

Abstract
B cells and their progeny that produce and release anti-neutrophil cytoplasmic autoantibodies (ANCA) are the primary cause for an aggressive form of necrotizing small vessel vasculitis. Cytoplasmic ANCA antigens are released at the surface and in the microenvironment of cytokine-primed neutrophils. Binding of ANCA to ANCA antigens activates neutrophils by both Fc receptor engagement and direct Fab’2 binding to antigen on the cell surface. ANCA-activated neutrophils release factors that induce alternative complement pathway activation, which establishes a potent inflammatory amplification loop that causes severe necrotizing vascular inflammation. The origin of the ANCA autoimmune response is unknown but appears to involve genetically determined HLA specificities that allow the autoimmune response to develop. One putative immunogenic mechanisms begins with an immune response to a peptide that is complementary to the autoantigen and evolves through an anti-idiotypic network to produce autoantibodies to the autoantigen. Another putative immunogenic mechanism begins with an immune response to a microbe-derived molecular mimic of the autoantigen resulting in antibodies that cross-react with the autoantigen. Release of neutrophil extracellular traps, apoptosis and increased granule protein expression of ANCA antigens may facilitate the initiation of an ANCA autoimmune response, augment established pathogenic ANCA production, or both. The ANCA B cell autoimmune response is facilitated by quantitatively and qualitatively impaired T cell and B cell suppression, and by release from activated neutrophils of B cell activating factors that enhance B cell proliferation and retard B cell apoptosis.
Keywords: Antineutrophil Cytoplasmic Autoantibodies, MPO-ANCA, PR3-ANCA, Vasculitis, Microscopic Polyangiitis, Granulomatosis with Polyangiitis
Overview of ANCA and ANCA Disease
Anti-neutrophil cytoplasmic autoantibodies (ANCA) are specific for proteins in the cytoplasm of neutrophils and monocytes. They were first discovered in serum by indirect immunofluorescence microscopy, which demonstrates cytoplasmic (C-ANCA) or perinuclear (P-ANCA) staining of normal human neutrophils (Fig. 1). C-ANCA were discovered serendipitously by a pathologist in Australia (David Davies) who was using normal human neutrophils as a substrate to detect anti-nuclear antibodies. He observed that a subset of patients with focal necrotizing and crescentic glomerulonephritis had circulating antibodies that bound to the cytoplasm of normal neutrophils [1]. Davis 1982 article [1] was largely overlooked until a European collaborative group lead by van der Woude reported in 1985 that that C-ANCA were closely associated with Wegener’s granulomatosis (now called granulomatosis with polyangiitis) and that ANCA titers diminished or disappeared with response to treatment [2]. The spectrum of ANCA and ANCA disease was extended in 1988 when Falk and Jennette described P-ANCA and reported that both C-ANCA and P-ANCA occurred not only in patients with granulomatosis with polyangiitis (Wegener’s) but also in patients with microscopic polyangiitis and renal limited necrotizing and crescentic glomerulonephritis that lacked well defined immunoglobulin deposits [3].
Figure 1.
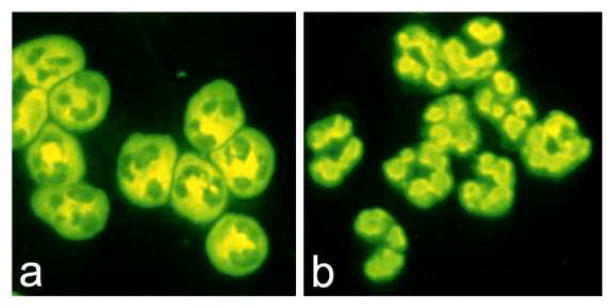
Indirect immunofluorescence microscopy showing the patterns of staining of cytospin preparations of alcohol-fixed normal human caused by ANCA. 1a: Cytoplasmic (C-ANCA) staining pattern caused by PR3-ANCA. 1b: Perinuclear (P-ANCA) staining caused by MPO-ANCA. (Second antibody is FITC-labeled rabbit anti-human IgG).
Numerous studies have confirmed that ANCA are associated with a distinctive category of small vessel inflammation that is characterized by necrotizing inflammation of vessels and absence or paucity of vessel wall localization of immunoglobulin and complement. This paucity of immunoglobulin distinguishes pauci-immune ANCA-associated vasculitis and glomerulonephritis from vasculitis and glomerulonephritis caused by extensive immune complex accumulation in vessel walls (i.e. immune complex vasculitis), and from vasculitis caused by in situ formation of immune complexes between vessel wall basement membrane antigens and anti-basement membrane autoantibodies, i.e. anti-glomerular basement membrane (anti-GBM) disease) [4]. This immunopathologic classification of small vessel vasculitis can be accomplished using direct immunofluorescence microscopy to determine the extent, pattern and composition of immunoglobulin deposits in vessel walls, for example in the walls of renal glomerular capillaries (Fig. 2), dermal venules or pulmonary alveolar capillaries.
Figure 2.
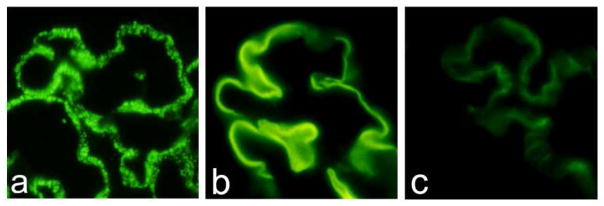
Direct immunofluorescence microscopy of glomerular capillaries demonstrating granular staining of capillary walls for IgG indicative of immune complex disease (2a), linear staining of glomerular basement membranes (GBM) for IgG indicative of anti-GBM disease (2b), and a paucity of staining of capillary walls for IgG in a patient with ANCA glomerulonephritis (2c). (FITC-labeled rabbit anti-human IgG)
According to the 2012 International Chapel Hill Consensus Conference Nomenclature of Vasculitides, ANCA-associated vasculitis is defined as necrotizing vasculitis, with few or no immune deposits, predominantly affecting small vessels, i.e., capillaries, venules, arterioles and small arteries [4] (Fig. 3). In the acute phase, the vasculitis is characterized by predominantly neutrophilic infiltration with extensive apoptosis and necrosis of neutrophils resulting in extensive nuclear fragmentation (leukocytoclasia) (Fig. 3a). If this feature is conspicuous, the descriptive term leukocytoclastic angiitis can be applied. When arteries are affected, the acute lesions often include the spillage of plasma into necrotic vessel walls and perivascular tissue where the coagulation cascade contacts thrombogenic stimuli and forms fibrin, resulting in a pattern of fibrinoid necrosis (Fig. 3c). All variants of ANCA disease can have glomerulonephritis that is characterized by segmental fibrinoid necrosis and crescent formation (Fig. 3b), both of which result from segmental rupture of glomerular capillaries by the inflammatory process.
Figure 3.

Vascular inflammation is patients with ANCA disease. 1a: Leukocytoclastic angiitis affecting a venule in the renal medulla. Note the prominent karyorrhexis of neutrophils (leukocytoclasia). 2b: Segmental necrotizing glomerulonephritis with a focus of deeply eosinophilic fibrinoid material with an adjacent cellular reaction (crescent). 3c. Small artery with segmental fibrinoid necrosis in the upper right portion of the vessel wall with intramural and perivascular infiltration of neutrophils and mononuclear leukocytes. 4d: Pulmonary alveolar capillaritis with hemorrhage into the air spaces and numerous neutrophils within alveolar capillaries. (Hematoxylin and eosin stain)
ANCA vasculitis patients are further subclassified on the basis of clinical and pathologic features into microscopic polyangiitis (MPA), granulomatosis with polyangiitis (GPA) (formerly known as Wegener’s granulomatosis) and eosinophilic granulomatosis with polyangiitis (EGPA) (formerly known as Churg-Strauss syndrome) [4]. MPA is necrotizing vasculitis with few or no immune deposits predominantly affecting small vessels (i.e., capillaries, venules, or arterioles) in the absence of granulomatosis or a history of asthma. Arteries may also be affected. GPA has the features of MPA with the additional feature of necrotizing granulomatous inflammation that usually involves the upper and/or lower respiratory tract. The vasculitic phase of EGPA is characterized by eosinophil-rich inflammation and necrotizing granulomatous inflammation often involving the respiratory tract, and necrotizing vasculitis predominantly affecting small to medium vessels, and associated with asthma and eosinophilia. The early prodromal phase of EGPA has eosinophilic inflammation in the lungs or gastrointestinal tract. ANCA only become frequent if patients develop overt vasculitis or glomerulonephritis. The pauci-immune necrotizing and crescentic glomerulonephritis of MPA, GPA, EGPA and renal limited ANCA vasculitis is pathologically and immunopathologically indistinguishable.
Organ limited expressions of ANCA disease occur, for example isolated pauci-immune necrotizing crescentic glomerulonephritis (renal-limited vasculitis) or GPA limited to the upper respiratory tract. However, patients who initially have organ-limited disease may subsequently develop multisystem disease.
In patients with vasculitis and glomerulonephritis, the two best characterized autoantigen targets for ANCA are myeloperoxidase (MPO) [3] and proteinase 3 (PR3) [5–7], although additional autoantigen targets have been detected or proposed, such as elastase [8] and lysosome associated membrane protein 2 (LAMP2) [9]. In indirect immunofluorescence assays, PR3-ANCA typically produce a cytoplasmic (C-ANCA) staining pattern whereas MPO-ANCA typically produce a perinuclear (P-ANCA) staining pattern. The P-ANCA pattern is caused by the artifactual relocation of MPO from the primary granules to the nucleus when substrate neutrophils are fixed in alcohol prior to drying onto slides [3]. This solubilizes the cationic MPO from the primary granules and allows it to diffuse to a stick on the anionic nuclei (Figure 1b). PR3-ANCA and MPO-ANCA produce identical cytoplasmic staining of formalin-fixed neutrophils in which the MPO has been covalently bound in position by the fixative.
In North America and Europe, patients with GPA more often have PR3-ANCA than MPO-ANCA, MPA patients have similar frequency of MPO-ANCA and PR3-ANCA, and EGPA and renal-limited vasculitis patients have predominantly MPO-ANCA [10]. In Asia, MPO-ANCA is more frequent than PR3-ANCA in patients with GPA as well as MPA [11]. A genome wide association study has shown that autoantigen specificities for PR3 versus MPO correlate better with genetic associations than the clinicopathologic phenotypes of MPA and GPA [12]. In addition, as noted earlier, the clinical manifestations of ANCA disease, including the patterns of organ system involvement, has different trends in patients with MPO-ANCA compared to those with PR3-ANCA [13]. The best approach to classifying and diagnosing ANCA disease is to specify both the serologic ANCA specificity as well as the clinicopathologic phenotype (e.g. MPO-ANCA MPA, MPO-ANCA GPA, PR3-ANCA MPA, PR3-ANCA GPA, MPO-ANCA EGPA, etc.) [4]. It is important to note that approximately 10% of patients with active untreated MPA and GPA are negative for ANCA in current conventional ANCA serologic assays, although at least some of these patients have ANCA that can be detected by more sensitive assays [15] or assays for other autoantigen targets [16] currently available only in research settings. Interestingly, EGPA patients have a high frequency of ANCA, usually MPO-ANCA, only when there is evidence for active vasculitis and glomerulonephritis as a component of the syndrome [17]. Concurrent PR3-ANCA and MPO-ANCA is rare except in patients with some forms of drug-induced ANCA disease [18].
Clinical Evidence that ANCA Cause Disease
As already noted, the very close association of a pathologically distinctive form of small vessel vasculitis with ANCA at least raises the possibility of a pathogenetic link. However, there is controversy about how well ANCA titers correlate with disease activity using the current generation of clinical laboratory tests [19]. However, more sophisticated analytically methods can discern the titers of pathogenic versus nonpathogenic ANCA with restricted epitope specificities that correlate better with ANCA disease activity. For example, Roth et al [15] used a sensitive epitope excision mass spectroscopy peptide identification technique to identify specific epitopes on MPO that were targeted only in patients with active MPO-ANCA disease [15]. Overall, over 20 different MPO epitopes were identified. Patients in disease remission had MPO-ANCA directed against nonpathogenic epitopes, and even healthy controls had very low titers of MPO-ANCA (natural autoantibodies) that reacted with these nonpathogenic epitopes, but only patients with active disease had detectable titers of MPO-ANCA specific for the pathogenic epitopes. Conventional ELISA using pathogenic linear epitopes showed a strong correlation between titers of MPO-ANCA and disease activity, whereas ELISA using nonpathogenic epitopes showed a poor correlation between titer of MPO-ANCA and disease activity [15]. Thus, although conventional serologic assays that use whole MPO molecules as substrate do not correlate with disease activity, epitope specific assays do. The basis for this epitope diversity may be epitope spreading that modulates the epitope specificity of ANCA during different phases of disease.
Another clinical observation that supports ANCA pathogenicity is the phenomenon of drug-induced ANCA disease [18]. This illustrate the concurrent induction of the autoantibodies and disease, and the resolution of the disease with resolution of the autoantibodies. A mechanism for hydralazine-induced ANCA disease that has been propose [18] involves dysregulation of ANCA antigen expression by neutrophils, which mimics a phenomenon that has been observed in ANCA disease patients who have aberrant epigenetically deregulated overexpression of MPO and PR3 in circulating neutrophils [20,21]. Hydralazine is a DNA methylation inhibitor that could reverses epigenetic silencing of PR3 and MPO, resulting in increased expression of both autoantigens in neutrophils to break tolerance and enhance neutrophil activation by ANCA [14]. The induction of ANCA and of ANCA disease by drugs is indirect evidence for the pathogenicity of ANCA.
Finally, the treatment response of ANCA disease supports an antibody mediated process. In addition to anti-inflammatory therapy with corticosteroids, therapy that targets B cells (e.g. rituximab) or reduces ANCA levels by plasma exchange is effective in ameliorating ANCA disease [22].
In Vitro Evidence that ANCA Can Cause Disease
If ANCA are pathogenic, they must be able to activate neutrophils and cause them to attack small vessels. Numerous in vitro experiments document that ANCA IgG can activate primed neutrophils causing them to release toxic oxygen radicals and destructive enzymes [23–25]. Neutrophil activation by ANCA is facilitated by priming of neutrophils, for example with cytokines (e.g. tumor necrosis factor alpha) or bacterial products (e.g. lipopolysaccharide), which results in the release of ANCA target antigens (e.g. MPO and PR3) at the cell surface where they can interact with ANCA. The interaction of ANCA with ANCA antigens on the surface of neutrophils and in the microenvironment of an inflammatory lesion activates neutrophils predominantly by engagement of Fc receptors by ANCA complexed to antigen [26,27] and also by ANCA Fab’2 binding to antigens on neutrophil surfaces [28,29]. ANCA also binds to ANCA antigens that have been release into the inflammatory microenvironment, including ANCA antigens adsorbed onto matrix and cells, such asendothelial cells [30]. Activation of neutrophils by ANCA in vitro causes injury and death of nearby cultured monolayers of endothelial cells [30–32], which conceptually supports a role for ANCA-induced neutrophil activation in the pathogenesis of vasculitis. Injury to endothelium by activated neutrophils also is likely to involve the release of neutrophil extracellular traps (NETs) that contain many cytotoxic factors [33,34].
In Vivo Evidence that ANCA Cause Disease
The most compelling evidence that ANCA are pathogenic derives from studies in animal models of ANCA disease. Although there are several convincing animal models of MPO-ANCA disease that closely mimic human ANCA disease [35], there are no unequivocal models of PR3-ANCA disease possibly because of quantitative and qualitative differences between human and PR3in the experimental animals used to date [36]. Xiao et al, developed the first convincing animal model of ANCA disease by injecting mouse anti-mouse anti-MPO IgG into mice [37], which resulted in the induction of pauci-immune necrotizing and crescentic glomerulonephritis in all mice and multiorgan necrotizing small vessel vasculitis in some mice. The anti-mouse MPO was derived from MPO−/− mice that were immunized with purified mouse MPO, thus the model is not a model of the genesis of an ANCA autoimmune response, but is a model of the pathogenesis of vascular inflammation mediated by anti-MPO IgG. Disease can be induced by injecting anti-MPO IgG into Rag2−/− mice that lack immune-competent B-cells and T-cells, demonstrating that antibody alone is sufficient to cause vasculitis and glomerulonephritis, and T-cells are not required [37]. Neutrophils are required for anti-MPO induced disease because depletion of neutrophils prevents disease [38]. The importance of neutrophils in the pathogenic process was further substantiated using a bone marrow transplantation approach to position MPO-positive neutrophils in the bodies of MPO knockout mice [39], which demonstrated that bone marrow-derived cells are sufficient and necessary targets to mediate glomerulonephritis and vasculitis induced by anti-MPO antibodies. As noted earlier, in vitro activation of neutrophils by ANCA IgG is facilitated by priming of neutrophils (e.g. with tumor necrosis factor α), which causes release of small amounts of ANCA antigens at the cell surface [23–25]. As predicted by this in vitro observation, injecting mice with bacterial lipopolysaccharide, which induces increased circulating tumor necrosis factor α, enhances the severity of glomerulonephritis induced by injection of anti-MPO [40], probably by priming circulating neutrophils to be more activatable by anti-MPO. This is the likely explanation for the clinical observation that onset and exacerbation of ANCA disease in patients often corresponds to flu-like symptoms (which are evidence for elevated circulating cytokines), for example caused by a concurrent viral respiratory tract infection.
The importance of the functional integrity of anti-MPO IgG Fc regions has been demonstrated in the anti-MPO mouse model by showing an effect of altered Fc glycosylation on disease induction [41]. Hydrolysis of anti-MPO IgG glycans by EndoS attenuates anti-MPO induced neutrophil activation in vitro and prevents induction of glomerulonephritis in mice. Systemic treatment with EndoS after disease induction ameliorates the development of disease. The precise mechanism for this effect has not been determined.
As noted earlier, immunohistology of glomeruli and other vessels in patients with ANCA glomerulonephritis and vasculitis typically reveals a paucity of staining for immunoglobulin or complement. However, most specimens have at least some deposition of immunoglobulin and complement components, especially at the sites of inflammation. When complement components are identified in biopsies from patients with ANCA disease, they appear to be the result of activation of the alternative pathway of complement activation rather than the classic or lectin pathway [42]. Additional support for the involvement of the alternative complement pathway in human ANCA disease is the reported finding by Gou et al [43] of elevated plasma levels of C3a, C5a, soluble C5b-9 and Bb in patients with active ANCA disease compared to patients in remission, whereas levels of properdin were lower in active disease than in remission and C4d levels were the same in both. These finding indicate involvement of the alternative pathway and not the classic or lectin pathways. This conclusion also is supported by the mouse model of ANCA disease that is induced by anti-MPO [44–47]. In this model, ANCA disease is prevented by depleting complement with Cobra venom factor [44] or inhibiting factor 5 [45], both of which block complement activation by all three pathways. The absence of C4, which blocks the classic and lectin pathways but not the alternative pathway, has no effect on disease induction, whereas the absence of complement factor B, which blocks the alternative pathway, prevents disease induction [44]. The apparent critical event in the participation of alternative complement pathway activation in the pathogenesis of ANCA disease is the engagement of C5a receptors (CD88) on neutrophils by C5a [46,47]. In fact, Anti-MPO induced disease does not occur in mice with absence of C5a and disease can be prevented by pretreatment with a small molecule inhibitor of the C5a receptor [47].
In vitro studies show that human and mouse neutrophils that are activated by ANCA release an as yet unidentified factor (possibly properdin) that activates the alternative complement pathway [44,46,48,49]. C5a is released by complement activation and binds to C5a receptors on neutrophils, resulting in priming of neutrophils with increased release of ANCA antigens at the cell surface. This is mediated by intracellular activation of p38 mitogen activated protein kinase (p38MAPK), extracellular signal-regulated kinase (ERK) and phosphoinositol 3-kinase (PI3 K), and can be inhibited with bisindolylmaleimide (BIS) [48,49].
Based on in vitro and animal model studies as well as observations in patients, Figure 4 summarizes the putative pathogenic events in ANCA vasculitis. Neutrophils that have been primed by cytokines display ANCA antigens at the cell surface where they can be accessed by ANCA. Binding of ANCA to ANCA antigens results in Fc receptor and Fab’2 mediated activation of neutrophils, which in turn releases factors that activate the alternative complement pathway. C5a generated by complement activation attracts and primes more neutrophils resulting in amplification of the inflammatory process, and induction of the necrotizing glomerulonephritis and vasculitis that are characteristic of ANCA disease (Fig. 3).
Figure 4.

Depiction of pathogenic events in ANCA vasculitis. 1) Normal unprimed neutrophils have ANCA antigens (e.g. MPO and PR3) sequestered within cytoplasmic granules. 2) Neutrophils that have been primed by cytokines release small amounts of ANCA antigens at the cell surface and in the microenvironment where they are targeted by ANCA. Fc receptor engagement by ANCA bound to ANCA antigens as well as Fab’2 binding to ANCA antigens on the cell surface activates neutrophils. 3) Activated neutrophils release factors that activate the alternative complement pathway with the generation of C5a. C5a is a potent chemoattractant for neutrophils and attracts more neutrophils to the nidus of inflammation, and C5a engages C5a receptor (CD88) resulting in additional priming of neutrophils for activation by ANCA. 4) A potent inflammatory amplification loop is established that drives severe necrotizing inflammation in vessel walls.
Origin of the ANCA Autoimmune Response
The prior sections of this review propose mechanisms by which ANCA activate neutrophils to cause vasculitis. The remaining sections will propose how the underlying autoimmune response arises and thrives to allow B cells to produce pathogenic ANCA. The first question to address is the initial antigenic stimulus for an immune response that results in the production of autoantibodies specific for MPO or PR3, or other neutrophil autoantigens that mediated ANCA disease.
An innovative theory of the genesis of the ANCA autoimmune response posits that the initial immune response is against complementary peptides (e.g. antisense peptides or mimics of antisense peptides) of the autoantigen peptides, and that the autoimmune response derives from this initial response as a consequence of anti-idiotypic responses (Fig. 4) [50,51]. A sense peptide has an amino acid sequence that is coded for by the nucleotide sequence of the sense strand of DNA (5′ to 3′) or by mRNA whose sequence contains the same coding information as the sense strand of DNA. The complementary peptide has an amino acid sequence that is coded by the complementary (antisense) strand of DNA or by complementary mRNA with the same sequence information as the complementary strand of DNA [52]. The definition of complementary peptides may be expanded to include peptides whose sequence is determined by reading the nucleoside sequence of complementary mRNA in the alternative 3′ to 5′ direction because this peptide should have a similar hydropathic profile to a 5′ to 3′ complementary peptide because only the middle nucleoside base of a codon or complementary codon defines the hydropathic character of an amino acid residue [52]. Codon-directed amino acid residues in a sense peptide have specific pair-wise interactions with the corresponding complementary codon-directed residues in complementary peptides.
A sense protein and its complementary protein have a natural affinity for binding to each other [52,53]. Importantly, a T cell or B cell (antibody) immune response to a complementary protein can result in the production of T cells and immunoglobulin that reacts with the sense protein [51,52]. In other words, immunization with an antisense complementary peptide can induce an antibody response to the sense peptide.
PR3-ANCA patients have circulating antibodies reactive with a protein produced from PR3-antisense RNA [50,51]. These antibodies may have been induced by an immune response against either PR3 antisense peptides, or by peptides that mimic the antisense peptides, for example derived from microbes. When the amino acid sequence of a protein translated 5′–3′ from antisense RNA from the PR3 gene (PRTN3) is entered into the basic local alignment search tool (BLAST), multiple microbial homologues of complementary PR3 are identified, including proteins from microbes that are known to be associated with PR3-ANCA, such as Ross River virus, Staphylococcus aureus and Entamoeba histolytica [51]. All of the patients reported by Davis in the first published report on ANCA disease had serologic evidence for Ross River virus infection [1] and Staph aureus infection is closely linked with disease activity and disease phenotype in patients with GPA (Wegener’s) [54]. Theoretically, mimics of PR3 antisense peptides derived from Ross River Virus or Staphylococcus aureus could initiate an immune response that evolves into an autoimmune anti-PR3 response.
Patients with PR3-ANCA disease who have IgG antibodies that react with complementary PR3 peptides would be predicted to have T cells that have antigen binding sites that react with complementary PR3 peptides. Yang at el. reported evidence that patients with PR3-ANCA disease have anti-complementary PR3 antibodies as well as anti-complementary PR3-specific memory T cells directed against PR3 complementary peptide 138–169 [55]. T-cells responsive to complementary PR3-peptide were not detected in MPO-ANCA disease patients.
The HLA DRB1*15 allele was overrepresented in PR3-ANCA disease patients with an immune response against sense/complementary 138–169 [55]. Interestingly, DRB1*1501 binds with high affinity to the amino acid sequences of the sense 138–169 PR3 peptide as well as the amino acid sequence of its complementary peptide [55,56]. An important causal role for immune recognition of this peptide epitope is suggested by the observation that African Americans with PR3-ANCA disease have a 73.3-fold higher odds of having HLA-DRB1*15 alleles than community-based controls [56]. In addition, sense PR3 peptides as well as complementary PR3 peptides bind to TNF-α-induced surface expression of DRB1*1501 on neutrophils [56]. Not all ANCA disease patients have antibodies for this particular sense/complementary PR3 peptide, probably because different ANCA patients have a different repertoire of epitope specificities.
Conceptually, in a patient with a predisposing antigen recognition capability, an infection by a pathogen that has a protein that has peptides that act as mimics of PR3 or MPO antisense peptides would initiate an appropriate antimicrobial immune response that would be transformed into an autoimmune response by an anti-idiotypic response (Fig. 4). This concept is supported by observations in multiple animal models of autoimmune disease in which anti-idiotypic antibodies raised against autoantibodies induce anti-anti-idiotypes that possess characteristics of the initial autoantibodies and caused disease after immunization [57].
An alternative to the theory that complementary proteins or their mimics induce the ANCA autoimmune response is that microbe-derived mimics of an autoantigen itself induce pathogenic autoantibodies. This theory has been proposed in the context of the putative ANCA specificity for lysosomal membrane protein-2 (LAMP2) [9], although the importance of this specificity in patients with ANCA disease is not completely resolved [58]. Anti-LAMP2 antibodies have been detected in patients with MPO-ANCA and PR3-ANCA as well as pauci-immune small vessel vasculitis that is negative for MPO-ANCA and PR3-ANCA [9]. The human LAMP-2 epitope that is recognized has100% homology with the bacterial adhesin FimH, and human anti-LAMP2 antibodies cross-react with bacterial FimH. One research group reported that rats immunized with FimH develop antibodies to rat and human LAMP-2 and develop pauci-immune necrotizing glomerulonephritis [9]. Another research group was not able to reproduce this observation [58]. If the pathogenicity of anti-LAMP2 antibodies is confirmed, this supports the concept that an infection with a fimbriated bacteria could initiate an immune response against FimH that cross reacts with human LAMP2 and mediates ANCA disease.
ANCA Antigen Presentation to B Cells
A genomewide association study has revealed HLA associations for both PR3-ANCA disease and MPO-ANCA disease [12]. The study was performed in a discovery cohort of 1233 United Kingdom patients with ANCA-associated vasculitis and 5884 controls, and was replicated in 1454 Northern European patients and 1666 controls. Both major-histocompatibility-complex (MHC) and non-MHC associations were found in ANCA disease patients. Although associations were identified with the MPA and GPA disease phenotypes, the strongest genetic associations were with the antigenic specificity of ANCA for PR3 versus MPO. Anti-proteinase 3 ANCA was associated with HLA-DP (P=6.2×10−89) and the genes encoding the major inhibitor of PR3 α1-antitrypsin (SERPINA1) (P=5.6×10−12) and proteinase 3 (PRTN3) (P=2.6×10−7). Anti-myeloperoxidase ANCA was associated with HLA-DQ (P=2.1×10−8). These strong associations between PR3-ANCA disease and MPO-ANCA disease and distinct HLA molecules suggest that HLA-determined immune responses against PR3 and MPO is a central pathogenic feature of ANCA disease.
The role of HLA in antigen presentation is supported further by the observation discussed earlier that the HLA DRB1*15 allele is overrepresented in African American PR3-ANCA disease patients with an immune response against sense/complementary 138–169 peptide, and that HLA DRB1*15 binds with high affinity to the amino acid sequences of the sense 138–169 PR3 peptide of its complementary peptide [55,56].
ANCA Antigen Presentation by Neutrophils
As discussed earlier, the antigenic stimulus that sets in motion a sequence of events that leads to ANCA disease may derive from an endogenous or exogenous source. However, there is no doubt that endogenous PR3, MPO and other potential ANCA antigen are endogenous self antigens that are targets for ANCA. Under healthy homeostatic circumstances, most MPO and PR3 is sequestered within the primary granules of neutrophils and the peroxidase-positive lysosomes of monocytes. Low level stimulation (priming) of neutrophils (and monocytes) causes low level release of MPO/PR3 at the cell surface. Full inflammatory activation of neutrophils results in extensive extracellular release of MPO/PR3 from granules (degranulation) as well as extrusion of intracellular contents as so-called neutrophil extracellular traps (NETs) containing chromatin strands decorated with cytoplasmic proteins including MPO and PR3 [59]. NETs are particularly effective at displaying antigens to dendritic cells for internalization and subsequent presentation on HLA molecules to facilitate immune responses, including autoimmune responses [59].
Although less explosive than full neutrophil activation with NETosis, neutrophil apoptosis also can enhance display of ANCA antigens for an immune response, although apoptosis may be more effective at displaying PR3 than MPO [60,61]. Neutrophil apoptosis often is very conspicuous at sites of acute ANCA vasculitis (contributing to the leukocytoclasia) and attracts an influx of monocytes and T cells that could result in modulation of the ANCA autoimmune response.
Expression of the genes for the ANCA antigens MPO and PR3 is increased in the peripheral blood neutrophils of patients with ANCA disease, which could contribute to the initiation or modulation of the ANCA immune response [20,21,62,63]. MPO and PR3 are synthesized and packaged into cytoplasmic granules in neutrophils and monocytes during myelogenesis in the bone marrow. Normally, there is little or no expression of the MPO or PR3 genes in mature peripheral blood leukocytes; however, in patients with active ANCA disease, the expression of both proteins, as well as other neutrophil granule proteins, is increased irrespective of whether they have MPO-ANCA or PR3-ANCA [20,21,62,63]. This abnormal gene expression is mediated by epigenetic events that allow gene expression rather than the usual silencing that is typical in mature circulating neutrophils [21].
NETosis, apoptosis and increased granule protein expression may facilitate the initiation of an ANCA autoimmune response, augment established pathogenic ANCA production, or both.
B Cell Regulation in ANCA Disease
Production of ANCA by B cells is regulated up and down by other cell types, including T cells, B cells and neutrophils. In patients with ANCA disease, the B cell autoimmune response appears to be facilitated by impaired T cell and B cell suppression, and by B cell stimulation by activated neutrophils.
Multiple studies have reported that patients with ANCA disease have a deficiency in and dysfunction of T regulatory cells (Tregs) that could contribute to the loss of tolerance that allows the emergence and persistence of a pathogenic ANCA autoimmune response [64–66]. Major CD4+ T helper (Th) effector cell types are distinguished according to surface markers expressed and the effector cytokines synthesized, for example Th1 characterized by interferon gamma production, Th2 characterized by IL-4 production, Th17 characterized by IL-17 production, and regulatory T cells (Tregs) characterized by high expression of CD25 and FoxP3. Morgan et al observed that patients with active ANCA disease (GPA, Wegener’s) had an increased proportion of CD4+ CD25+ T cells but the percentage of Foxp3-positive cells was decreased [64]. They also noted that that increased circulating CD4+ Foxp3+ cells was associated with more rapid disease remission [64]. The suppressive function of Tregs from ANCA disease patients was markedly decreased in ANCA disease patients with active disease but more normal during remission [65]. Free et al. reported abnormalities in the suppressive Treg network as well as increased frequency of a distinct proinflammatory suppression-resistant effector CD4+ T cell population in patients with active ANCA disease [66]. Although peripheral blood Treg cell frequency was increased in patients with active disease the Treg cells had decreased suppressive function because of frequent expression of a FoxP3 isoform lacking exon 2, which appeared to alter Treg function. In addition, they identified elevated levels of a CD4+ T cell population that was resistant to Treg suppression and produced proinflammatory cytokines. Thus, multiple quantitative and qualitative disturbances in Tregs may contribute to insufficient suppression of ANCA-producing B cells in patients with ANCA disease.
As with T cells, there are B cells with different functional phenotypes. High CD5 surface expression is characteristic of some B cells with regulatory function (Bregs), which also can be characterized by production of IL-10 [67]. In animal models of autoimmunity, Bregs appear to ameliorate immune responses in autoimmunity [68]. In accord with this, Bunch et al. observed that patients with active ANCA disease had lower percentages of peripheral blood CD5+ B cells whereas patients in remission had a percentage of CD5+ B cells no different from healthy controls [69]. Normalization of peripheral blood CD5+ B cells after targeted B cell therapy with rituximab correlated with more effective remission [69].
Normally functioning regulatory T cells and regulatory B cells downregulate B cells that are producing pathogenic ANCA. An opposite upregulating effect may be induced by neutrophils that are activated by ANCA [70–74]. Activated neutrophils release many mediators that modulate innate and adaptive immune responses, including proinflammatory, immunoregulatory, angiogenic and fibrogenic cytokines, chemokines, and ligands of the TNF superfamily including B-cell-activating factor (BAFF)/B lymphocyte stimulator (BLyS) and a proliferation-inducing ligand (APRIL), which are important stimuli for B cells that enhance proliferation and retard apoptosis [70]. Krumbholz reported that patients with ANCA disease (GPA, Wegener’s) had elevated levels of circulating BAFF/BlyS that were diminished by treatment [71]. Sanders et al also observed that plasma levels of BAFF were significantly higher in patients with ANCA disease and were higher at diagnosis than at 12 months after diagnosis [72]. Nagai et al measured serum levels of APRIL and BAFF/BlyS in patients with MPO-ANCA disease [73]. Elevated BAFF/BlyS levels, but not APRIL levels, correlated with active disease and ANCA titer. Bader et al confirmed that BAFF/BlyS levels are increased in patients with ANCA disease but the levels did not correlate with increased ANCA titers [74].
Thus, in patients with ANCA disease, the B cell autoimmune response appears to be facilitated by permissive T cell and B cell regulation, and by B cell stimulating factors released by activated neutrophils.
Conclusion
The evidence in this review supports the primary importance of ANCA-producing B cells and their plasma cell progeny in the pathogenesis of ANCA disease. Priming of neutrophils with cytokines causes the release of ANCA antigen on the cell surface and in the microenvironment. Binding of ANCA to ANCA antigens activates neutrophils by both Fc receptor engagement and direct Fab’2 binding to antigen on the cell surface. ANCA-activated neutrophils release factors that induce alternative complement pathway activation, which establishes a potent inflammatory amplification loop that causes severe necrotizing vascular inflammation. The origin of the ANCA autoimmune response is unknown but appears to involve genetically determined HLA specificities that allow the autoimmune response to develop. One putative immunogenic mechanisms begins with an immune response to a peptide that is complementary to the autoantigen and evolves through an anti-idiotypic network to produce autoantibodies to the autoantigen. Another putative immunogenic mechanism begins with an immune response to a microbe-derived molecular mimic of the autoantigen resulting in antibodies that cross-react with the autoantigen. Release of neutrophil extracellular traps, apoptosis and increased granule protein expression of ANCA antigens may facilitate the initiation of an ANCA autoimmune response, augment established pathogenic ANCA production, or both. The ANCA B cell autoimmune response is facilitated by quantitatively and qualitatively impaired T cell and B cell suppression, and by release from activated neutrophils of B cell activating factors that enhance B cell proliferation and retard B cell apoptosis.
Figure 5.
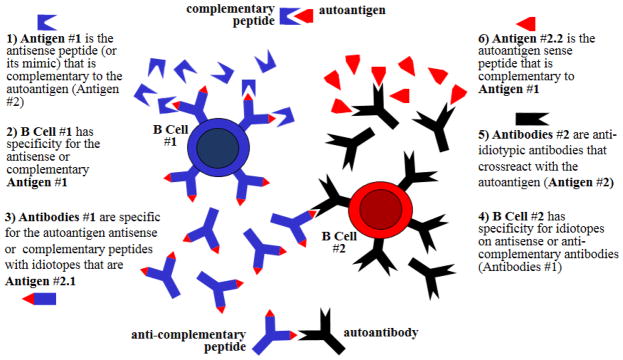
Diagram of the putative induction of an autoimmune response by an immune response to a peptide that is complementary to the autoantigen (e.g. autoantigen antisense peptide or mimic of the antisense peptide). An endogenous complementary peptide (e.g. 3′to 5′ antisense peptide or 5′ to 3′ sense peptide) or an exogenous peptide (e.g. a mimic of the antisense peptide brought in by a microbial organism) could be the target antigen #1 of an appropriate immune response resulting in the generation of B cells #1 that produce antibodies #1. The idiotopes of antibodies #1 are the target antigens #2.1 for antibodies #2 that are produced by B cells #2. Antibodies #2 act as both anti-idiotype antibodies for idiotope antigens #2.1 but also as autoantibodies against the autoantigen #2.2.
Figure 6.

Diagram depicting the genesis, regulation and amplification of B cells that produce pathogenic ANCA. Beginning in the upper left, genetically determined HLA antigen binding sites present antigen that initiate T cell and B cell recognition of antigens that set in motion the evolution of a B cell response that results in B cells and plasma cells that produce pathogenic ANCA. This requires inefficient down regulation by T cells and B cells that normally would suppress a pathogenic autoimmune response. Neutrophil activation by ANCA may augment and sustain this autoimmune response by enhanced display of ANCA autoantigens on neutrophil extracellular traps and on apoptotic neutrophils, overexpression of ANCA antigens in neutrophils, and release of B cell activating factors. This sets up amplification loops (curved arrows) with ANCA production activating neutrophils and activated neutrophils in turn stimulating B cells to make more ANCA.
Acknowledgments
Funded by NIH NIDDK P01-DK058335.
References
- 1.Davies DJ, Moran JE, Niall JF, Ryan GB. Segmental necrotizing glomerulonephritis with antineutrophil antibody: possible arbovirus aetiology? Br Med J. 1982;285:606. doi: 10.1136/bmj.285.6342.606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van der Woude FJ, Rasmussen N, Lobatto S, Wiik A, Permin H, van Es LA, van der Giessen M, van der Hem GK, The TH. Autoantibodies against neutrophils and monocytes: tool for diagnosis and marker of disease activity in Wegener’s granulomatosis. Lancet. 1985;1:425–429. doi: 10.1016/s0140-6736(85)91147-x. [DOI] [PubMed] [Google Scholar]
- 3.Falk RJ, Jennette JC. Anti-neutrophil cytoplasmic autoantibodies with specificity for myeloperoxidase in patients with systemic vasculitis and idiopathic necrotizing and crescentic glomerulonephritis. N Engl J Med. 1988;318:1651–1657. doi: 10.1056/NEJM198806233182504. [DOI] [PubMed] [Google Scholar]
- 4.Jennette JC, Falk RJ, Bacon PA, et al. Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 2013. 2012;65:1–11. doi: 10.1002/art.37715. [DOI] [PubMed] [Google Scholar]
- 5.Goldschmeding R, van der Schoot CE, ten Bokkel Huinink D, Hack CE, van den Ende ME, Kallenberg CGM, von dem Borne AEGK. Wegener’s granulomatosis autoantibodies identify a novel diisopropylfluorophosphate-binding protein in the lysosomes of normal human neutrophils. J Clin Invest. 1988;4:1577–1579. doi: 10.1172/JCI114335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Niles JL, McCluskey T, Ahmad MF, Amin Arnaout MA. Wegener’s granulomatosis autoantigen is a novel neutrophil serine proteinase. Blood. 1989;74:1888–1893. [PubMed] [Google Scholar]
- 7.Jennette JC, Hoidal JH, Falk RJ. Specificity of anti-neutrophil cytoplasmic autoantibodies for proteinase 3. Blood. 1990;75:2263–2264. [PubMed] [Google Scholar]
- 8.Nässberger L, Sjöholm AG, Jonsson H, Sturfelt G, Akesson A. Autoantibodies against neutrophil cytoplasm components in systemic lupus erythematosus and in hydralazine-induced lupus. Clin Exp Immunol. 1990;81:380–3. doi: 10.1111/j.1365-2249.1990.tb05342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kain R, Exner M, Brandes R, et al. Molecular mimicry in pauci-immune focal necrotizing glomerulonephritis. Nat Med. 2008;14:1088–96. doi: 10.1038/nm.1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scott DG, Watts RA. Epidemiology and clinical features of systemic vasculitis. Clin Exp Nephrol. 2013;17:607–10. doi: 10.1007/s10157-013-0830-8. [DOI] [PubMed] [Google Scholar]
- 11.Xu PC, Chen M, Zhao MH. Antineutrophil cytoplasmic autoantibody-associated vasculitis in Chinese patients. Clin Exp Nephrol. 2012;17:705–7. doi: 10.1007/s10157-012-0702-7. [DOI] [PubMed] [Google Scholar]
- 12.Lyons PA, Rayner TF, Trivedi S, et al. Genetically distinct subsets within ANCA-associated vasculitis. N Engl J Med. 2012;367:214–23. doi: 10.1056/NEJMoa1108735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lionaki S, Blyth ER, Hogan SL, Hu Y, Senior JBA, Jennette CE, Nachman PH, Jennette JC, Falk RJ. Classification of antineutrophil cytoplasmic autoantibody vasculitides: The role of antineutrophil cytoplasmic autoantibody specificity for myeloperoxidase or proteinase 3 in disease recognition and prognosis. Arthritis Rheum. 2012;64:3452–62. doi: 10.1002/art.34562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roth AJ, Ooi J, Hess JJ, et al. ANCA Epitope Specificity Determines Pathogenicity, Detectability and Clinical Predictive Value. J Clin Invest. 2013;123:1773–83. doi: 10.1172/JCI65292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peschel A, Basu N, Benharkou A, Brandes R, Brown M, Dieckmann R, Rees AJ, Kain R. Autoantibodies to hLAMP-2 in ANCA-Negative Pauci-Immune Focal Necrotizing GN. J Am Soc Nephrol. 2013 doi: 10.1681/ASN.2013030320. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sinico RA, Di Toma L, Maggiore U, et al. Prevalence and clinical significance of antineutrophil cytoplasmic antibodies in Churg-Strauss syndrome. Arthritis Rheum. 2005;52:2926–35. doi: 10.1002/art.21250. [DOI] [PubMed] [Google Scholar]
- 18.Pendergraft WF, Niles JL. Trojan horses: drug culprits associated with antineutrophil cytoplasmic autoantibody (ANCA) vasculitis. Curr Opin Rheumatol. 2014;26:42–9. doi: 10.1097/BOR.0000000000000014. [DOI] [PubMed] [Google Scholar]
- 19.Thai LH, Charles P, Resche-Rigon M, Desseaux K, Guillevin L. Are anti-proteinase-3 ANCA a useful marker of granulomatosis with polyangiitis (Wegener’s) relapses? Results of a retrospective study on 126 patients. Autoimmun Rev. 2013;S1568–9972(13):00212–7. doi: 10.1016/j.autrev.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 20.Yang JJ, Pendergraft WF, Alcorta DA, et al. Circumvention of normal constraints on granule protein gene expression in peripheral blood neutrophils and monocytes of patients with antineutrophil cytoplasmic autoantibody-associated glomerulonephritis. J Am Soc Nephrol. 2004;15:2103–2114. doi: 10.1097/01.ASN.0000135058.46193.72. [DOI] [PubMed] [Google Scholar]
- 21.Ciavatta DJ, Yang J, Preston GA, et al. Epigenetic basis for aberrant upregulation of autoantigen genes in humans with ANCA vasculitis. J Clin Investig. 2010;120:3209–3219. doi: 10.1172/JCI40034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Geetha D, Seo P. Advances in therapy for ANCA-associated vasculitis. Curr Rheumatol Rep. 2012;14:509–15. doi: 10.1007/s11926-012-0284-0. [DOI] [PubMed] [Google Scholar]
- 23.Falk RJ, Terrell RS, Charles LA, Jennette JC. Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proc Natl Acad Sci USA. 1990;87:4115–4119. doi: 10.1073/pnas.87.11.4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Charles LA, Caldas ML, Falk RJ, Terrell RS, Jennette JC. Antibodies against granule proteins activate neutrophils in vitro. J Leuk Biol. 1991;50:539–546. doi: 10.1002/jlb.50.6.539. [DOI] [PubMed] [Google Scholar]
- 25.Csernok E, Ernst M, Schmitt W, Bainton DF, Gross WL. Activated neutrophils express proteinase 3 on their plasma membrane in vitro and in vivo. Clin Exp Immunol. 1994;95:244–50. doi: 10.1111/j.1365-2249.1994.tb06518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Porges AJ, Redecha PB, Kimberly WT, et al. Anti-neutrophil cytoplasmic antibodies engage and activate human neutrophils via Fc gamma RIIa. J Immunol. 1994;153:1271–1280. [PubMed] [Google Scholar]
- 27.Kocher M, Siegel ME, Edberg JC, Kimberly RP. Cross-linking of Fc gamma receptor IIa and Fc gamma receptor IIIb induces different proadhesive phenotypes on human neutrophils. J Immunol. 1997;159:3940–8. [PubMed] [Google Scholar]
- 28.Kettritz R, Jennette JC, Falk RJ. Cross-linking of ANCA-antigens stimulates superoxide release by human neutrophils. J Am Soc Nephrol. 1997;8:386–394. doi: 10.1681/ASN.V83386. [DOI] [PubMed] [Google Scholar]
- 29.Williams JM, Ben Smith A, Hewins P, et al. Activation of the G(i) heterotrimeric G protein by ANCA IgG F(ab’)2 fragments is necessary but not sufficient to stimulate the recruitment of those downstream mediators used by intact ANCA IgG. J Am Soc Nephrol. 2003;14:661–669. doi: 10.1097/01.asn.0000050223.34749.f4. [DOI] [PubMed] [Google Scholar]
- 30.Savage CO, Gaskin G, Pusey CD, Pearson JD. Myeloperoxidase binds to vascular endothelial cells, is recognized by ANCA and can enhance complement dependent cytotoxicity. Adv Exp Med Biol. 1993;336:121–123. doi: 10.1007/978-1-4757-9182-2_20. [DOI] [PubMed] [Google Scholar]
- 31.Ewert BH, Jennette JC, Falk RJ. Anti-myeloperoxidase antibodies stimulate neutrophils to damage human endothelial cells. Kidney Int. 1992;41:375–383. doi: 10.1038/ki.1992.52. [DOI] [PubMed] [Google Scholar]
- 32.Lu X, Garfield A, Rainger GE, Savage CO, Nash GB. Mediation of endothelial cell damage by serine proteases, but not superoxide released from antineutrophil cytoplasmic antibody-stimulated neutrophils. Arthritis Rheum. 2006;54:1619–28. doi: 10.1002/art.21773. [DOI] [PubMed] [Google Scholar]
- 33.Kessenbrock K, Krumbholz M, Schönermarck U, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med. 2009;15:623–5. doi: 10.1038/nm.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gupta AK, Joshi MB, Philippova M, et al. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Let. 2010;584:3193–7. doi: 10.1016/j.febslet.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 35.Jennette JC, Xiao H, Falk R, Gasim AM. Experimental models of vasculitis and glomerulonephritis induced by antineutrophil cytoplasmic autoantibodies. Contrib Nephrol. 2011;169:211–20. doi: 10.1159/000314776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Korkmaz B, Jenne DE, Gauthier F. Relevance of the mouse model as a therapeutic approach for neutrophil proteinase 3-associated human diseases. Int Immunopharmacol. 2013;17:1198–205. doi: 10.1016/j.intimp.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 37.Xiao H, Heeringa P, Hu P, Liu Z, Zhao M, Aratani Y, Maeda N, Falk RJ, Jennette JC. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J Clin Invest. 2002;110:955–963. doi: 10.1172/JCI15918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xiao H, Heeringa P, Liu Z, Huugen D, Hu P, Falk RJ, Jennette JC. The role of neutrophils in the induction of glomerulonephritis by anti-myeloperoxidase antibodies. Am J Pathol. 2005;167:39–45. doi: 10.1016/S0002-9440(10)62951-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schreiber A, Xiao H, Falk RJ, Jennette JC. Bone marrow-derived cells are sufficient and necessary targets to mediate glomerulonephritis and vasculitis induced by anti-myeloperoxidase antibodies. J Am Soc Nephrol. 2006;17:3355–64. doi: 10.1681/ASN.2006070718. [DOI] [PubMed] [Google Scholar]
- 40.Huugen D, Xiao H, an Esch A, Falk RJ, Peutz-Kootstra CJ, Buurman WA, Cohen Tervaert JW, Jennette JC, Heeringa P. Aggravation of anti-myeloperoxidase antibody induced glomerulonephritis by bacterial lipopolysaccharide: role of tumor necrosis factor α. Am J Pathol. 2005;167:47–58. doi: 10.1016/s0002-9440(10)62952-5. [DOI] [PMC free article] [PubMed] [Google Scholar]; 25(1 Suppl 44):52–6. [Google Scholar]
- 41.van Timmeren MM, van der Veen BS, Stegeman CA, Petersen AH, Hellmark T, Collin M, Heeringa P. IgG glycan hydrolysis attenuates ANCA-mediated glomerulonephritis. J Am Soc Nephrol. 2010;21:1103–14. doi: 10.1681/ASN.2009090984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xing GQ, Chen M, Liu G, Heeringa P, Zhang JJ, Zheng XEJ, Kallenberg CG, Zhao MH. Complement activation is involved in renal damage in human antineutrophil cytoplasmic autoantibody associated pauci-immune vasculitis. J Clin Immunol. 2009;29:282–91. doi: 10.1007/s10875-008-9268-2. [DOI] [PubMed] [Google Scholar]
- 43.Gou SJ, Yuan J, Chen M, Yu F, Zhao MH. Circulating complement activation in patients with anti-neutrophil cytoplasmic antibody-associated vasculitis. Kidney Int. 2012;83:129–37. doi: 10.1038/ki.2012.313. [DOI] [PubMed] [Google Scholar]
- 44.Xiao H, Schreiber A, Heeringa P, Falk RJ, Jennette JC. Alternative complement pathway in the pathogenesis of disease mediated by antineutrophil cytoplasmic autoantibodies. Am J Pathol. 2007;170:52–64. doi: 10.2353/ajpath.2007.060573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huugen D, van Esch A, Xiao H, Peutz-Kootstra CJ, Buurman WA, Cohen Tervaert JW, Jennette JC, Heeringa P. Inhibition of complement factor C5 protects against anti-myeloperoxidase antibody-mediated glomerulonephritis in mice. Kidney Int. 2007;71:646–654. doi: 10.1038/sj.ki.5002103. [DOI] [PubMed] [Google Scholar]
- 46.Schreiber A, Xiao H, Jennette JC, Schneider W, Luft FC, Kettritz R. C5a receptor mediates neutrophil activation and ANCA-induced glomerulonephritis. J Am Soc Nephrol. 2009;20:289–98. doi: 10.1681/ASN.2008050497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xiao H, Dairaghi DJ, Powers JP, Ertl LS, Baumgart T, Wang Y, Seitz LC, Penfold MET, Gao L, Hu1 P, Lu B, Gerard NP, Gerard C, Schall TJ, Jaen JC, Falk RJ, Jennette JC. C5a receptor (CD88) blockade protects against MPO-ANCA glomerulonephritis. J Am Soc Nephrol. 2013 doi: 10.1681/ASN.2013020143. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hao J, Meng LQ, Xu PC, Chen M, Zhao MH. p38MAPK, ERK and PI3K signaling pathways are involved in C5a-primed neutrophils for ANCA-mediated activation. PLoS One. 2012;7:e38317. doi: 10.1371/journal.pone.0038317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hao J, Chen M, Zhao MH. Involvement of protein kinase C in C5a-primed neutrophils for ANCA-mediated activation. Mol Immunol. 2013;54:68–73. doi: 10.1016/j.molimm.2012.10.041. [DOI] [PubMed] [Google Scholar]
- 50.Pendergraft WF, Preston GA, Shah RR, Tropsha A, Jennette JC, Falk RJ. cPR3105-206, a protein complementary to the autoantigen proteinase 3, triggers autoimmunity. Nature Med. 2004;10:72–79. doi: 10.1038/nm968. [DOI] [PubMed] [Google Scholar]
- 51.Preston GA, Pendergraft WF, 3rd, Falk RJ. New insights that link microbes with the generation of antineutrophil cytoplasmic autoantibodies: the theory of autoantigen complementarity. Curr Opin Nephrol Hypertens. 2005;14:217–22. doi: 10.1097/01.mnh.0000165886.93427.b1. [DOI] [PubMed] [Google Scholar]
- 52.Heal JR, Roberts GW, Raynes JG, Bhakoo A, Miller AD. Specific interactions between sense and complementary peptides: the basis for the proteomic code. Chembiochem. 2002;3:136–51. doi: 10.1002/1439-7633(20020301)3:2/3<136::AID-CBIC136>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 53.Mekler LB. On the specific mutual interaction of amino acid residues of polypeptide chains and amino acid residues with codons. Oncology. 1973;27:286–288. doi: 10.1159/000224740. [DOI] [PubMed] [Google Scholar]
- 54.Laudien M, Gadola SD, Podschun R, et al. Nasal carriage of Staphylococcus aureus and endonasal activity in Wegener s granulomatosis as compared to rheumatoid arthritis and chronic Rhinosinusitis with nasal polyps. Clin Exp Rheumatol. 2010;28(1 Suppl 57):51–5. [PubMed] [Google Scholar]
- 55.Yang JJ, Bautz DJ, Lionaki S, et al. ANCA patients have T cells responsive to complementary PR-3 autoantigen. Kidney Intern. 2008;74:1159–69. doi: 10.1038/ki.2008.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang JJ, Cao Y, Schmitz JL, et al. DRB1*15 allele is a risk factor for PR3-ANCA disease in African Americans. Clin Exp Immunol. 2011;164(Suppl 1):131. doi: 10.1681/ASN.2010101058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shoenfeld Y. Idiotypic induction of autoimmunity: a new aspect of the idiotypic network. FASEB J. 1994;8:1296–301. doi: 10.1096/fasebj.8.15.8001742. [DOI] [PubMed] [Google Scholar]
- 58.Roth AJ, Brown MC, Smith RN, et al. Anti-LAMP-2 antibodies are not prevalent in patients with antineutrophil cytoplasmic autoantibody glomerulonephritis. J Am Soc Nephrol. 2012;23:545–55. doi: 10.1681/ASN.2011030273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sangaletti S, Tripodo C, Chiodoni C, et al. Neutrophil extracellular traps mediate transfer of cytoplasmic neutrophil antigens to myeloid dendritic cells toward ANCA induction and associated autoimmunity. Blood. 2012;120:3007–18. doi: 10.1182/blood-2012-03-416156. [DOI] [PubMed] [Google Scholar]
- 60.Durant S, Pederzoli M, Lepelletier Y, et al. Apoptosis-induced proteinase 3 membrane expression is independent from degranulation. J Leukoc Biol. 2004;75:87–98. doi: 10.1189/jlb.0203079. [DOI] [PubMed] [Google Scholar]
- 61.Kantari C, Pederzoli-Ribeil M, Amir-Moazami O, et al. Proteinase 3, the Wegener autoantigen, is externalized during neutrophil apoptosis: evidence for a functional association with phospholipid scramblase 1 and interference with macrophage phagocytosis. Blood. 2007;110:4086–95. doi: 10.1182/blood-2007-03-080457. [DOI] [PubMed] [Google Scholar]
- 62.Ohlsson S, Hellmark T, Pieters K, et al. Increasedmonocyte transcription of the proteinase 3 gene in small vessel vasculitis. Clin Exp Immunol. 2005;141:174–82. doi: 10.1111/j.1365-2249.2005.02819.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Alcorta DA, Barnes DA, Dooley MA, et al. Leukocyte gene expression signatures in antineutrophil cytoplasmic autoantibody and lupus glomerulonephritis. Kidney Int. 2007;72:853–64. doi: 10.1038/sj.ki.5002371. [DOI] [PubMed] [Google Scholar]
- 64.Morgan MD, Day CJ, Piper KP, et al. Patients with Wegener’s granulomatosis demonstrate a relative deficiency and functional impairment of T-regulatory cells. Immunology. 2010;130:64–73. doi: 10.1111/j.1365-2567.2009.03213.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rimbert M, Hamidou M, Braudeau C, et al. Decreased numbers of blood dendritic cells and defective function of regulatory T cells in antineutrophil cytoplasmic antibody-associated vasculitis. PLoS One. 2011;6:e18734. doi: 10.1371/journal.pone.0018734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Free ME, Bunch DO, McGregor JA, et al. Patients with antineutrophil cytoplasmic antibody-associated vasculitis have defective Treg cell function exacerbated by the presence of a suppression-resistant effector cell population. Arthritis Rheum. 2013;65:1922–33. doi: 10.1002/art.37959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Soldevila G, Raman C, Lozano F. The immunomodulatory properties of the CD5 lymphocyte receptor in health and disease. Curr Opin Immunol. 2011;23:310–318. doi: 10.1016/j.coi.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mauri C. Regulation of immunity and autoimmunity by B cells. Curr Opin Immunol. 2010;22:761–767. doi: 10.1016/j.coi.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 69.Bunch DO, McGregor JG, Khandoobhai NB, et al. Decreased CD5+ B cells in active ANCA vasculitis and relapse after rituximab. Clin J Am Soc Nephrol. 2013;8:382–91. doi: 10.2215/CJN.03950412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Scapini P, Bazzoni F, Cassatella MA. Regulation of B-cell-activating factor (BAFF)/B lymphocyte stimulator (BLyS) expression in human neutrophils. Immunol Lett. 2008;15;116:1–6. doi: 10.1016/j.imlet.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 71.Krumbholz M, Specks U, Wick M, et al. BAFF is elevated in serum of patients with Wegener’s granulomatosis. J Autoimmun. 2005;25:298–302. doi: 10.1016/j.jaut.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 72.Sanders JS, Huitma MG, Kallenberg CG, Stegeman CA. Plasma levels of soluble interleukin 2 receptor, soluble CD30, interleukin 10 and B cell activator of the tumour necrosis factor family during follow-up in vasculitis associated with proteinase 3-antineutrophil cytoplasmic antibodies: associations with disease activity and relapse. Ann Rheum Dis. 2006;65:1484–1489. doi: 10.1136/ard.2005.046219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nagai M, Hirayama K, Ebihara I, Shimohata H, Kobayashi M, Koyama A. Serum levels of BAFF and APRIL in myeloperoxidase anti-neutrophil cytoplasmic autoantibody-associated renal vasculitis: association with disease activity. Nephron Clin Pract. 2011;118:c339–45. doi: 10.1159/000323393. [DOI] [PubMed] [Google Scholar]
- 74.Bader L, Koldingsnes W, Nossent J. B-lymphocyte activating factor levels are increased in patients with Wegener’s granulomatosis and inversely correlated with ANCA titer. Clin Rheumatol. 2010;29:1031–1035. doi: 10.1007/s10067-010-1526-z. [DOI] [PubMed] [Google Scholar]