 15.Hullo M-F, Moszer I, Danchin A, Martin-Verstraete I. CotA of Bacillus subtilis is a copper-dependent laccase. J Bacteriol. 2001;183(18):5426–5430. doi: 10.1128/JB.183.18.5426-5430.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Childs RE, Bardsley WG. The steady-state kinetics of peroxidase with 2,2′-azino-di-(3-ethyl-benzthiazoline-6-sulphonic acid) as chromogen. Biochem J. 1975;145(1):93–103. doi: 10.1042/bj1450093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choi O, et al. Biosynthesis of plant-specific phenylpropanoids by construction of an artificial biosynthetic pathway in Escherichia coli. J Ind Microbiol Biotechnol. 2011;38(10):1657–1665. doi: 10.1007/s10295-011-0954-3. [DOI] [PubMed] [Google Scholar]
- 18.Ippen-Ihler KA, Minkley EG., Jr The conjugation system of F, the fertility factor of Escherichia coli. Annu Rev Genet. 1986;20(1):593–624. doi: 10.1146/annurev.ge.20.120186.003113. [DOI] [PubMed] [Google Scholar]
- 19.Thiel T, Wolk CP. Conjugal transfer of plasmids to cyanobacteria. Methods Enzymol. 1987;153:232–243. doi: 10.1016/0076-6879(87)53056-7. [DOI] [PubMed] [Google Scholar]
- 20.Elhai J, Wolk CP. Developmental regulation and spatial pattern of expression of the structural genes for nitrogenase in the cyanobacterium Anabaena. EMBO J. 1990;9(10):3379–3388. doi: 10.1002/j.1460-2075.1990.tb07539.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elhai J, Wolk CP. Conjugal transfer of DNA to cyanobacteria. Methods Enzymol. 1988;167:747–754. doi: 10.1016/0076-6879(88)67086-8. [DOI] [PubMed] [Google Scholar]
- 22.Ng W-O, Zentella R, Wang Y, Taylor J-SA, Pakrasi HB. PhrA, the major photoreactivating factor in the cyanobacterium Synechocystis sp. strain PCC 6803 codes for a cyclobutane-pyrimidine-dimer-specific DNA photolyase. Arch Microbiol. 2000;173(5-6):412–417. doi: 10.1007/s002030000164. [DOI] [PubMed] [Google Scholar]
- 23.Gianfreda L, Xu F, Bollag J-M. Laccases: A useful group of oxidoreductive enzymes. Bioremediat J. 1999;3(1):1–26. [Google Scholar]
- 24.Thurston CF. The structure and function of fungal laccases. Microbiology. 1994;140(1):19–26. [Google Scholar]
- 25.Claus H. Laccases: Structure, reactions, distribution. Micron. 2004;35(1-2):93–96. doi: 10.1016/j.micron.2003.10.029. [DOI] [PubMed] [Google Scholar]
- 26.Ducat DC, Way JC, Silver PA. Engineering cyanobacteria to generate high-value products. Trends Biotechnol. 2011;29(2):95–103. doi: 10.1016/j.tibtech.2010.12.003. [DOI] [PubMed] [Google Scholar]
- 27.Fellay R, Frey J, Krisch H. Interposon mutagenesis of soil and water bacteria: A family of DNA fragments designed for in vitro insertional mutagenesis of gram-negative bacteria. Gene. 1987;52(2-3):147–154. doi: 10.1016/0378-1119(87)90041-2. [DOI] [PubMed] [Google Scholar]
- 28.Rippka R, Deruelles J, Waterbury JB, Herdman M, Stanier RY. Generic assignments, strain histories and properties of pure cultures of cyanobacteria. J Gen Microbiol. 1979;111(1):1–61. [Google Scholar]
- 29.He Q, Vermaas W. Chlorophyll a availability affects psbA translation and D1 precursor processing in vivo in Synechocystis sp. PCC 6803. Proc Natl Acad Sci USA. 1998;95(10):5830–5835. doi: 10.1073/pnas.95.10.5830. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.