

Significance
Muscle insulin resistance is a major factor in the pathogenesis of type 2 diabetes, but the underlying cellular mechanisms are yet unclear. This study found that muscle diacylglycerol content was temporally associated with protein kinase Cθ activation and impairment of insulin signaling in human skeletal muscle. A similar relationship between muscle diacylglycerol content and protein kinase Cθ activation was observed in insulin-resistant obese and type 2 diabetic individuals. In contrast, we observed no relationship between other putative mediators of muscle insulin resistance including ceramides, acylcarnitines, or circulating adipocytokines. These data support the hypothesis that diacylglycerol activation of protein kinase Cθ and subsequent impairment of insulin signaling plays a major role in the pathogenesis of muscle insulin resistance in humans.
Keywords: lipotoxicity, insulin signaling
Abstract
Muscle insulin resistance is a key feature of obesity and type 2 diabetes and is strongly associated with increased intramyocellular lipid content and inflammation. However, the cellular and molecular mechanisms responsible for causing muscle insulin resistance in humans are still unclear. To address this question, we performed serial muscle biopsies in healthy, lean subjects before and during a lipid infusion to induce acute muscle insulin resistance and assessed lipid and inflammatory parameters that have been previously implicated in causing muscle insulin resistance. We found that acute induction of muscle insulin resistance was associated with a transient increase in total and cytosolic diacylglycerol (DAG) content that was temporally associated with protein kinase (PKC)θ activation, increased insulin receptor substrate (IRS)-1 serine 1101 phosphorylation, and inhibition of insulin-stimulated IRS-1 tyrosine phosphorylation and AKT2 phosphorylation. In contrast, there were no associations between insulin resistance and alterations in muscle ceramide, acylcarnitine content, or adipocytokines (interleukin-6, adiponectin, retinol-binding protein 4) or soluble intercellular adhesion molecule-1. Similar associations between muscle DAG content, PKCθ activation, and muscle insulin resistance were observed in healthy insulin-resistant obese subjects and obese type 2 diabetic subjects. Taken together, these data support a key role for DAG activation of PKCθ in the pathogenesis of lipid-induced muscle insulin resistance in obese and type 2 diabetic individuals.
Decreased insulin-stimulated muscle glycogen synthesis, attributable to reduced insulin-stimulated glucose transport activity (1–3), plays a major role in the pathogenesis of type 2 diabetes (T2D) but the cellular and molecular mechanisms responsible for this abnormality remain unknown (4–6).
Increased intramyocllular lipid content is a strong predictor of muscle insulin resistance in sedentary adults and children (7–9). Previous studies in lipid-infused rodents have implicated lipid-induced increases in muscle diacylglycerol (DAG) content, leading to activation of PKCθ and subsequent decreased insulin signaling at the level of insulin receptor substrate (IRS)-1 tyrosine phosphorylation (10, 11). Other studies have implicated increases in muscle ceramide (12) and acylcarnitine content attributable to incomplete fatty acid oxidation as causal factors in muscle insulin resistance (13). Alternatively, inflammation and alterations in plasma and tissue adipocytokines have also been implicated in causing lipid-induced muscle insulin resistance (14). Translating these findings to humans has been problematic and has led to inconsistent results (15, 16). However, these studies did not provide a comprehensive analysis for all of these putative pathways under conditions of acute and chronic insulin resistance. Furthermore, it is possible that study-related differences regarding age and physical activity of the respective control groups could have contributed to the discrepant results.
In this study, we took a comprehensive view of all of the putative mechanisms that have been proposed to cause lipid-induced muscle insulin resistance by measuring these parameters in serial muscle biopsies in healthy, young, lean, sedentary humans before and during a lipid infusion to induce acute muscle insulin resistance. In addition, we also examined these same parameters in healthy insulin resistant obese subjects, as well as in insulin-resistant patients with T2D.
Results
Lipid-Induced Muscle Insulin Resistance.
Plasma metabolites and hormones.
Plasma fatty acid (FA) levels were comparable at baseline before glycerol (GLY) and lipid (LIP) infusion (LIP: 0.46 ± 0.07 mmol/L; GLY: 0.41 ± 0.05 mmol/L). Mean plasma FA concentrations increased during LIP (2.30 ± 0.14 mmol/L at 360 min vs. 0 min; P < 0.001) and remained unchanged during GLY (0.31 ± 0.06 mmol/L at 360 min vs. 0 min: not significant; LIP vs. GLY at 360 min: P < 0.001). Thus, during low-insulin clamps, fasting plasma insulin was replaced and lipolysis was not suppressed, as assessed from steady-state plasma FA levels during GLY. During high-insulin clamps, plasma FA increased during LIP (1.65 ± 0.10 mmol/L; P < 0.001: 510 min vs. 0 min) and decreased during GLY (0.10 ± 0.04 mmol/L; P < 0.01: 510 min vs. 0 min; P < 0.001: GLY vs. LIP at 510 min). Plasma triglycerides increased during LIP and remained unchanged during GLY compared with baseline (290 ± 25 vs. 67 ± 13 mg/dL; P < 0.001). Plasma insulin concentrations were comparable at baseline (9.5 ± 3.3 vs. 13.3 ± 4.3 pmol/L) and after 4 h of LIP and GLY (9.8 ± 2.9 vs. 11.2 ± 2.6 pmol/L) but increased to 60 ± 2 and 61 ± 4 pmol/L (LIP and GLY) during the high-dose insulin clamp. C-peptide was suppressed to undetectable levels in nearly all subjects within 120 min of the clamp.
Glucose metabolism, energy expenditure, and substrate oxidation.
Insulin-stimulated whole-body glucose disposal was 61% lower (P < 0.001) during LIP than during GLY (3.8 ± 1.3 vs. 9.7 ± 3.8 mg⋅kg−1⋅min−1) (Fig. S1). During LIP, glucose disposal was 50% and 63% lower in females and males (P < 0.005 and P < 0.001 each vs. GLY), respectively, without difference between the sexes. The respiratory quotient (RQ) did not change during LIP (0.79 ± 0.02) but increased during GLY (0.98 ± 0.03; basal vs. clamp; P < 0.005). During insulin stimulation, lipid oxidation fell by 32 ± 9% and 78 ± 22% in LIP and GLY (P < 0.001 vs. fasting), remaining higher in LIP (P < 0.01 vs. GLY). LIP reduced nonoxidative glucose use by 49% (P < 0.05 vs. GLY). Total plasma acylcarnitines did not change after 4 h of LIP (10.9 ± 0.9 vs. 12.1 ± 1.5 μM) and decreased by 45% after 4 h of the GLY (9.7 ± 0.8 vs. 5.4 ± 0.4 μM; P < 0.001).
Myocellular lipid metabolites.
During LIP, total muscle DAG doubled at 2.5 h (287 ± 45 nmol/g; P < 0.005 vs. baseline) and decreased at 4 h (215 ± 23 nmol/g; P < 0.05 at 2.5 vs. 4 h), remaining higher compared with baseline (P < 0.005, baseline vs. 4 h). Total DAG did not change during GLY. During LIP, total membrane DAG rose by 80% at 2.5 h (P < 0.01 vs. basal) and remained 40% higher at 4 h compared with baseline (P < 0.05, baseline vs. 4 h) (Fig. 1A). Total membrane DAG did not change during GLY (Fig. S2A). Certain membrane DAG species (C18:1, C16:0; C16:0, C20:4; C18:2, C18:0; C18:1, C18:0; C18:1, C18:1; C18:1, C18:2; C18:2, C18:2; C16:0, C18:2) were increased during LIP (Fig. 2A) but remained unchanged during GLY (Fig. S3A and Tables S1 and S2). In LIP, total cytosolic DAG content increased by 80% at 2.5 h (P < 0.01 vs. basal) (Fig. 1A), decreased by 36% at 4 h (P < 0.05 vs. 2.5 h), and remained higher compared with baseline (P < 0.01). During GLY, total cytosolic DAG did not change (Fig. S2A). Certain cytosolic DAG species increased during LIP (C16:0, C20:4; C18:2, C18:0; C18:1, C18:1; C18:1, C18:2; C18:2, C18:2; C16:0, C18:2; Fig. 2B) but remained unchanged during GLY (Fig. S3B and Tables S1 and S2).
Fig. 1.

(A) Myocellular DAG concentrations in the membrane and cytosolic fraction and myocellular ceramide concentrations during lipid infusion in young lean healthy controls (CON) (n = 10). (B) Activation of myocellular PKCθ, -δ, and -ε during lipid infusion in CON (n = 10–14). (C) Phosphorylation of serine 1101 residue at IRS1-Ser1101-Px at baseline and its relative increase after 4 h glycerol (white and light gray columns) or lipid (dark gray and black columns) infusion in young lean healthy controls (CON) (n = 7). (D and E) PI3K-Px (E) and membrane/cytosolic ratio of Akt-Ser473 phosphorylation (Akt-Ser473-Px) (D) at baseline and after 4.5 h of glycerol (light gray column) or lipid (black column) infusion during insulin stimulation for 30 min in CON (n = 7). Data are given as means ± SEM. *P < 0.05; #P < 0.01; §P < 0.001. (F) Increased plasma FAs lead to myocellular accumulation of DAGs and consequent IRS1-Ser1101-Px, impaired PI3K-Px, and blunted insulin stimulation of Akt-Ser473-Px.
Fig. 2.

Concentration of membrane (A) and cytosolic (B) DAG species in healthy, lean controls (CON) (n = 16) at baseline (white columns), after 2.5 h (light gray columns), and after 4 h (dark gray columns) of lipid infusion. (C and D) Membrane (C) and cytosolic (D) DAG species concentrations in CON (white columns; n = 16), in young obese humans (OBE) (light gray columns; n = 10), and in elderly obese patients with T2D (black columns; n = 10). Data are given as means ± SEM. *P < 0.05; #P < 0.01; §P < 0.001 (2.5 or 4 h of lipid infusion vs. 0 h and OBE and T2D vs. CON).
Total ceramides (Fig. 1A) and individual ceramide species (Fig. S4 A and B) did not change during LIP or GLY. Likewise, we observed no changes in myocellular acylcarnitines after 4 h of LIP or GLY (207 ± 27 vs. 181 ± 18 or 133 ± 5 nmol/g; n = 7), with a tendency for lower acylcarnitines during GLY (P = 0.051: basal vs. 4 h GLY; P = 0.071: GLY vs. LIP after 4 h).
Activation of myocellular PKCθ.
Translocation of PKCθ from cytosol to plasma membrane, reflecting activation of PKCθ, was not altered at 2.5 h but increased after 4 h of LIP (P < 0.05: LIP vs. GLY; P < 0.005: basal and 2.5 h vs. 4 h of LIP; Fig. 1B). GLY did not affect activation of PKCθ (Fig. S2B). Activation of PKC isoforms δ and ε did not occur (Fig. 1B and Fig. S2B).
Myocellular insulin signaling.
Phosphorylation at serine residues 1101 of insulin receptor substrate 1 (IRS1-Ser1101-Px) increased twofold (P < 0.005) at 4 h of LIP but did not change during GLY (Fig. 1C). Hyperinsulinemia increased the membrane/cytosol ratio of Akt phosphorylation at serine 473 (Akt-Ser473-Px) by threefold during GLY (P < 0.001 basal vs. insulin) but not during LIP (Fig. 1D). Accordingly, hyperinsulinemia stimulated phosphoinositide-3 kinase phosphorylation (PI3K-Px) by 184% (P < 0.05) during GLY but not during LIP (Fig. 1E).
Circulating adipokines and adhesion molecules.
Plasma concentrations of interleukin (IL)-6 (2.09 ± 1.21 vs. 1.80 ± 1.02 pg/mL), adiponectin (7.7 ± 2.2 vs. 8.2 ± 1.0 µg/mL), retinol-binding protein (RBP)4 (21.3 ± 5.7 vs. 21.6 ± 4.6 µg/mL), and soluble intercellular adhesion molecule (sICAM)-1 (142 ± 26 vs. 153 ± 142 ng/mL) were comparable after 4 h of LIP and GLY.
Plasma and Tissue Parameters in Insulin-Resistant Obese and Insulin-Resistant T2D Subjects.
Plasma metabolites and hormones.
Fasting plasma FA concentrations were comparable in lean healthy human subjects (CON) (0.43 ± 0.05 mmol/L), obese subjects (0.50 ± 0.05 mmol/L), and in T2D patients (0.68 ± 0.06 mmol/L) and decreased during hyperinsulinemic–euglycemic clamps to 0.09 ± 0.02 in obese and to 0.13 ± 0.03 mmol/L in T2D (P < 0.001 vs. baseline). Fasting plasma triglycerides were higher in obese and T2D (112 ± 15 and 172 ± 15 mg/dL) compared with CON (97 ± 7 mg/dL; P < 0.05). During hyperinsulinemic–euglycemic clamps, plasma insulin concentrations were similar in all groups (control: 78 ± 7 pmol/L; obese: 73 ± 5 pmol/L; T2D: 60 ± 5 pmol/L).
Glucose uptake, energy expenditure, and substrate oxidation.
Physical activity was comparable between obese, T2D, and CON (Baecke index: 2.6 ± 0.6, 2.5 ± 0.7, 2.4 ± 0.5). Insulin-stimulated glucose disposal was 78% lower in obese (10.6 ± 3.8 vs. 1.6 ± 0.7 mg⋅kg−1⋅min−1; P < 0.005) and 88% lower in T2D (1.5 ± 1.0 mg⋅kg−1⋅min−1; P < 0.001) compared with CON (Fig. S5). RQ did not change during the hyperinsulinemic–euglycemic clamps compared with fasting (obese: 0.79 ± 0.02 vs. 0.87 ± 0.04; T2D: 0.71 ± 0.06 vs. 0.88 ± 0.02). At baseline, lipid oxidation was 22% and 36% lower in obese and T2D participants than in CON. During the clamp, nonoxidative glucose utilization was 76% and 41% (P < 0.001) lower compared with CON. Plasma acylcarnitines did not differ between the groups (CON: 10.9 ± 0.8 μM; obese: 8.9 ± 0.9 μM; T2D: 11.3 ± 0.8 μM).
Myocellular lipid metabolites.
Total DAGs were 1.5- and 2-fold higher in obese and T2D participants compared with CON (P < 0.001 vs. CON) and did not differ between obese and T2D. Membrane DAGs were not increased in obese but 30% higher in T2D than in CON and obese (P < 0.001) (Fig. 3A). Cytosolic DAGs were 85% and 120% greater in obese (P < 0.001 vs. CON) and in T2D (P < 0.005 vs. CON) (Fig. 3A). Specific membrane (Fig. 2C and Tables S1 and S2) and cytosolic DAG species (Fig. 2D and Tables S1 and S2) were increased in obese and T2D patients. Neither total DAG or subcellular fractions differed between female and male participants. Total ceramides (Fig. 3A) and ceramide species (Fig. S4C) were comparable in all groups. Acylcarnitines did not differ between the groups [CON: 203 ± 25 nmol/g (n = 7); obese: 176 ± 22 nmol/g (n = 10); T2D: 175 ± 22 nmol/g (n = 10)]; however, given the limited number of samples in each group, a type 2 error cannot be excluded.
Fig. 3.
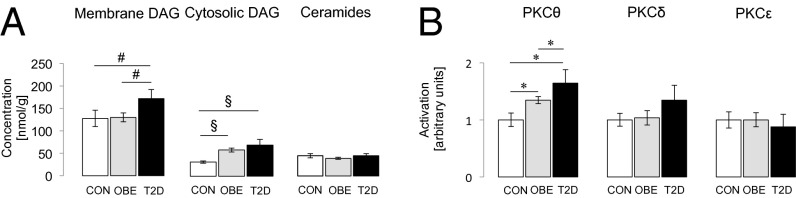
Myocellular concentration of membrane and cytosolic DAGs and myocellular total ceramide concentrations (A) and activation of PKCθ, -δ, and -ε (B) in young lean healthy controls (CON) (white columns; n = 16), in young obese humans (OBE) (dark gray columns; n = 10), and in elderly obese patients with T2D (black column; n = 10). Data are given as means ± SEM. *P < 0.05; #P < 0.01; §P < 0.001.
Correlation analyses of basal biopsy samples of the whole study population revealed that total cytosolic DAG correlated negatively with insulin sensitivity (M value) (r = −0.566; P < 0.001) but positively with body mass index (BMI) (r = 0.509; P < 0.005). After adjustment for BMI, C18:0 and C18:2 (r = −0.441; P < 0.05), C16:0- and C18:2-containing (r = −0.479; P < 0.05) cytosolic DAG negatively correlated with M value. In the membrane fraction, C18:0 and C20:4 (r = −0.416; P < 0.05), C18:0 and C18:2 (r = −0.492; P < 0.05), C18:1 and C18:2 (r = −0.403; P < 0.05), C18:2 and C18:2 (r = −0.410; P < 0.05), as well as C16:0- and C18:2-containing (r = −0.619; P < 0.005) DAG correlated negatively with M value, even after adjustment for BMI. C20:4- and C20:5-containing membrane DAG correlated negatively with BMI and positively with M value. Adjustment for M value abolished the associations of DAG species with BMI. Some cytosolic DAG species [C18:0, C20:4 (r = 0.379; P = 0.025); C18:2, C18:0 (r = 0.417; P = 0.013); C18:1, C18:2 (r = 0.340; P = 0.045); and C16:0, C18:2 (r = 0.348; P = 0.041)] and the following membrane species correlated with age: C18:2, C18:0 (r = 0.480; P = 0.004); C18:2, C18:2 (r = 0.445; P = 0.007); C16:0, C20:4 (r = 0.352; P = 0.038); C18:0, C20:4 (r = 0.352; P = 0.037); C18:1, C18:2 (r = 0.394; P = 0.019); and C16:0, C18:2 (r = 0.0365; P = 0.031). Adjustment for HbA1c and M value abolished these correlations.
Activation of myocellular PKCθ.
Activation of PKCθ was 35% and 64% (P < 0.05 vs. CON) higher in obese and T2D than in CON and 22% (P < 0.05) greater in T2D than in obese. In contrast, there were no differences in activation of PKCδ and PKCε between groups (Fig. 3B). PKCθ activation correlated negatively with M value (r = −0.575; P < 0.005) but positively with plasma FA (r = 0.428; P < 0.05), BMI (r = 0.436; P < 0.05), total cytosolic (r = 0.441; P < 0.05), and membrane DAG (r = 0.410; P < 0.05) and with specific DAG species (Table S3). C18:2- or C20:4-containing DAG exhibited the strongest relationships with PKCθ activation and were increased in both the lipid infusion studies, as well as in the insulin-resistant obese subjects and T2D subjects. In contrast to the strong relationship between muscle DAG content, PKCθ activation, and muscle insulin resistance, there was no relationship between muscle insulin resistance and total or any specific ceramide species in skeletal muscle.
Circulating adipokines and adhesion molecules.
In obese and T2D, plasma concentrations of IL-6 (1.84 ± 1.02 and 1.70 ± 0.68 pg/mL), RBP4 (29.5 ± 10.3 and 33.2 ± 65.4 µg/mL), and sICAM-1 (191 ± 64 and 245 ± 58 ng/mL) were comparable to CON (1.78 ± 1.16 pg/mL; 21.4 ± 5.3 µg/mL; 145 ± 47 ng/mL). Plasma adiponectin concentrations were slightly lower in obese and T2D [3.70 ± 1.83 and 2.78 ± 0.85 µg/mL; P < 0.05 vs. CON (7.9 ± 1.4 µg/mL)].
Discussion
This study found that increasing plasma FA, by lipid infusion, leads to a transient increase in intramyocellular DAG species (C16:0, C18:0, C18:1, C18:2, and C20:4), which were temporally related to PKCθ activation, increased IRS1-Ser1101-Px, and impairment of insulin-stimulated PI3K activation and Akt-Ser473-Px (Fig. 1F). In contrast, lipid-induced muscle insulin resistance in these subjects was not associated with increases in intramuscular ceramide or acylcarnitine content or changes in circulating adipocytokines. Furthermore, muscle insulin resistance in obese and T2D individuals exhibited similarly increased cytosolic DAG content and PKCθ activation and no relationship with muscle ceramide or acylcarnitine content. Similar to the lipid infusion studies, the DAG species containing either C16:0, C18:0, C18:1, C18:2, or C20:4 FA showed the strongest relationship with PKCθ activation and insulin resistance in obese and T2D individuals.
Short-term elevation of plasma FA concentrations similarly reduced M values in both male and female volunteers, which argues against sex-related differences in the metabolic response to an acute lipid infusion (15, 16). The decrease in M values induced by lipid infusion could mostly be attributed to decreased insulin-stimulated rates of nonoxidative glucose metabolism, which is consistent with previous studies demonstrating lipid-induced reductions in insulin-stimulated glucose transport/phosphorylation activity, leading to reductions in muscle glycogen synthesis (3, 7, 17, 18). The early blunting of the increase in glucose-6-phosphate during combined lipid and insulin infusion (1) argues against the putative role of lipid-induced reductions of pyruvated dehydrogenase activity and glucose oxidation as being responsible for the observed insulin resistance (19). It has been postulated that impaired lipid oxidation (20), interference of lipids with insulin-stimulation of ATP synthesis (21), or relative deficiency of glycolytic precursors of the tricarboxylic acid cycle leading to accumulation of acylcarnitines (13) could underlie lower glucose uptake. However, plasma and myocellular acylcarnitines were not increased during the lipid infusion or in the insulin-resistant obese and T2D subjects, and mitochondrial ATP synthesis only decreased after induction of muscle insulin resistance (22), indicating that altered oxidative capacity is not a prerequisite for lipid-induced muscle insulin resistance. Overreplacement of insulin is unlikely to underlie differences of acylcarnitines between lipid and glycerol infusion protocols because plasma insulin concentrations were low and similar in both protocols.
Previous studies in rodent models have implicated increases in myocellular lipid metabolites, such as ceramides and DAGs, with subsequent inhibition of insulin signaling as causal factors in the pathogenesis of lipid-induced muscle insulin resistance (5, 6, 10, 11, 23). Studies in humans have been less conclusive because of conflicting results of studies with different experimental protocols, resulting in varying plasma FA concentrations and compositions (15, 16, 24). By performing serial muscle biopsies before and during a lipid infusion, we found that total muscle DAG content increased transiently within 2.5 h and subsequently declined 4 h after starting the lipid infusion. These data are consistent with previous rodent studies demonstrating a transient increase in muscle DAG content during a lipid infusion (11). This transient increase in muscle DAG content may explain the inability of some previous studies that failed to observe an increase in muscle DAG content in relation to muscle insulin resistance during a lipid infusion (25). It is also possible that higher lipid oxidation rates or increased conversion of DAG to triglyceride by increased DAG acyltransferase (DGAT) activity in physically active persons might have prevented a lipid-induced increase in muscle DAG content (26). In this regard, the present study was comprised of only sedentary participants, thereby avoiding possible confounding effects resulting from different levels of physical activity on lipid-induced muscle insulin resistance.
In this study, we infused somatostatin, to inhibit endogenous insulin secretion, and insulin, to maintain fasting plasma insulin concentrations, to avoid any potential confounding effects mediated by lipid-induced endogenous insulin secretion. Nevertheless, even subtle changes of plasma insulin levels can affect endogenous FA release (27). We cannot distinguish between endogenous and exogenous FA but during this short-term lipid-induced muscle insulin resistance; the majority of the FA is provided by the infused lipid. Moreover, DAG did not increase during glycerol infusion experiments; thus, FAs from endogenous sources do not seem to play a major role in our model of short-term muscle insulin resistance. Chronic insulin resistance was examined in fasting muscle biopsies obtained from insulin resistant obese and T2D subjects; thus, endogenous FAs played a prominent role for the increased muscle DAG content in these individuals. Although steady-state plasma FA and insulin concentrations did not change during the glycerol infusion in the low dose hyperinsulinemic–euglycemic clamp study, we cannot rule out that insulin might have been overreplaced in these experiments. This, together with the absence of growth-hormone replacement, might account for lower FA flux rates during the low-dose hyperinsulinemic–euglycemic clamp study with glycerol infusion (28). However, replacement of plasma insulin was the same during glycerol and lipid infusions; thus, any effects of potentially suppressed endogenous flux rates of plasma FA on RQ and M values during the high-insulin clamp are rather unlikely.
This study also reports for the first time to our knowledge the temporal sequence of changes in muscle DAG composition and localization and insulin signaling in humans. Lipid infusion increased mainly DAG containing C18:0, C18:1, or C18:2 FAs and in addition membrane DAG with C16:0 and C20:4 FAs. Although composition of DAG species likely reflects the availability of FAs from circulating sources, we found that C16:0-, C18:1-, C18:2-, and C20:4-containing DAG were also increased in obese and T2D subjects, with C18:2 representing the most abundant residue in DAG species.
The rise in myocellular DAG content during the lipid infusion preceded the membrane translocation of PKCθ, which was observed at 4 h of lipid infusion in the absence of changes in PKCβ, PKCε, and PKCδ activities. These data are consistent with previous studies in rodents that have shown that lipid infusion leads to a transient increase in intramyocellular DAG content, PKCθ activation, and muscle insulin resistance (10, 11). In vitro activation of different nPKC isoforms vary in response to different DAG species with different time courses and competitive effects of specific DAG species (29). This might explain the difference between the present and previous findings of increased activity of membrane-associated PKCβII and PKCδ isoforms but unchanged PKCθ, PKCε, and PKCζ after a 6-h lipid infusion combined with a hyperinsulinemic-euglycemic clamp (15). Furthermore, the present study also found that lipid-induced muscle insulin resistance was associated with increased myocellular IRS1-Ser1101-Px at 4 h of lipid infusion, which was associated with blunted insulin-stimulation of PI3K activation and Akt-Ser473-PX in the membrane/cytosolic fraction. These data are consistent with a previous in vitro study demonstrating that increased IRS1-Ser1101-Px will lead to reduced insulin-stimulated Akt-Ser473-PX (30).
Previous lipid infusion studies in humans provided evidence for impaired IRS1-Tyr-Px and PI3K-Px combined with reduced (31) or unchanged Akt-Px but did not assess lipid metabolites or nPKC activation (32). Other studies failed to show impairment of insulin-stimulated IRS1-Tyr-Px, PI3K-Px IRS1, and Akt-Ser473-Px despite reduction of insulin-stimulated glucose uptake (16, 17). This inconsistency could result from differences in study protocols, such as combined lipid and insulin infusions, duration and dosage of lipid infusion, and timing of muscle biopsies, as well as from individual variation of the study participants (e.g., physical activity, age, and glucose tolerance).
Lipid infusion has also been shown to increase circulating cytokines and in turn induce a proinflammatory status and insulin resistance (33). It has been proposed that saturated FAs can activate the innate immune system and inflammatory pathways via binding to toll-like receptor (TLR)4, which in turn leads to an increase in myocellular ceramide content, which will lead to insulin resistance by direct inhibition of Akt (23). In this study, myocellular concentrations of total, as well as ceramide species, were comparable before and after lipid infusion and not related to the induction of muscle insulin resistance. This is consistent with some (15, 16) but not other studies in humans (34). Although it is possible that the low content of saturated FAs in the lipid infusion used in the current study might explain this discrepancy, a recent study found that saturated fat-induced insulin resistance can occur independently of TLR4 activation and increased in tissue ceramide content (35). Finally, markers of systemic inflammation, IL-6, adiponectin, RBP4, and sICAM-1, which have all been implicated in causing insulin resistance in obese and T2D (14) individuals, remained unchanged during the lipid infusion, thus dissociating these factors from lipid-induced muscle insulin resistance in this study (24).
This study also found that increased muscle DAG content and PKCθ activation was strongly associated with muscle insulin resistance in obese and T2D individuals, independent of dyslipidemia. Enzymes involved in DAG synthesis (adipose triglyceride lipase) are negatively related to insulin sensitivity (36), whereas enzymes controlling DAG hydrolysis (hormone-sensitive lipase) or conversion to triacylglycerols are decreased in obese humans (36). On the other hand, obese individuals with normal DAG content but increased triacylglycerols had higher muscle DGAT expression (37). Thus, individual lipolytic control in skeletal muscle could modulate lipotoxic effects and thereby explain the difference between reports of unchanged (38, 39) or increased total muscle DAG in obese humans (40). Previous studies found that the degree of DAG saturation associated positively (39), negatively (41), or not (38) with insulin resistance. We found that several cytosolic and membrane DAG species containing C16:0, C18:0, C:18:1, C18:2, and C20:4 were associated with both BMI and M value. After adjustment for BMI, the correlations were more pronounced in the membrane fraction and mainly driven by unsaturated FAs (C:18:1, C18:2, and C20:4) at one or two binding sites. Increased DAG species containing C:18:1, C18:2, and C20:4 were also associated with acute induction of muscle insulin resistance during the lipid infusion. We also found that C20:4, C20:5 membrane DAG species correlated positively with insulin sensitivity, which is consistent with previous findings in fat-fed animals (42). Polyunsaturated FAs of muscle phospholipids, which were not measured in the present study, have previously been shown to positively correlate with insulin sensitivity and have been suggested to increase membrane fluidity and increased movement of lipid drafts and insulin signaling mediators (43).
This study also addressed alternative mechanisms that have been proposed to cause muscle insulin resistance. We found no relationship between myocellular acylcarnitine concentrations and muscle insulin resistance during the lipid infusion or in insulin-resistant obese and T2D subjects, thus dissociating acylcarnitines from muscle insulin resistance under these conditions. Plasma acylcarnitines can be increased in insulin resistant humans, specifically during hyperglycemia, which has been attributed to intracellular deficiency of glycolytic precursors needed for complete lipid oxidation (44). The participants in the current study were nondiabetic or metabolically well-controlled T2D, which might explain the normal plasma acylcarnitines. Finally, we found no differences in plasma concentrations of IL-6, RBP4, and sICAM-1 in our obese and T2D subjects, which is in accordance with some (45) but not all (46) studies. Only plasma adiponectin concentrations were slightly lower in obese and T2D subjects compared with control subjects.
In summary, by performing sequential biopsies during a lipid infusion, we were able to delineate the temporal sequence of events responsible for acute lipid-induced insulin resistance in human skeletal muscle. We found that raising plasma fatty acid concentrations led to a transient increase in myocellular DAG content (mostly C18:0, C18:1, or C18:2 species) at ∼2.5 h, which was temporally related to PKCθ activation, increased IRS1-Ser1101-Px, and decreased insulin activation of AKT2. We found a similar strong relationship between muscle DAG content and PKCθ activation with muscle insulin resistance in obese and T2D subjects. In contrast, we observed no relationship between myocellular acylcarnitine content or ceramide content, thus dissociating these metabolites from muscle insulin resistance under these conditions. Taken together, these data support the hypothesis that DAG activation of PKCθ plays an important role in mediating lipid-induced insulin resistance in human skeletal muscle and identifies this pathway as a relevant therapeutic target to prevent and reverse muscle insulin resistance in humans.
Experimental Procedures
Participants gave written informed consent before inclusion, which was registered (Clinicaltrials.gov Identifier no. NCT01229059) and approved by the local institutional ethics board of Heinrich Heine University. They underwent a screening procedure including clinical examination, and a standardized 75-g oral glucose tolerance test. We included the following: (i) young (20–40 y) lean (BMI of 20–25 kg/m2) glucose-tolerant subjects without family history of diabetes; (ii) age-matched glucose-tolerant insulin-resistant obese subjects; and (iii) obese subjects with T2D (Table S4). Exclusion criteria were smoking, medication affecting insulin sensitivity, lipid metabolism or immune system (acute or common) diseases, and a history of cancer. Female subjects were studied between days 5–9 of their menstrual cycle. For 3 d before every study day, participants refrained from physical exercise, consumed isocaloric diet, and fasted for 12 h before the start of the experiments. Subjects with T2D were on treatment with metformin (n = 8) or sitagliptin (n = 1). Glucose-lowering medication was withdrawn for 1 wk before the study.
Lipid-Induced Muscle Insulin-Resistance Study.
The protocol is depicted in Fig. S6. Healthy young lean participants underwent two experimental conditions in random order. A pancreatic clamp was performed (−240 to 0 min) with continuous infusion of somatostatin (0.1 µg⋅kg−1⋅min−1; UCB Pharma) to inhibit insulin secretion. Insulin (Actrapid; Novo Nordisc) was applied to standardize fasting insulin levels (6 mU⋅m−2 body surface per minute, –240 to 0 min) and combined with infusion of d-[6,6-2H2]glucose (−360 to +150 min) as previously described (22). Intravenous infusion of lipids (lipid infusion: n = 36; 20% (vol/vol) Intralipid; Fresenius Kabi) was administered (−240 to −230 min: 10 mL/h; −229 to +150 min: 90 mL/h) to raise plasma FAs. On another occasion spaced by 4- to 8-wk intervals, 2.5% (wt/vol) glycerol dissolved in 0.9% saline (glycerol infusion: n = 24; Fresenius Kabi) was infused (−240 to −230 min: 10 mL/h; −229 to +150 min: 90 mL/h).
Obese and T2D Subject Studies.
The design is depicted in Fig. S7. Common insulin resistance was investigated in insulin resistant obese (n = 10) and patients with T2D (n = 10) without severe dyslipidemia (triglyceride levels >300 mg/dL). Insulin resistance was defined by oral glucose insulin sensitivity values below 462.8 mL⋅min−1⋅m−2, as previously reported (47). All participants underwent a high-insulin euglycemic clamp (0−150 min) combined with infusion of d-[6,6-2H2]glucose (−180 to +150 min).
Hyperinsulinemic–Euglycemic Clamp Studies.
A primed-continuous intravenous infusion of d-[6,6-2H2]glucose (98% enriched; Cambridge Isotope Laboratories) at (3.6 mg⋅kg−1 body weight) × (fasting glucose [mg/dL])/90 [mg/dL] for 5 min and at 0.036 mg⋅min−1⋅kg−1 body weight was started before the start of the high-insulin clamp (40 mU⋅m−2 body surface per minute, 0 to +150 min) during the studies on lipid-induced insulin resistance (−360 min) and 180 min before the start of the high-insulin euglycemic clamp in all studies (−180) until the end of the experiments (+150 min).
Indirect Calorimetry.
Indirect calorimetry was performed using Vmax Encore 29n (CareFusion) at baseline (−330 to −300 min) and during steady-state high-insulin clamp conditions (120 to 150 min). Physical activity index was assessed according to Baecke et al. (48).
Metabolites and Hormones.
Blood samples were chilled and centrifuged, and supernatants were stored at −20 °C. Blood glucose was measured using the glucose oxidase method (EKF biosen C-Line glucose analyzer; EKF Diagnostic). Serum triglycerides were analyzed by enzymatic assays on a Hitachi analyzer (Roche Diagnostics) and free FAs with the microfluorimetric method [intraassay coefficient of variation (CV), <1%; interassay CV, 2.4%; Wako] after prevention of lipolysis using orlistat (21). High-molecular-weight adiponectin was determined using the Adiponectin (Multimeric) ELISA kit from ALPCO Diagnostics. Intra- and interassay CVs were 5.1% and 8.2%. Plasma concentrations of TNFα and IL-6 were determined by Quantikine HS (TNFα and IL-6) and Quantikine (IL-1ra) ELISA kits from R&D Systems, as described (49). For assessment of acylcarnitines, 50 µL of plasma or ∼200 mg of tissue was homogenized and centrifuged. The supernatant was passed through a reversed-phase cartridge (Sep-Pak C18; 500 mg; Waters), and the eluate was concentrated to 100 µL and injected into an API 6500 Qtrap (ABSCIEX). The limit of detection is 1 × 10−6 nmol acetylcarnitine injection, corresponding to 3 pmol/g in tissue and 4.5 × 10−4 pmol/µL in plasma, with a CV of 22%. The CV is 12% at 1 × 10−5 nmol and 0.3% at 1 × 10−4 nmol acetylcarnitine injections.
Skeletal Muscle Biopsy Samples.
All study participants had muscle biopsies at baseline. Twenty-three of the CON underwent biopsies at baseline and during LIP after 2.5 and 4 h, 11 of those also during glycerol infusion. Thirteen CON had muscle biopsies at baseline and after 4.5 h of lipid and glycerol infusion after 30 min of the high-insulin clamp to examine insulin signaling during short-term insulin resistance. The region above the musculus vastus lateralis was anesthetized with local anesthetics (Xylocain 2%) (24), a muscle biopsy of ∼70–400 mg was obtained using a Bergstrom needle. Samples were immediately stored in liquid nitrogen and then frozen at −80 °C.
Muscle tissue was homogenized in a buffer solution (20 mM Tris⋅HCl, 1 mM EDTA, 0.25 mM EGTA, 250 mM sucrose, 2 mM PMSF) containing a protease inhibitor mixture (Roche), and samples were centrifuged at 100,000 × g for 1 h. The supernatants containing the cytosolic fraction and the pellet consisting of the membrane fraction were collected. DAG and ceramide concentration were measured as previously described (50).
Membrane translocation for PKC isoforms was assessed as described (51). PKC translocation was expressed as ratio of membrane over cytosol bands on the same film.
For phosphorylation of IRS1-Ser1101, the membrane was probed with anti phospho IRS1-Ser1101 first and then stripped and probed again with antibodies for total IRS-1 (Cell Signaling). The membrane and cytosolic fractions were prepared using differential centrifugation to assess Akt-Ser473-Px (Cell Signaling). IRS1-associated PI3K activity was determined in muscle extracts after immunoprecipitation with IRS1 antibody/agarose conjugate overnight at 4 °C.
Calculations and Statistics.
All statistical analyses were performed using SPSS 6.0 software (SPSS). Data are presented as means ± SD throughout the text and as means ± SEM in the figures. Statistical comparisons between study groups were performed using ANOVA and repeated-measurements ANOVA with Tukey post hoc testing when appropriate. Within-group differences were determined using two-tailed Student t tests. Differences were considered significant at the 5% level.
Supplementary Material
Acknowledgments
We thank Christel Führer, Myrko Esser, Kai Tinnes, Gabi Gornitzka, Irena Latta, Ulrike Poschen, Mario Kahn, and Karin Röhrig for excellent technical assistance. This work was supported by the Ministry of Science and Research of the State of North Rhine-Westphalia and the German Federal Ministry of Health. This study was supported by European Foundation for the Study of Diabetes Grant 12/2009; German Research Foundation Grant Sonderforschungsbereich 575; the German Diabetes Association; Schmutzler-Stiftung; Skröder-Stiftung; the German Center for Diabetes Research; US Public Health Service Grants R01 DK-49230, R24 DK-085638, and P30 DK-45735; and the Novo Nordisk Foundation Center for Basic Metabolic Research at the University of Copenhagen.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1409229111/-/DCSupplemental.
References
- 1.Roden M, et al. Mechanism of free fatty acid-induced insulin resistance in humans. J Clin Invest. 1996;97(12):2859–2865. doi: 10.1172/JCI118742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cline GW, et al. Impaired glucose transport as a cause of decreased insulin-stimulated muscle glycogen synthesis in type 2 diabetes. N Engl J Med. 1999;341(4):240–246. doi: 10.1056/NEJM199907223410404. [DOI] [PubMed] [Google Scholar]
- 3.Dresner A, et al. Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J Clin Invest. 1999;103(2):253–259. doi: 10.1172/JCI5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roden M. How free fatty acids inhibit glucose utilization in human skeletal muscle. News Physiol Sci. 2004;19:92–96. doi: 10.1152/nips.01459.2003. [DOI] [PubMed] [Google Scholar]
- 5.Shulman GI. Cellular mechanisms of insulin resistance. J Clin Invest. 2000;106(2):171–176. doi: 10.1172/JCI10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Samuel VT, Shulman GI. Mechanisms for insulin resistance: Common threads and missing links. Cell. 2012;148(5):852–871. doi: 10.1016/j.cell.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roden M, et al. Rapid impairment of skeletal muscle glucose transport/phosphorylation by free fatty acids in humans. Diabetes. 1999;48(2):358–364. doi: 10.2337/diabetes.48.2.358. [DOI] [PubMed] [Google Scholar]
- 8.Weiss R, et al. Prediabetes in obese youth: A syndrome of impaired glucose tolerance, severe insulin resistance, and altered myocellular and abdominal fat partitioning. Lancet. 2003;362(9388):951–957. doi: 10.1016/S0140-6736(03)14364-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perseghin G, Petersen K, Shulman GI. Cellular mechanism of insulin resistance: Potential links with inflammation. Int J Obes Relat Metab Disord. 2003;27(Suppl 3):S6–S11. doi: 10.1038/sj.ijo.0802491. [DOI] [PubMed] [Google Scholar]
- 10.Griffin ME, et al. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes. 1999;48(6):1270–1274. doi: 10.2337/diabetes.48.6.1270. [DOI] [PubMed] [Google Scholar]
- 11.Yu C, et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem. 2002;277(52):50230–50236. doi: 10.1074/jbc.M200958200. [DOI] [PubMed] [Google Scholar]
- 12.Chavez JA, Holland WL, Bär J, Sandhoff K, Summers SA. Acid ceramidase overexpression prevents the inhibitory effects of saturated fatty acids on insulin signaling. J Biol Chem. 2005;280(20):20148–20153. doi: 10.1074/jbc.M412769200. [DOI] [PubMed] [Google Scholar]
- 13.Koves TR, et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008;7(1):45–56. doi: 10.1016/j.cmet.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 14.Glass CK, Olefsky JM. Inflammation and lipid signaling in the etiology of insulin resistance. Cell Metab. 2012;15(5):635–645. doi: 10.1016/j.cmet.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Itani SI, Ruderman NB, Schmieder F, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes. 2002;51(7):2005–2011. doi: 10.2337/diabetes.51.7.2005. [DOI] [PubMed] [Google Scholar]
- 16.Høeg LD, et al. Lipid-induced insulin resistance affects women less than men and is not accompanied by inflammation or impaired proximal insulin signaling. Diabetes. 2011;60(1):64–73. doi: 10.2337/db10-0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Storgaard H, et al. Dissociation between fat-induced in vivo insulin resistance and proximal insulin signaling in skeletal muscle in men at risk for type 2 diabetes. J Clin Endocrinol Metab. 2004;89(3):1301–1311. doi: 10.1210/jc.2003-031243. [DOI] [PubMed] [Google Scholar]
- 18.Boden G, Chen X, Ruiz J, White JV, Rossetti L. Mechanisms of fatty acid-induced inhibition of glucose uptake. J Clin Invest. 1994;93(6):2438–2446. doi: 10.1172/JCI117252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963;1(7285):785–789. doi: 10.1016/s0140-6736(63)91500-9. [DOI] [PubMed] [Google Scholar]
- 20.Lanza IR, et al. Chronic caloric restriction preserves mitochondrial function in senescence without increasing mitochondrial biogenesis. Cell Metab. 2012;16(6):777–788. doi: 10.1016/j.cmet.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brehm A, et al. Increased lipid availability impairs insulin-stimulated ATP synthesis in human skeletal muscle. Diabetes. 2006;55(1):136–140. [PubMed] [Google Scholar]
- 22.Brehm A, et al. Acute elevation of plasma lipids does not affect ATP synthesis in human skeletal muscle. Am J Physiol Endocrinol Metab. 2010;299(1):E33–E38. doi: 10.1152/ajpendo.00756.2009. [DOI] [PubMed] [Google Scholar]
- 23.Holland WL, et al. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J Clin Invest. 2011;121(5):1858–1870. doi: 10.1172/JCI43378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nowotny B, et al. Mechanisms underlying the onset of oral lipid-induced skeletal muscle insulin resistance in humans. Diabetes. 2013;62(7):2240–2248. doi: 10.2337/db12-1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vistisen B, et al. Effect of gender on lipid-induced insulin resistance in obese subjects. Eur J Endocrinol. 2008;158(1):61–68. doi: 10.1530/EJE-07-0493. [DOI] [PubMed] [Google Scholar]
- 26.Perseghin G, et al. Normal insulin sensitivity and IMCL content in overweight humans are associated with higher fasting lipid oxidation. Am J Physiol Endocrinol Metab. 2002;283(3):E556–E564. doi: 10.1152/ajpendo.00127.2002. [DOI] [PubMed] [Google Scholar]
- 27.Jensen MD, Caruso M, Heiling V, Miles JM. Insulin regulation of lipolysis in nondiabetic and IDDM subjects. Diabetes. 1989;38(12):1595–1601. doi: 10.2337/diab.38.12.1595. [DOI] [PubMed] [Google Scholar]
- 28.Krusenstjerna-Hafstrøm T, et al. Growth hormone (GH)-induced insulin resistance is rapidly reversible: An experimental study in GH-deficient adults. J Clin Endocrinol Metab. 2011;96(8):2548–2557. doi: 10.1210/jc.2011-0273. [DOI] [PubMed] [Google Scholar]
- 29.Madani S, Hichami A, Legrand A, Belleville J, Khan NA. Implication of acyl chain of diacylglycerols in activation of different isoforms of protein kinase C. FASEB J. 2001;15(14):2595–2601. doi: 10.1096/fj.01-0753int. [DOI] [PubMed] [Google Scholar]
- 30.Li Y, et al. Protein kinase C Theta inhibits insulin signaling by phosphorylating IRS1 at Ser(1101) J Biol Chem. 2004;279(44):45304–45307. doi: 10.1074/jbc.C400186200. [DOI] [PubMed] [Google Scholar]
- 31.Belfort R, et al. Dose-response effect of elevated plasma free fatty acid on insulin signaling. Diabetes. 2005;54(6):1640–1648. doi: 10.2337/diabetes.54.6.1640. [DOI] [PubMed] [Google Scholar]
- 32.Kruszynska YT, et al. Fatty acid-induced insulin resistance: Decreased muscle PI3K activation but unchanged Akt phosphorylation. J Clin Endocrinol Metab. 2002;87(1):226–234. doi: 10.1210/jcem.87.1.8187. [DOI] [PubMed] [Google Scholar]
- 33.Krogh-Madsen R, et al. Effect of short-term intralipid infusion on the immune response during low-dose endotoxemia in humans. Am J Physiol Endocrinol Metab. 2008;294(2):E371–E379. doi: 10.1152/ajpendo.00507.2007. [DOI] [PubMed] [Google Scholar]
- 34.Straczkowski M, et al. Relationship between insulin sensitivity and sphingomyelin signaling pathway in human skeletal muscle. Diabetes. 2004;53(5):1215–1221. doi: 10.2337/diabetes.53.5.1215. [DOI] [PubMed] [Google Scholar]
- 35.Galbo T, et al. Saturated and unsaturated fat induce hepatic insulin resistance independently of TLR-4 signaling and ceramide synthesis in vivo. Proc Natl Acad Sci USA. 2013;110(31):12780–12785. doi: 10.1073/pnas.1311176110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Badin PM, et al. Altered skeletal muscle lipase expression and activity contribute to insulin resistance in humans. Diabetes. 2011;60(6):1734–1742. doi: 10.2337/db10-1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Coen PM, et al. Reduced skeletal muscle oxidative capacity and elevated ceramide but not diacylglycerol content in severe obesity. Obesity (Silver Spring) 2013;21(11):2362–2371. doi: 10.1002/oby.20381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Coen PM, et al. Insulin resistance is associated with higher intramyocellular triglycerides in type I but not type II myocytes concomitant with higher ceramide content. Diabetes. 2010;59(1):80–88. doi: 10.2337/db09-0988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van Hees AM, et al. Skeletal muscle fatty acid handling in insulin resistant men. Obesity (Silver Spring) 2011;19(7):1350–1359. doi: 10.1038/oby.2011.10. [DOI] [PubMed] [Google Scholar]
- 40.Bergman BC, Hunerdosse DM, Kerege A, Playdon MC, Perreault L. Localisation and composition of skeletal muscle diacylglycerol predicts insulin resistance in humans. Diabetologia. 2012;55(4):1140–1150. doi: 10.1007/s00125-011-2419-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Amati F, et al. Skeletal muscle triglycerides, diacylglycerols, and ceramides in insulin resistance: Another paradox in endurance-trained athletes? Diabetes. 2011;60(10):2588–2597. doi: 10.2337/db10-1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Figueras M, Olivan M, Busquets S, López-Soriano FJ, Argilés JM. Effects of eicosapentaenoic acid (EPA) treatment on insulin sensitivity in an animal model of diabetes: Improvement of the inflammatory status. Obesity (Silver Spring) 2011;19(2):362–369. doi: 10.1038/oby.2010.194. [DOI] [PubMed] [Google Scholar]
- 43.Borkman M, et al. The relation between insulin sensitivity and the fatty-acid composition of skeletal-muscle phospholipids. N Engl J Med. 1993;328(4):238–244. doi: 10.1056/NEJM199301283280404. [DOI] [PubMed] [Google Scholar]
- 44.Mihalik SJ, et al. Increased levels of plasma acylcarnitines in obesity and type 2 diabetes and identification of a marker of glucolipotoxicity. Obesity (Silver Spring) 2010;18(9):1695–1700. doi: 10.1038/oby.2009.510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Erion DM, Shulman GI. Diacylglycerol-mediated insulin resistance. Nat Med. 2010;16(4):400–402. doi: 10.1038/nm0410-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kado S, Nagata N. Circulating intercellular adhesion molecule-1, vascular cell adhesion molecule-1, and E-selectin in patients with type 2 diabetes mellitus. Diabetes Res Clin Pract. 1999;46(2):143–148. doi: 10.1016/s0168-8227(99)00083-2. [DOI] [PubMed] [Google Scholar]
- 47.Prikoszovich T, et al. Body and liver fat mass rather than muscle mitochondrial function determine glucose metabolism in women with a history of gestational diabetes mellitus. Diabetes Care. 2011;34(2):430–436. doi: 10.2337/dc10-1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baecke JA, Burema J, Frijters JE. A short questionnaire for the measurement of habitual physical activity in epidemiological studies. Am J Clin Nutr. 1982;36(5):936–942. doi: 10.1093/ajcn/36.5.936. [DOI] [PubMed] [Google Scholar]
- 49.Herder C, et al. Elevated levels of the anti-inflammatory interleukin-1 receptor antagonist precede the onset of type 2 diabetes: The Whitehall II study. Diabetes Care. 2009;32(3):421–423. doi: 10.2337/dc08-1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Neschen S, et al. Prevention of hepatic steatosis and hepatic insulin resistance in mitochondrial acyl-CoA:glycerol-sn-3-phosphate acyltransferase 1 knockout mice. Cell Metab. 2005;2(1):55–65. doi: 10.1016/j.cmet.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 51.Kumashiro N, et al. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc Natl Acad Sci USA. 2011;108(39):16381–16385. doi: 10.1073/pnas.1113359108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.