

Significance
A major question in biology is how information is regulated during passage from one generation to the next. Here we show that histone methylation is regulated by a cooperative mechanism between the H3K4me2 demethylase LSD1 and the H3K9me2 methyltransferase MET-2. Without these enzymes, the transgenerational inheritance of H3K4me2 results in spermatogenesis genes being expressed somatically and in progeny being sterile. This indicates that H3K4me2 may function in the heritable maintenance of cell fate and that LSD1 and MET-2 may maintain fertility by reestablishing an epigenetic ground state between generations. In addition, by manipulating parental H3K4me2 levels, we demonstrate that chromatin states in the progeny are dependent on parental histone methylation levels. This provides a potential mechanism for the transgenerational inheritance of traits.
Abstract
The Caenorhabditis elegans LSD1 H3K4me2 demethylase SPR-5 reprograms epigenetic transcriptional memory during passage through the germ line. Here we show that mutants in the H3K9me2 methyltransferase, met-2, result in transgenerational epigenetic effects that parallel spr-5 mutants. In addition, we find that spr-5;met-2 double mutants have a synergistic effect on sterility, H3K4me2, and spermatogenesis expression. These results implicate MET-2 as a second histone-modifying enzyme in germ-line reprogramming and suggest a model in which SPR-5 and MET-2 function cooperatively to reestablish an epigenetic ground state required for the continued immortality of the C. elegans germ line. Without SPR-5 and MET-2, we find that the ability to express spermatogenesis genes is transgenerationally passed on to the somatic cells of the subsequent generation. This indicates that H3K4me2 may act in the maintenance of cell fate. Finally, we demonstrate that reducing H3K4me2 causes a large increase in H3K9me2 added by the SPR-5;MET-2 reprogramming mechanism. This finding suggests a novel histone code interaction in which the input chromatin environment dictates the output chromatin state. Taken together, our results provide evidence for a broader reprogramming mechanism in which multiple enzymes coordinately regulate histone information during passage through the germ line.
Since the discovery of histone modifications and the initial proposal of the histone code (1), a number of experiments have implicated histone modifications in the maintenance of transcriptional memory (2–8). Despite this implicit association, an understanding of how histone modifications are regulated developmentally, as well as the exact relationship between certain histone modifications, remains elusive. For example, at fertilization, the highly differentiated transcriptional program of the germ line may be reprogrammed by maternal factors in the oocyte to reestablish the epigenetic ground state of the zygote. This reprogramming capacity of the oocyte is implied by the ability of oocyte factors to reprogram a differentiated cell during somatic cell nuclear transfer (SCNT) (9, 10). However, the low efficiency observed in SCNT and in the similar induction of pluripotent stem cells highlights the complexity involved in reprogramming differentiated fates back to a ground pluripotent state (11, 12). Moreover, embryos derived from SCNT have been shown to continue to express genes from their previous differentiated state (13). These results indicate that germ-line reprogramming requires the resetting of an epigenetic transcriptional memory, but the mechanism of this reprogramming is not well understood.
One emerging player in transcriptional memory is dimethylation of histone 3 at lysine 4 (H3K4me2). During transcription, H3K4 methyltransferases, such as mixed-lineage leukemia (MLL), associate with a core COMPASS complex consisting of WDR5, RbBP5, and ASH2L (14). This complex promotes H3K4 methyltransferase activity and may enable the methyltransferase to interact with elongating RNA polymerase II (14). As a result, H3K4me2 is acquired at expressed genes during RNA polymerase II elongation. Deposition of this modification could serve to mark previously transcribed genes and maintain expression patterns through cell division as a form of transcriptional memory. Consistent with this hypothesis, the continued expression of somatic cell lineage-specific genes in embryos derived from SCNT is dependent on lysine 4 of histone H3.3 (4).
On the basis of this transcriptional memory model, it was previously hypothesized that the histone lysine-specific demethylase 1 (LSD1) is required to remove germ-line H3K4me2 and prevent the transgenerational maintenance of germ-line transcription (3). Loss of the C. elegans LSD1 ortholog spr-5 results in a germ-line mortality phenotype in which sterility increases across generations. This increasing sterility correlates with an increase in both the expression level of spermatogenesis genes and the accumulation of H3K4me2 at these genes. These data suggest that H3K4me2 can serve as an epigenetic transcriptional memory and that erasure of this modification by SPR-5 is critical for reprogramming this memory (3).
As H3K9me2 is associated with repressed chromatin, H3K4me2 and H3K9me2 have been observed to occupy mutually exclusive regions of the genome (15). In addition, mutations in enzymes that oppositely regulate these two histone modifications can have similar effects on transcription (16, 17). As a result, it is possible that enzymes that oppositely regulate these modifications may function together to regulate key developmental transitions. However, at this time, there is no direct evidence for this. In C. elegans, the SETDB family H3K9 methyltransferase MET-2 is expressed in the germ line (http://nematode.lab.nig.ac.jp/) and is responsible for the deposition of H3K9me2 in adult germ cells (18). Similar to spr-5 mutants, a null mutation in met-2 results in a germ-line mortality phenotype (19). Thus, it is possible that spr-5 and met-2 function cooperatively in the same reprogramming mechanism.
Here we examine the functional interaction between SPR-5 and MET-2 and test the hypothesis that they act together to reprogram epigenetic transcriptional memory during passage through the germ line in C. elegans. If SPR-5 and MET-2 function coordinately in germ-line reprogramming, we would expect to observe some or all of the following: met-2 mutants would have a germ-line mortality phenotype with kinetics that parallel spr-5 mutants, spermatogenesis genes that are targets for SPR-5 demethylation should also be targets for MET-2 H3K9 methylation, spermatogenesis genes that transgenerationally accumulate H3K4me2 and are misregulated across multiple generations in spr-5 mutants should be similarly affected in met-2 mutants, spr-5;met-2 double mutants should have an additive effect on H3K4me2 and expression at SPR-5 spermatogenesis targets, and the specificity for spermatogenesis targets originally observed in genome-wide expression studies in spr-5 mutants should be recapitulated in spr-5;met-2 mutants. Alternatively, if SPR-5 and MET-2 do not function in the same reprogramming pathway, we would expect that some or all of these predictions would not be met. In this article, we examine the functional interaction between SPR-5 and MET-2 and show that aspects of all these predictions are met. On the basis of these data, we propose a model in which SPR-5 and MET-2 function together in a coordinated germ-line reprogramming mechanism by erasing active H3K4me2 and replacing it with H3K9me2. Furthermore, we demonstrate that this reprogramming mechanism reacts to decreasing the levels of H3K4me2 by adding excess H3K9me2. This suggests that germ-line reprogramming functions through a responsive mechanism in which the input chromatin environment dictates the output chromatin state.
Results
met-2 Germ-line Mortality Phenotype.
Mutants in the H3K9me2 methyltransferase, MET-2, have a germ-line mortality phenotype where sterility increases across generations (19). However, this phenotype was analyzed differently than spr-5(by101) mutants (3). As a result, it is difficult to compare the kinetics of the two germ-line mortality phenotypes. Therefore, to determine whether the met-2(n4256) germ-line mortality phenotype has kinetics that are similar to spr-5 mutants, we reanalyzed met-2 mutants in our laboratory, using the same methods previously used to score the spr-5 germ-line mortality phenotype. Compared with wild-type animals, which maintain a relatively constant brood size of ∼325 progeny over 28 generations (Fig. S1A), met-2 mutant animals show a precipitous decline from an average of 250 progeny over the same time frame (Fig. S1B). Moreover, the trajectory of this brood size decline is similar to the decline that was previously reported in spr-5 mutant animals (3).
SPR-5 Spermatogenesis Targets Acquire MET-2-Dependent H3K9me2 in the Embryo.
Previous genome-wide analysis demonstrated that predominantly spermatogenesis genes are misregulated in spr-5 mutants (3). If MET-2 acts in the SPR-5 epigenetic reprogramming mechanism, then these same spermatogenesis genes may be targeted for H3K9me2 silencing in the embryo by MET-2. To test this hypothesis, we examined H3K9me2 by chromatin immunoprecipitation (ChIP) with an antibody against H3K9me2. In wild-type mixed-stage populations, we find a 3.5-fold and 5.0-fold (average, 4.25-fold) enrichment at two SPR-5 spermatogenesis targets, along with a 4.9-fold and 6.9-fold (average, 5.9-fold) enrichment at two other germ-line expressed genes compared with no antibody controls (Fig. 1A). This demonstrates that these genes are targets for H3K9 methylation. To verify that this H3K9me2 is being acquired during embryogenesis, we confirmed the results on purified embryonic samples. These experiments demonstrated a larger (7- and 8.2-fold) enrichment in embryos at the two SPR-5 spermatogenesis targets (Fig. 1B). This indicates that these germ-line genes may be predominantly targeted for H3K9me2 in the embryo. Importantly, we detect no enrichment of H3K9me2 in met-2 mutants compared with no antibody controls (Fig. 1 A and B). This indicates that the acquisition of H3K9me2 in the embryo is MET-2-dependent.
Fig. 1.

SPR-5 spermatogenesis targets acquire MET-2-dependent H3K9me2 in the embryo. (A) ChIP-qPCR-measured enrichment of H3K9me2 at four germ-line-expressed genes in mixed-stage wild-type (N2) and met-2 mutant strains. (B) Chip-qPCR measured enrichment of H3K9me2 at the indicated genes in embryos from wild-type (N2) and met-2 animals. The fold enrichment of H3K9me2 Ab over no Ab is shown. The individual measurements from which the fold enrichments were calculated are shown in Fig. S2.
Heritable Accumulation of H3K4me2 and Spermatogenesis Expression in met-2 Mutants.
Because met-2 and spr-5 mutants both have germ-line mortality phenotypes as well as overlapping targets, we hypothesized that MET-2 and SPR-5 may be working, at least partially, together. Our previous analysis demonstrated an increase in H3K4me2 at spermatogenesis genes over generations in spr-5 mutant animals (3). If this increased H3K4me2 results in the germ-line mortality phenotype in spr-5 animals, we reasoned that it may also occur in met-2 mutants. Therefore, we performed ChIP assays with anti-H3K4me2 antibodies on spermatogenesis genes that we have previously found to accumulate H3K4me2 in spr-5 mutant animals (3). These analyses were performed on mixed-stage met-2 mutants from early (F2), middle (F12), and late (F24) generations. These experiments show that, as in spr-5 mutants (3), H3K4me2 accumulates from early to late generations at spermatogenesis genes in met-2 animals, although not as highly as in spr-5 mutants (Fig. 2A). In addition, it was previously found in spr-5 mutant animals that the expression of spermatogenesis genes is misregulated in a highly coordinated fashion, where the expression increases for many generations before decreasing at late generations when the population is highly sterile (3). To determine whether these changes also occur in met-2 mutants, we performed quantitative reverse-transcription PCR (qRT-PCR) on spermatogenesis genes that were previously found to be misregulated in spr-5 mutants (3). Remarkably, this analysis demonstrates that expression of spermatogenesis is misregulated in a pattern that is similar to spr-5 mutants (Fig. 2B and ref. 3).
Fig. 2.
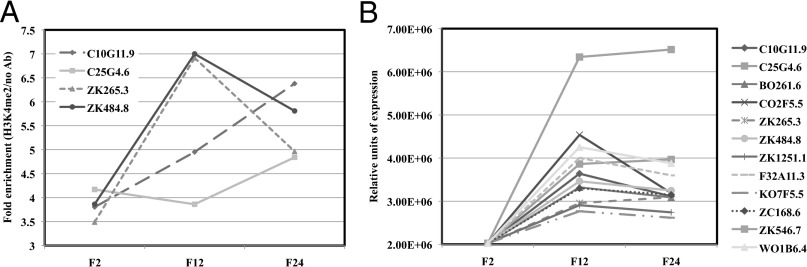
Heritable accumulation of H3K4me2 and the expression of spermatogenesis genes across generations in met-2 mutants. (A) ChIP-qPCR-measured enrichment of H3K4me2 at four spermatogenesis genes in mixed-stage met-2 mutants from generations 2, 12, and 24. The fold enrichment of H3K4me2 Ab over no Ab is shown. The individual measurements from which the fold enrichments were calculated are shown in Fig. S3. (B) Quantitative RT-PCR showing the relative expression of 12 spermatogenesis genes in mixed-stage met-2 mutants over 24 generations. The expression is normalized to actin (act-1).
spr-5;met-2 Double Mutants Have a Synthetic Sterile Phenotype.
The increase in H3K4me2 at spermatogenesis genes in met-2 mutants suggests a possible functional relationship between H3K4me2 and H3K9me2. To directly test this possibility, we examined spr-5(by101);met-2(n4256) double-mutant animals. If SPR-5 and MET-2 are part of a cooperative reprogramming mechanism to remove H3K4me2 and add H3K9me2, then double-mutant animals might have a synergistic effect on fertility. In contrast to spr-5 and met-2 single-mutant animals, which require ∼30 generations to reach a severely sterile phenotype, spr-5;met-2 double-mutant animals become completely sterile in a single generation (Fig. 3). This phenotype occurs earlier and is more severe than either single mutant, as all double-mutant animals are 100% sterile, whereas some individual single-mutant animals retain some fertility, even at late generations (Fig. 3 and ref. 3). Importantly, first-generation homozygous double mutants that came from hT2[bli-4(e937) let-?(q782) qIs48]-balanced mothers are fertile with normal brood sizes (Fig. 3), presumably as a result of maternal enzymatic activity rescuing these animals. This finding is consistent with SPR-5 and MET-2 being at least predominantly expressed in the gonad (ref. 3; http://nematode.lab.nig.ac.jp/).
Fig. 3.

spr-5;met-2 synthetic sterile phenotype. The average brood size of spr-5, met-2, and spr-5;met-2 strains in progressive generations. spr-5;met-2 mutants display complete sterility in one generation.
To investigate why spr-5;met-2 animals are sterile, we examined the gonads of these animals by differential interference contrast imaging. Previous analysis of late-generation sterile spr-5 mutants found that these animals had fairly normal numbers of proliferating germ cells and sperm but very few oocytes (3). In addition, the proximal gonad of late-generation spr-5 animals was highly disorganized with ongoing spermatogenesis in adult worms (as indicated by the presence of residual bodies) and a delay in larval progression (3). In sterile spr-5;met-2 adult mutants, we observe short, highly disorganized gonads with some proliferating germ cells; ongoing spermatogenesis (as indicated by the presence of residual bodies); and very few oocytes with abnormal morphology and larval delay (Fig. S4 A and B). This is reminiscent of the late-generation spr-5 phenotype (3). In addition, spr-5;met-2 mutants have multiple protruding vulvas (Fig. S4B). It is unclear why this occurs. However, spr-5 mutants were originally identified on the basis of their ability to suppress a presenilin vulval defect, so it is possible that spr-5;met-2 mutants may have multiple protruding vulvas because of defects in the Notch pathway (20, 21). In addition, met-2 has previously been shown to produce a synthetic multivulva (synMuv) phenotype in combination with other mutations (19, 22).
spr-5;met-2 Mutants Have a Synergistic Accumulation of H3K4me2 and Increase in Spermatogenesis Gene Expression.
The synthetic sterility between spr-5 and met-2 indicates that these two enzymatic activities may be acting at least partially cooperatively. If this is correct, and the double-mutant sterile phenotype is a result of a synergistic effect on the same spermatogenesis targets, then spr-5;met-2 double mutants should have an additive effect on H3K4me2 and expression levels at these genes. To test this possibility, we performed H3K4me2 ChIP and qRT-PCR on mixed-stage double-mutant animals. Compared with early-generation single-mutant animals, spr-5;met-2 double-mutant animals show a large increase in H3K4me2 at spermatogenesis genes in individual biological replicates (Fig. 4A). This equates to a large increase (56%–4,208%; average, >1,000%) in the overall average fold enrichment (Fig. 4B). Furthermore, this increase in H3K4me2 correlates with a large increase (61%–9,871%; average, >1,800%) in spermatogenesis gene expression levels in double-mutant versus single-mutant animals (Fig. 4C). Taken together, these results (Figs. 3 and 4) indicate that the spr-5;met-2 double-mutant animals recapitulate in one generation the phenotypes that take ∼30 generations to be realized in single-mutant animals.
Fig. 4.

Synergistic accumulation of H3K4me2 and expression of spermatogenesis genes in spr-5;met-2 mutants. (A) ChIP-qPCR-measured enrichment of H3K4me2 at four spermatogenesis genes in mixed-stage met-2, spr-5, and spr-5;met-2 mutants. Data from three independent biological replicates (series 1, 2, and 3) are shown. The fold enrichment of H3K4me2 Ab over no Ab is shown. The individual measurements from which the fold enrichments were calculated are shown in Fig. S5. (B) Average fold enrichment from series 1, 2, and 3 (from A) of H3K4me2 Ab over no Ab. (C) qRT-PCR analysis of 12 spermatogenesis genes was performed on mixed-stage met-2, spr-5, and spr-5;met-2 mutants. Data from two independent biological replicates (series 1 and 2) are shown. The expression is normalized to actin (act-1). The exact value and error for the two highest values are shown to accommodate scale. ZK546.7 and W01B6.4 are inset because of scale. The error bars represent SEM. (D) qRT-PCR showing the relative expression of two spermatogenesis genes in wild-type (N2) and spr-5;met-2 mutant L1 larvae. The expression is normalized to ama-1, which is unchanged in spr-5;amx-1 mutants. The error bars represent SEM.
In previous analyses of spr-5 mutants (3), it could not be determined whether loss of spr-5 results in spermatogenesis genes being ectopically expressed somatically in the subsequent generation. To determine whether this is the case, we examined the expression of spermatogenesis-enriched genes in spr-5;met-2 mutant L1 larvae. The germ line of L1 larvae consists of only the two germ-line precursor cells, Z2 and Z3, which have not yet entered spermatogenesis. As a result, these larvae are composed almost completely of somatic tissue. qPCR expression analysis demonstrated a large (443% and 12,367%, respectively) increase in spermatogenesis gene transcripts in spr-5;met-2 double-mutant L1 animals compared with wild-type L1 animals (Fig. 4D). This increase (Fig. 4C) suggests that spermatogenesis gene expression in spr-5;met-2 double mutants occurs outside of spermatogenesis, likely in somatic lineages.
spr-5;met-2 Mutants Have a Synergistic Accumulation of H3K4me2, but Not Expression, at Nonspermatogenesis Targets.
Previous genome-wide expression analysis demonstrated that SPR-5 predominantly affects the expression of spermatogenesis genes (3). To determine whether this is also the case in spr-5;met-2 mutants, we examined H3K4me2 and expression levels of an oocyte (rme-2), gut (ges-1), and muscle (unc-98) gene in these mutants. We also examined H3K4me2 levels at an intergenic region 3′ of the spermatogenesis gene C25G4.6. We detect no H3K4me2 enrichment at the intergenic region in spr-5;met-2 mutants compared with spr-5 or met-2 individual mutants (Fig. 5A). This demonstrates that the detected H3K4me2 enrichment at other loci is specific (Fig. 4 A and B and Fig. 5A). Surprisingly, H3K4me2 levels are highly elevated (554%–5,288%; average, 2,810%) at the oocyte (rme-2), gut (ges-1), and muscle (unc-98) genes in spr-5;met-2 mutants compared with spr-5 or met-2 individual mutants (Fig. 5A). This suggests that SPR-5;MET-2 activity is not confined to spermatogenesis targets. However, unlike at spermatogenesis loci, the elevation in H3K4me2 does not lead to an increase in the expression of these other genes (Fig. 5B). Thus, just as was originally observed in spr-5 mutants (3), spr-5;met-2 mutants only misexpress spermatogenesis genes.
Fig. 5.

Synergistic accumulation of H3K4me2, but not expression, at nonspermatogenesis targets. (A) ChIP-qPCR-measured enrichment of H3K4me2 at an oocyte (rme-2), gut (ges-1), and muscle (unc-98) gene, but no enrichment at an intergenic region 3′ of the spermatogenesis gene C25G4.6, in mixed-stage spr-5;met-2 mutants compared with met-2 and spr-5 mutants. The fold enrichment of H3K4me2 Ab over no Ab is shown. The individual measurements from which the fold enrichments were calculated are shown in Fig. S6. (B) qRT-PCR analysis of rme-2, ges-1, unc-98, and C25G4.6 was performed on mixed-stage met-2, spr-5, and spr-5;met-2 mutants. C25G4.6 was included as a positive control and for comparison with Fig. 4C. The expression is normalized to actin (act-1).
SPR-5;MET-2 Respond to Lowered H3K4me2 Input.
The synthetic effect on sterility, H3K4me2 levels, and spermatogenesis-enriched expression argue for a functional interplay between SPR-5 and MET-2 in mediating germ-line reprogramming. To determine whether this epigenetic reprogramming mechanism can functionally respond to changes in input information, we took advantage of wdr-5(ok1417) mutants to manipulate the input levels of H3K4me2. WDR-5 is a critical part of the H3K4me2 machinery (14, 23–25). As a result, wdr-5 mutants should have decreased levels of H3K4me2. To confirm that H3K4me2 levels are reduced in wdr-5 mutants at SPR-5;MET-2 targets, we performed H3K4me2 ChIP on mixed-stage wild-type and wdr-5 mutant worms. This analysis demonstrated a decrease in H3K4me2 at these loci in wdr-5 mutant worms (Fig. 6A). Therefore, because wdr-5 mutant worms have decreased levels of germ-line-acquired H3K4me2, but wild-type copies of both spr-5 and met-2, these worms can be used to determine how the SPR-5;MET-2 mechanism functionally reacts to decreases in H3K4me2 input levels. To examine this response, we performed H3K9me2 ChIP on mixed-stage wdr-5 mutant worms. These experiments demonstrated a large increase (613%–3,005%; average, >1,800%) in H3K9me2 compared with wild type in all four SPR-5;MET-2 targets we examined (Fig. 6B). This result indicates that the germ-line reprogramming mechanism, mediated by SPR-5 and MET-2, is sensitive to levels of input H3K4me2. Finally, to confirm the specificity of the SPR-5;MET-2 reprogramming mechanism, we performed H3K9me2 ChIP in wdr-5 mutants on the oocyte, gut, and muscle genes that show increases in H3K4me2 in spr-5;met-2 double mutants. Consistent with SPR-5 and MET-2 acting outside of spermatogenesis, we observe a large increase (587%–8,620%; average, 2,335%) in H3K9me2 at these genes in wdr-5 mutants (Fig. 6C).
Fig. 6.

Decreased H3K4me2 and increased H3K9me2 in wdr-5 mutants. (A) ChIP-qPCR-measured enrichment of H3K4me2 at four germ-line-expressed genes in mixed-stage wild-type (N2) and wdr-5 mutant strains. Data from two independent biological replicates (series 1 and 2) are shown. The fold enrichment of H3K4me2 Ab over no Ab is shown. The individual measurements from which the fold enrichments were calculated are shown in Fig. S7. (B) ChIP-qPCR-measured enrichment of H3K9me2 at four germ-line-expressed genes in mixed-stage wild-type, met-2 and wdr-5 mutant strains. The fold enrichment of H3K9me2 Ab over no Ab is shown. (C) ChIP-qPCR-measured enrichment of H3K9me2 at an oocyte (rme-2), gut (ges-1), and muscle (unc-98) gene in mixed-stage wild-type, met-2, and wdr-5 mutant strains. The fold enrichment of H3K9me2 Ab over no Ab is shown. The individual measurements from which the fold enrichments were calculated are shown in Fig. S7. (D) SPR-5;MET-2 model.
Discussion
The H3K4me2 demethylase SPR-5 is required to demethylate H3K4me2 in spermatogenesis genes during passage through the germ line (3). Without SPR-5, this H3K4me2 accumulates across generations, resulting in a germ-line mortality phenotype where the population of mutant worms becomes increasingly sterile over time (3). Here we report that a second histone-modifying enzyme, the H3K9 methyltransferase MET-2, functions cooperatively in this epigenetic reprogramming mechanism.
Consistent with previous results (19), we find that met-2 mutant animals exhibit a germ-line mortality defect with kinetics that parallel spr-5 mutant animals (3). The similarity of the two phenotypes raises the possibility that SPR-5 and MET-2 act together in an epigenetic switch that reprograms genes from an active (H3K4me2) to inactive (H3K9me2) chromatin environment. To address this possibility we first asked whether germ-line genes are enriched for H3K9me2 in the embryo. Using H3K9me2 ChIP, we find that previously identified SPR-5 spermatogenesis targets are also targets for MET-2-dependent H3K9me2 (Fig. 1). Next, we asked whether met-2 mutants have transgenerational effects at these loci that are similar to what has previously been observed in spr-5 mutants. Loss of SPR-5 results in the heritable accumulation of H3K4me2 at spermatogenesis genes (3). met-2 mutants also have an accumulation of H3K4me2 at these loci, albeit to a lesser extent than in spr-5 mutants. This is likely because wild-type SPR-5 activity antagonizes the accumulation of H3K4me2. In spr-5 mutants, the accumulation of H3K4me2 correlates with increases in the expression of these spermatogenesis genes for multiple generations. After increasing for multiple generations, the expression of spermatogenesis genes declines again at late generations, when spr-5 mutants are highly sterile (3). Strikingly, we observe similar effects at these same spermatogenesis loci in met-2 mutants over multiple generations (Fig. 2).
spr-5 and met-2 mutants have some differences in sterility and accumulation of H3K4me2 across generations. As a result, it is possible, or even likely, that SPR-5 and MET-2 have some activity that is independent of one another. Nevertheless, our finding that decreasing fertility, along with the accumulation of H3K4me2 and the misexpression of spermatogenesis genes, tracks in a similar fashion across many generations in both spr-5 and met-2 mutants (compare with ref. 3) argues that SPR-5 and MET-2 act, at least to some extent, cooperatively. To directly test the interaction between SPR-5 and MET-2, we generated spr-5;met-2 double mutants. If SPR-5 and MET-2 function cooperatively, we might expect that double mutants would have a synergistic affect on fertility, H3K4me2, and expression. Whereas spr-5 and met-2 are mostly sterile after multiple generations, we find that spr-5;met-2 double mutants have a synergistic 100% sterility phenotype in a single generation (Fig. 3). This enhanced sterility has also been observed recently by Greer and colleagues (26). Importantly, the spr-5;met-2 synergistic effect is specific to spr-5;met-2, as both we and others (19, 26) have found that many combinations of spr-5 and met-2 with other chromatin-related mutants do not have a synthetic sterility effect.
In spr-5;met-2 mutants, we observe developmental defects in the gonad that are reminiscent of late-generation spr-5 defects (Fig. S4). These findings are consistent with the sterility being a result of an exasperated effect on the spr-5 pathway. Furthermore, we observe a synergistic effect on H3K4me2 and expression (Fig. 4). Compared with spr-5 and met-2, single mutants that have a heritable accumulation of H3K4me2 and corresponding misexpression of spermatogenesis genes over many generations, we find that spr-5;met-2 mutants have a much larger accumulation of H3K4me2 and a larger increase in the expression of spermatogenesis genes in a single generation (Fig. 4). Thus, we propose that the observed germ-line defects in spr-5;met-2 mutant may be a result of the overexpression of spermatogenesis genes during spermatogenesis and oogenesis. This is analogous to what has been previously suggested in spr-5 mutants (3).
In addition to the expression analysis performed on mixed-stage populations, we also observe a large increase in the expression of spermatogenesis genes in spr-5;met-2 L1 larvae (Fig. 4D). As these L1 larvae have only two germ-line precursor cells and no ongoing spermatogenesis, this suggests that spermatogenesis genes are also being expressed outside of spermatogenesis in somatic lineages. Thus, we propose that without SPR-5 and MET-2, the transgenerational inheritance of H3K4me2 results in spermatogenesis genes being expressed ectopically in the somatic cells of the subsequent generation. This indicates that H3K4me2 may function in the heritable maintenance of cell fate. However, we do not detect any embryonic lethality or other overt somatic phenotypes. This implies that the ectopic expression of spermatogenesis genes does not significantly impair the ability to establish or maintain somatic cell fates.
Previous genome-wide expression analysis classified SPR-5 as predominantly affecting spermatogenesis genes (3). As a result, we initially focused on these targets to determine whether SPR-5 and MET-2 function together. To determine whether the SPR-5;MET-2 reprogramming mechanism is also confined to these loci, we examined H3K4me2 and expression at an oocyte (rme-2), gut (ges-1), and muscle (unc-98) gene in spr-5;met-2 mutants. These experiments show that H3K4me2 is highly increased at all of these loci in double mutants (Fig. 5A). This demonstrates that the SPR-5;MET-2 reprogramming mechanism is not specific to spermatogenesis loci. Thus, we propose that this reprogramming mechanism acts broadly in multiple lineages to reestablish an epigenetic ground state during passage through the germ line. However, in contrast to spermatogenesis loci, the increase in H3K4me2 at these other tissue-specific loci does not result in their increased expression (Fig. 5B). This is consistent with the specificity for spermatogenesis loci previously observed in spr-5 genome-wide expression data (3). Taken together, the accumulation of H3K4me2, without a corresponding increase in expression, indicates that the accumulation of H3K4me2 at these other loci (rme-2, ges-1, and unc-98) is not sufficient to cause their ectopic expression. Thus, we hypothesize there is an additional level of regulation at these other loci that prevents them from being ectopically expressed. Future studies will hopefully give us insight into this mechanism.
On the basis of the phenotypes and molecular effects observed, we propose the following model (Fig. 6D): In the germ line, H3K4me2 is acquired during transcription. SPR-5 erases this active chromatin (H3K4me2), and MET-2 reinforces the removal of this active chromatin by establishing repressive chromatin (H3K9me2). Perturbation of either component of the pathway results in heritable accumulation of H3K4me2 over many generations, whereas simultaneous loss of both components results in immediate accumulation of H3K4me2 in one generation at levels that result in immediate sterility. Thus, we propose that SPR-5 and MET-2 function cooperatively during passage through the germ line to maintain germ-line immortality. This SPR-5;MET-2 reprogramming pathway is likely part of a broader reprogramming system in which multiple enzymes coordinately regulate histone information during passage through the germ line. In addition, on the basis of the similarity between natural germ-line reprogramming and the artificial reprogramming that occurs during the induction of pluripotent stem cells, we speculate that the SPR-5;MET-2 reprogramming pathway may also function in these processes.
H3K4 and H3K9 are just 4 amino acids apart, so it is possible that methylation of H3K4me2 could block the acquisition of H3K9me2. In vitro biochemical evidence suggests that H3K4me blocks the acquisition of H3K9me via SU(VAR)3–9 (16, 27). However, H3K4me does not block the acquisition of H3K9me via G9a (27, 28) or SETDB1 (29). Our data are consistent with the SETDB1 biochemistry. If H3K4me2 were to completely block the ability to acquire H3K9me2, then the spr-5 mutant alone would be expected to result in the complete sterility phenotype because the H3K4me2 that was not removed by SPR-5 would block the activity of MET-2. Because both spr-5 and met-2 mutations are required to give rise to the complete sterility phenotype, it argues that H3K4me2 does not completely block the acquisition of H3K9me2 via MET-2 (a SETDB1 ortholog).
To fully understand the function of the histone code, it is important to understand the exact nature of the cooperation between SPR-5 and MET-2. Therefore, to further probe this relationship, we used wdr-5 mutants to manipulate H3K4me2 levels. WDR-5 is a critical part of the H3K4me2 machinery (14, 23–25), so we reasoned that we could use wdr-5 mutants to reduce the amount of H3K4me2 input in the germ line. Because both SPR-5 and MET-2 are functional in these wdr-5 mutants, we could then determine how the SPR-5;MET-2 reprogramming mechanism reacts to reduced input levels of H3K4me2. In wdr-5 mutants, we find that decreases in H3K4me2 result in large increases in the levels of H3K9me2 at four germ-line-expressed genes (Fig. 6 A and B). The increased addition of H3K9me2 in wdr-5 mutants could occur through enhancement of MET-2 activity or via increased recruitment of MET-2. Regardless, this result differs from the predictions of a simple model in which SPR-5 erases all H3Kme2, and MET-2 subsequently adds H3K9me2. If reprogramming occurred through this simple model, then decreasing H3K4me2 input levels should make SPR-5-mediated H3K4me2 erasure easier and would be expected to have no effect on H3K9me2 levels. Instead, our data suggest that H3K4me2, and its associated chromatin environment, normally reduces the MET-2-mediated acquisition of H3K9me2 and prevents the establishment of an increased repressed environment. Despite this, as stated earlier, H3K4me2 cannot completely block the ability to acquire H3K9me2. Otherwise, spr-5 mutants would be expected to have the full sterility phenotype by themselves. Thus, taken together, our data suggest a more complex and nuanced version of the histone code, in which the information contained in the state of the input chromatin environment has the ability to influence the level of output chromatin environment.
The increase in H3K9me2 observed in wdr-5 mutants also has significant consequences for our understanding of the epigenetic inheritance of traits and the epigenetic causes of disease. wdr-5 mutants live significantly longer than their wild-type counterparts (30). Amazingly, this increased longevity is epigenetically heritable, such that the progeny of wdr-5 mutants outcrossed to wild-type animals for multiple generations live longer than wild-type animals, despite the presence of normal WDR-5 activity (31). It is possible that the SPR-5;MET-2 germ-line reprogramming mechanism may explain the epigenetic inheritance of increased longevity in wdr-5 mutants. Our data indicate that decreased H3K4me2 in wdr-5 mutants results in increased H3K9me2. If this H3K9me2 is stably heritable and passed on to subsequent generations, then the accompanying repressed chromatin environment may block the ability to reacquire H3K4me2 and transcribe at normal levels, even in the presence of functional WDR-5 in outcrossed progeny. We propose that this may result in the observed transgenerational inheritance of longevity. A similar effect could also give rise to transgenerational effects in humans.
Materials and Methods
The strains are described in SI Materials and Methods. All relevant techniques are also included there. They include germ-line mortality scoring, chromatin immunoprecipitation, qRT-PCR, and differential interference contrast imaging.
Supplementary Material
Acknowledgments
We thank the Horvitz laboratory (Department of Biology, Massachusetts Institute of Technology, Cambridge, MA) for providing the met-2 mutant strain; members of the D.J.K. laboratory; and B. Kelly, M. Edwards, C. Bean, T. Caspary, and W. Sale for helpful discussions on the work and the manuscript. S.C.K. was supported by the Fellowships in Research and Science Teaching postdoctoral fellowship program. C.C.R. was supported by a Biochemistry, Cell and Molecular Biology Training Grant (5T32GM008367).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1321843111/-/DCSupplemental.
References
- 1.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403(6765):41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 2.Ng HH, Robert F, Young RA, Struhl K. Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol Cell. 2003;11(3):709–719. doi: 10.1016/s1097-2765(03)00092-3. [DOI] [PubMed] [Google Scholar]
- 3.Katz DJ, Edwards TM, Reinke V, Kelly WG. A C. elegans LSD1 demethylase contributes to germline immortality by reprogramming epigenetic memory. Cell. 2009;137(2):308–320. doi: 10.1016/j.cell.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ng RK, Gurdon JB. Epigenetic memory of an active gene state depends on histone H3.3 incorporation into chromatin in the absence of transcription. Nat Cell Biol. 2008;10(1):102–109. doi: 10.1038/ncb1674. [DOI] [PubMed] [Google Scholar]
- 5. Rechtsteiner A, Ercan S, Takasaki T, Phippen TM, Egelhofer TA, Wang W, Kimura H, Lieb JD, Strome S (2010) The histone H3K36 methyltransferase MES-4 acts epigenetically to transmit the memory of germline gene expression to progeny. PLoS Genet 2;6(9):e1001091. [DOI] [PMC free article] [PubMed]
- 6.Mito Y, Henikoff JG, Henikoff S. Genome-scale profiling of histone H3.3 replacement patterns. Nat Genet. 2005;37(10):1090–1097. doi: 10.1038/ng1637. [DOI] [PubMed] [Google Scholar]
- 7.Kundu S, Peterson CL. Role of chromatin states in transcriptional memory. Biochim Biophys Acta. 2009;1790(6):445–455. doi: 10.1016/j.bbagen.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Furuhashi H, et al. Trans-generational epigenetic regulation of C. elegans primordial germ cells. Epigenetics Chromatin. 2010;3(1):15. doi: 10.1186/1756-8935-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gurdon JB, Elsdale TR, Fischberg M. Sexually mature individuals of Xenopus laevis from the transplantation of single somatic nuclei. Nature. 1958;182(4627):64–65. doi: 10.1038/182064a0. [DOI] [PubMed] [Google Scholar]
- 10.Campbell KH, McWhir J, Ritchie WA, Wilmut I. Sheep cloned by nuclear transfer from a cultured cell line. Nature. 1996;380(6569):64–66. doi: 10.1038/380064a0. [DOI] [PubMed] [Google Scholar]
- 11.Lau F, Ahfeldt T, Osafune K, Akustsu H, Cowan CA. Induced pluripotent stem (iPS) cells: An up-to-the-minute review. F1000 Biol Rep. 2009;1:84. doi: 10.3410/B1-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hochedlinger K, Jaenisch R. Nuclear transplantation: Lessons from frogs and mice. Curr Opin Cell Biol. 2002;14(6):741–748. doi: 10.1016/s0955-0674(02)00380-0. [DOI] [PubMed] [Google Scholar]
- 13.Ng RK, Gurdon JB. Epigenetic memory of active gene transcription is inherited through somatic cell nuclear transfer. Proc Natl Acad Sci USA. 2005;102(6):1957–1962. doi: 10.1073/pnas.0409813102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shilatifard A. The COMPASS family of histone H3K4 methylases: Mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem. 2012;81:65–95. doi: 10.1146/annurev-biochem-051710-134100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2011;12(1):7–18. doi: 10.1038/nrg2905. [DOI] [PubMed] [Google Scholar]
- 16.Rudolph T, et al. Heterochromatin formation in Drosophila is initiated through active removal of H3K4 methylation by the LSD1 homolog SU(VAR)3-3. Mol Cell. 2007;26(1):103–115. doi: 10.1016/j.molcel.2007.02.025. [DOI] [PubMed] [Google Scholar]
- 17.Musri MM, et al. Histone demethylase LSD1 regulates adipogenesis. J Biol Chem. 2010;285(39):30034–30041. doi: 10.1074/jbc.M110.151209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bessler JB, Andersen EC, Villeneuve AM. Differential localization and independent acquisition of the H3K9me2 and H3K9me3 chromatin modifications in the Caenorhabditis elegans adult germ line. PLoS Genet. 2010;6(1):e1000830. doi: 10.1371/journal.pgen.1000830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andersen EC, Horvitz HR. Two C. elegans histone methyltransferases repress lin-3 EGF transcription to inhibit vulval development. Development. 2007;134(16):2991–2999. doi: 10.1242/dev.009373. [DOI] [PubMed] [Google Scholar]
- 20.Jarriault S, Greenwald I. Suppressors of the egg-laying defective phenotype of sel-12 presenilin mutants implicate the CoREST corepressor complex in LIN-12/Notch signaling in C. elegans. Genes Dev. 2002;16(20):2713–2728. doi: 10.1101/gad.1022402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eimer S, Lakowski B, Donhauser R, Baumeister R. Loss of spr-5 bypasses the requirement for the C.elegans presenilin sel-12 by derepressing hop-1. EMBO J. 2002;21(21):5787–5796. doi: 10.1093/emboj/cdf561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Poulin G, Dong Y, Fraser AG, Hopper NA, Ahringer J. Chromatin regulation and sumoylation in the inhibition of Ras-induced vulval development in Caenorhabditis elegans. EMBO J. 2005;24(14):2613–2623. doi: 10.1038/sj.emboj.7600726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li T, Kelly WG. A role for Set1/MLL-related components in epigenetic regulation of the Caenorhabditis elegans germ line. PLoS Genet. 2011;7(3):e1001349. doi: 10.1371/journal.pgen.1001349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fisher K, Southall SM, Wilson JR, Poulin GB. Methylation and demethylation activities of a C. elegans MLL-like complex attenuate RAS signalling. Dev Biol. 2010;341(1):142–153. doi: 10.1016/j.ydbio.2010.02.023. [DOI] [PubMed] [Google Scholar]
- 25.Simonet T, Dulermo R, Schott S, Palladino F. Antagonistic functions of SET-2/SET1 and HPL/HP1 proteins in C. elegans development. Dev Biol. 2007;312(1):367–383. doi: 10.1016/j.ydbio.2007.09.035. [DOI] [PubMed] [Google Scholar]
- 26. Greer EL, et al. (2014) A histone methylation network regulates transgenerational epigenetic memory in C. elegans. Cell Rep 7(1):113-126. [DOI] [PMC free article] [PubMed]
- 27.Nishioka K, et al. Set9, a novel histone H3 methyltransferase that facilitates transcription by precluding histone tail modifications required for heterochromatin formation. Genes Dev. 2002;16(4):479–489. doi: 10.1101/gad.967202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chin HG, et al. Sequence specificity and role of proximal amino acids of the histone H3 tail on catalysis of murine G9A lysine 9 histone H3 methyltransferase. Biochemistry. 2005;44(39):12998–13006. doi: 10.1021/bi0509907. [DOI] [PubMed] [Google Scholar]
- 29.Schultz DC, Ayyanathan K, Negorev D, Maul GG, Rauscher FJ., 3rd SETDB1: A novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002;16(8):919–932. doi: 10.1101/gad.973302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Greer EL, et al. Members of the H3K4 trimethylation complex regulate lifespan in a germline-dependent manner in C. elegans. Nature. 2010;466(7304):383–387. doi: 10.1038/nature09195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Greer EL, et al. Transgenerational epigenetic inheritance of longevity in Caenorhabditis elegans. Nature. 2011;479(7373):365–371. doi: 10.1038/nature10572. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.