

Abstract
The mechanism by which Aβ causes neuronal dysfunction/death in Alzheimer’s disease is unclear. Previously, we showed that Aβ inhibits several microtubule-dependent kinesin motors essential for mitosis and also present in mature neurons. Here we show that inhibition of kinesin 5 (Eg5) by Aβ blocks neuronal function by reducing transport of neurotrophin and neurotransmitter receptors to the cell surface. Specifically, cell-surface NGF/NTR(p75) and NMDA receptors decline in cells treated with Aβ or the Kin5 inhibitor monastrol, or expressing APP. Aβ and monastrol also inhibit NGF-dependent neurite outgrowth from PC12 cells and glutamate-dependent Ca++ entry into primary neurons. Like Aβ, monastrol inhibits long-term potentiation, a cellular model of NMDA-dependent learning and memory, and Kin5 activity is absent from APP/PS transgenic mice brain or neurons treated with Aβ. These data imply that cognitive deficits in AD may derive in part from inhibition of neuronal Eg5 by Aβ, resulting in impaired neuronal function/survival through receptor mis-localization. Preventing inhibition of Eg5 or other motors by Aβ may represent a novel approach to Alzheimer’s disease therapy.
1. Introduction
Genetic and biochemical studies have identified the Aβ peptide as playing a key role in the pathogenesis of Alzheimer’s disease, but the mechanism by which Aβ and other AD-related proteins, such as tau and apoE, cause neuronal degeneration is still being elucidated (Lee, 1996; Mandelkow and Mandelkow, 1998; Lee and Trojanowski, 2006; Hardy, 2009; Potter and Wisniewski, 2012). For example, neuronal function depends critically on the correct localization and function of neurotransmitter and neurotrophin receptors, which are disrupted in AD, but the mechanism of this disruption is unknown (Tong et al., 2004; Almeida et al., 2005; Snyder et al., 2005; Abisambra et al., 2010; Liu et al., 2010). Previous findings suggested that receptor dysfunction may be linked to microtubule defects. For example, APP over-expression or Aβ treatment disrupts the function and structure of the cellular MT network, requires Tau for its pathogenic effects (Geller and Potter, 1999; Pigino et al., 2001; Rapoport et al., 2002; Tezapsidis et al., 2003; Hamano et al., 2005; Roberson et al., 2007; Liu et al., 2008; Boeras et al., 2008; Liu et al., 2009; Shah et al., 2009; Abisambra et al., 2010; Granic et al., 2010 Borysov et al., 2011), and causes mis-localization of Low Density in Lipoprotein Receptor (LDLR) in cultured neurons (Abisambra et al., 2010). Furthermore, Aβ directly binds to and inhibits certain microtubule-dependent kinesin motors, including Eg5/kinesin5/kif11 (Borysov et al., 2011), which are necessary for mitotic spindle structure and function (Hsu et al., 1985; Mailes et al., 2004; Mazumdar et al., 2004; Heard and Walsczak 1999; Walczak and Heard, 2008). For example, studies of Michaelis-Menten kinetics revealed that Aβ competitively inhibits Eg5/kinesin 5, but has no effect on the classic KH1 kinesin motor or on CENP-E (Borysov et al., 2011). Furthermore, Aβ inhibits the binding of Eg5 to microtubules (Borysov et al., 2011). The fact that the several Aβ-inhibited motors Eg5/kinesin5, Kif11 and MCAK are also present and functional in mature neurons (Tekemura et al. 1996; Baas, 1998) and that Aβ expressed in transgenic mice carrying human AD-causing mutant APP reduces the activity of kinesin 5/Eg5 in mouse brain to undetectable levels (Borysov et al., 2011) suggested to us that MT motor inhibition by Aβ might cause much of the neuronal dysfunction of AD by disrupting microtubule-dependent movement of key cellular constituents. To test this hypothesis, we asked whether Aβ inhibition of kinesin 5/Eg5 disrupts the localization of neurotrophin and neurotransmitter receptors to the cell surface, leading to impaired neuronal function. Specifically, cell surface levels of NGF/NTR(p75) and NMDA receptors were found to be greatly reduced in cells treated with Aβ or expressing APP, or treated with monastol, a Eg5/kinesin 5 inhibitor (Kapoor et al., 2000). Both Aβ and monastrol consequently inhibit NGF-dependent neurite outgrowth from PC12 cells and reduce glutamate-dependent Ca++ entry into primary neurons. Furthermore, Eg5/kinesin 5 activity is absent from primary neurons treated with Aβ, as it is in APP/PS transgenic mice brain, as mentioned above (Borysov et al., 2011). Finally, like Aβ, monastrol inhibits long-term potentiation, a cellular model of NMDA-dependent learning and memory. These data imply that cognitive deficits in Alzheimer’s disease may derive in part from inhibition of neuronal Eg5/kinesin 5 by Aβ, resulting in impairment of neuronal function through neurotransmitter and neurotrophin receptor mis-localization.
2. Methods
2.1. Antibodies
The following primary antibodies were used: anti-NMDA NR1 (extracellular) antibody (Alomone labs, 3μg/ml); anti-NMDAR2B (Millipore, 2μg/ml); anti-extracellular p75 (gift from Dr. Moses Chao; Huber and Chao, 1995); anti-alpha tubulin, wheat germ agglutinin conjugates (WGA, Invitrogen). Goat anti-rabbit AlexaFluor 488, 594, and goat anti-mouse AlexaFluor 488 (Invitrogen, Molecular Probes) antibodies were diluted according to the manufacturer for immunohistochemistry. WGA was conjugated with AlexaFluor 633. Anti-human Eg5 was from Abcam (ab37009). CellMask plasma membrane stain (Invitrogen) was also used.
2.2. Animals
Non-transgenic (NTG) mice were used for preparing primary neuron cultures. Day 18 pregnant mice were anesthesized with 0.1 mg/g Nembutal, and the whole brains of E18 embryos were immediately removed for processing. Primary neurons were obtained from the cortex and hippocampus. Brain slices for electrophysiology were obtained from 18-day NTG mice as described above. All animal studies were approved by the University of South Florida’s Institutional Animal Care and Use Committee and abided by that Committee’s Policies on Animal Care and Use in accordance with the Guide for the Care and Use of Laboratory Animals, the Animal Welfare Regulations Title 9 Code of Federal Regulations Subchapter A, “Animal Welfare”, Parts 1–3, and the Public Health Service Policy on Humane Care and Use of Laboratory Animals.
2.3. Cell culture
H4 cells (ATCC) were cultured in Optimem, 10% FBS, 1% Penicillin/Streptomycin, H4APP cells (from Todd Golde) were supplemented with additional 0.1% Hygromycin. PC12 cells were plated at low density on collagen coated plastic in RPMI plus 1% horse serum and NGF (50ng/ml). Neurites were noticeable at 1–3 days. Medium was changed 3x/wk. Neurons were prepared as described in Padmanabhan et al (2006) with modifications., Timed pregnant C57/black mice were obtained from Harlan, the animals were anaesthetized with pentobarbital, the embryos were dissected out, and their cortices and hippocampi triturated in. The dissociated cortex was centrifuged and the cells resuspended in neurobasal primary media (Invitrogen) with B27 supplement, Penicillin/streptomycin, and Lglutamine (all Life technologies) and plated onto poly-L-lysine-coated (Sigma) culture plates. Unattached cells were removed after 4 hours. The cells were replenished with ½ fresh media every fourth day.
2.4. Aβ internalization
FITC-Aβ 1–42 (Anaspec; HiLyte Flour 488-labeled; 1 μM) was added to 1-week old neuronal cultures and the cells incubated overnight. Medium containing FITC-Aβ was removed, the cells washed with PBS, and fresh medium added before image acquisition. Cells were primarily neurons by morphology.
2.5. Immunolabelling of cell surface receptors
The immmunolabelling experiments were performed on 3-week-old cultures in poly-L-lysine coated 6-well-plates and 8-chamber slides. After treatment with either monastrol (50–100μM; Sigma), Aβsc or Aβ42 (1μM) for 24hrs (or 48hrs for PC12 cells) the cells were washed twice with PBS. Cells were fixed in 4% paraformaldehyde for 5 min at room temperature. After two rinses in PBS the non-specific binding was blocked with 10% normal goat serum, 0.2% Triton X-100, 0.02% NaN3 in Tris-buffered saline (or in blocking buffer without detergent for non-permeabilized cell experiments) for 45 min at room temperature, then incubated for 2 hrs at room temperature with primary antibody diluted in blocking buffer. After three, 5min washes in PBS, slides were incubated with secondary antibody diluted (1:1000) in blocking buffer for 45 min at room temperature in the dark and washed two times in PBS. Slides were stained with Hoechst (1mg/ml in PBS) for 3 min to reveal cellular nuclei, washed for 3 minutes three times in PBS, and mounted using GelMount (Fisher Scientific).
2.6. Eg-5 immunoprecipitation
Endogenous mouse Eg5/kinesin5 protein was immunoprecipitated with anti-human Eg5 (Abcam, ab37009, 1:5 dilution), and subjected to enzymatic kinetics assay based on Kinesin ELIPA Biochem kit (Cytoskeleton Inc.) as described (Borysov et al., 2011).
2.7 Live/dead assay
Primary neurons were grown for three weeks before treated with either monastrol (100μM; Sigma), Aβsc or Aβ42 (1μM) for 48hrs. Live dead assay (Molecular Probes) was used according to the manufacturer’s instructions and the reaction was measured by a Bio-Tek Synergy 2 plate reader. Data reported were obtained from six independent experiments. P-values were obtained from paired t-test analyses.
2.8. Signal quantification and statistical analyses
Immunofluorescence of samples was examined with Olympus FV1000 MPE laser scanning confocal microscope. The intensity and colocalization of fluorochrome signals was analyzed by measuring the fluorescence intensities of pixels using Olympus FV10-ASW 2.1 program. All data reported were obtained from at least three measurements on three independent experiments. Data were analyzed by One Way ANOVA with Tukey’s Multiple Comparison Test post hoc and results were considered significant when p<0.05. The figures were modified in Adobe Photoshop by adjusting the brightness and contrast to images equally and simultaneously. Three dimensional reconstructions of confocal microscope images were done by Slidebook software.
2.9. Ca++ Imaging
Primary neurons were treated for 24 hours with 1μM Aβ 42, 100 μM monastol or nothing (control). Fura-2 (Biotium) was added at 4μM for 40 min. Glutamate (1mM) was added at 5 min. DL-APV (1mM) (Tocris Bioscience) was added at 10 min to selectively block NMDA channels. 20 cell images and backgrounds were identified in each treatment groups on NIS Elements software. Calcium was measured repetitively for 2 seconds duration at 30 second intervals for 15 minutes on a Nikon TE 2000E microscope and were analyzed in Microsoft Excel.
2.10. Long Term Potentiation
An LTP protocol that has been shown previously to be sensitive to Aβ application (Townsend et al., 2006; Smith et al., 2009) was used to determine whether chemical inhibition of Eg5 by monastrol inhibits LTP. Hippocampal slices (400 um thickness) were cut with a vibratome from coronal sections of 18 days old mouse pups and given 2 hours of recovery in an recording chamber with interface-perfusion of artificial cerebrospinal fluid (ACSF) and maintained at 32° C. Field recordings of excitatory post-synaptic potentials (fEPSPs) were evoked through stimulation of the Schaffer collaterals (bipolar, 0.1 msec duration and at a strength that evoked 50% maximum response) and recording in area CA1 of the hippocampus. Baseline data were recorded before and then after 30 minutes of perfusion with or without 100μM monastrol. Potentiation was induced using a standard 2 train, 1 sec. 100 Hz electrical stimulation, known to be generated in an NMDA receptor-dependent mechanism. Data were recorded repetitively at 20-second intervals for 60 minutes after the induction of LTP and analyzed in pClamp 10 and Microsoft Excel.
3. Results
3.1. Aβ or monastrol inhibition of Eg5/kinesin 5 causes reduced cell surface localization of the p75 neurotrophin receptor
To test whether inhibition of Eg5 induces mis-localization of neurotrophin receptors, we used quantititative confocal microsopy to investigate the localization of the p75 receptor in H4 and PC12 cells treated with Aβ, expressing APP, or treated with the specific kinesin 5 inhibitor, monastrol.
In non-permeabilized H4 cells treated with monastrol and in H4APP cells overexpressing Aβ, fewer p75 receptors (green) were observed on the cell surface (Fig.1A,B), whereas, the total number of receptors assessed in permeabilized cells using the same assay for best comparison, did not change compared to control (Fig.1C), as we had previously found for the LDL receptor in these cells (Abisambra et al., 2010). The relative fluorescence intensity of cell surface wheat germ agglutinin (WGA) also did not change significantly between the treatment groups (not shown). The relative fluorescence intensity of surface p75 receptors was also lower in Aβ40, Aβ42 or monastrol treated PC12 cells (Fig.1D), which have been shown previously to bind and take up Aβ peptide (Puduslo et al., 2010). The involvement of kinesin 5 in the reduced cell surface p75 localization caused by Aβ or monastrol is indicated by the finding that the cell surface localization could be partly restored in PC12 cells by transfection of a kinesin 5-expressing plasmid (Fig. 1E).
Figure 1.
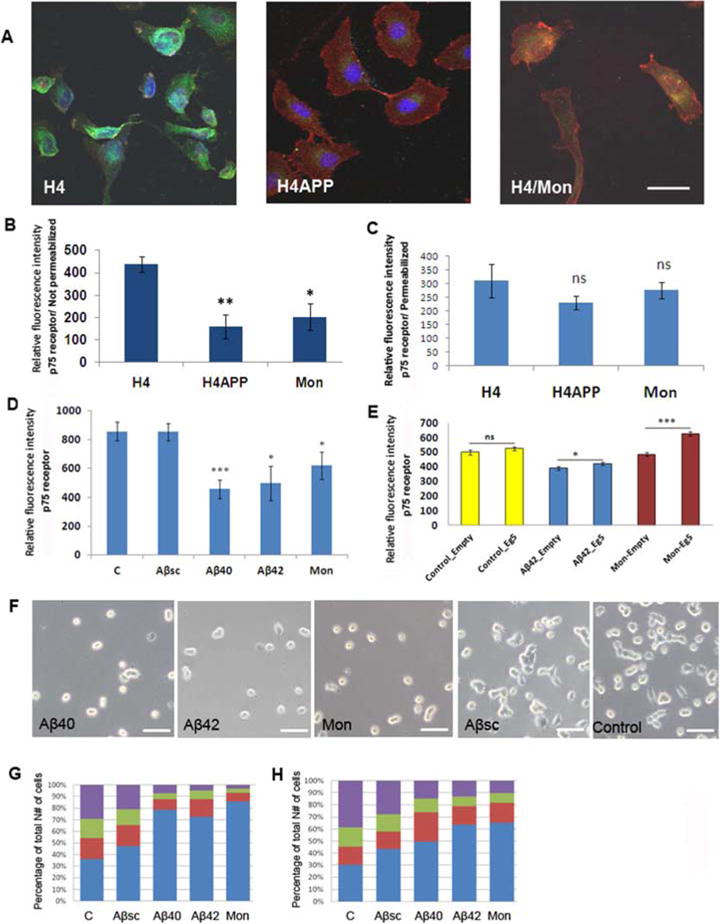
Aβ or monastrol reduces cell surface localization and function of neurotrophin p75 receptors. Confocal immune-microscopy revealed fewer p75 receptors (green) on the cell surface of non-permeabilized H4 cells treated with the Eg5/kinesin 5 inhibitor monastrol for 24 hrs and of non-permebilized H4APP cells overexpressing Aβ (A,B), whereas the total receptors staining did not change compared to control when the cells were permeabilized (C). WGA is shown by red. Treatment of PC12 cells with either Aβ1–40, Aβ 1–42, or monastrol also resulted in reduced cell surface expression of p75 receptors (D), which could be partly restored by over-expression of Eg5 from a transfected plasmid (E). PC12 cells treated with Aβ40, Aβ42 or monastrol grew none or a few, very short processes in response to NGF, as opposed to cells exposed to Aβ-scrambled peptide or to untreated control cells, which grew evident processes (F). Quantitation of process formation under different conditions is shown at 24 hr (G) and 48 hr (H). Blue: round cells with zero processes; Red: round cell with one or two processes shorter than cell body; Green: round or elongated cell with one or more processes longer than the cell body; Purple: star shaped, flattened cell with more processes.
3.2. Aβ or monastrol treatment causes reduced sensitivity to Nerve Growth Factor
The reduced cell surface expression of the p75 receptor induced in PC 12 cells by Aβ or monastrol suggests that p75 receptor function is also likely to be inhibited. Indeed, PC12 cells treated with Aβ40, Aβ42 or monastrol grew none or a few, very short processes in response to NGF, compared to control cells or cells treated with a scrambled Aβ peptide, which responded normally to NGF (Fig.1F). A graphical representation of the data shows the relative levels of differentiated cells after 24 hours (Fig.1G) and 48 hours (Fig.1H). More undifferentiated, round PC12 cells were found in Aβ and monastrol treated groups, while differentiated cells with star shaped, flattened cell bodies and multiple processes were more common in Aβsc-treated or control groups after NGF treatment.
3.3. Aβ or monastrol treatment causes reduced cell surface localization of NMDA receptors in H4 cells and disrupts the microtubule network in neurons
To function effectively in the brain, neurons must be able to respond to neurotransmitters as well as to neurotrophins. The NMDA receptor has been shown to be particularly important for learning and memory, and its expression and/or function is reduced in Alzheimer’s disease and transgenic mouse models thereof (Snyder et al., 2005; Wang et al., 2009; Gladding and Raymond, 2011). H4APP- or monastrol-treated H4 cells, expressed a reduced number of NMDA/NR1 (green) receptor subunits on the cell surface (Fig.2A, B). The surface expression level of WGA did not differ between the treatment groups (not shown). In permeabilized cells, there was no significant difference in the relative fluorescence intensity of NMDA/NR1 between the treatment groups, indicating that the reduced cell surface NMDA/NR1 protein was due to poor transport and not reduced expression (Fig.2C). Total NMDAR2B receptor subunits detected with an antibody to a cytosol-facing epitope were also reduced in H4APP and in H4 cells treated with monastrol (Fig.2D,E).
Figure 2.

Inhibition of Eg 5/kinesin5 by Aβ or monastrol treatment causes reduced cell surface localization of NMDA receptors in H4 cells and disrupts the microtubule network in neurons. Confocal immuno-microscopy showed reduced numbers of cell surface NMDA/NR1 (green) in H4APP cells or monastrol treated H4 cells (A, B). WGA is represented by red. When the cells were permeabilized, there was no significant difference in the relative fluorescence intensity between the treatment groups (C). Similarly, an antibody to a cytosol-facing epitope of the NMDAR2B subunit showed a significantly-reduced level of receptors in permeabilized H4APP and in H4 cells treated with monastrol (D,E).
To investigate the above finding in primary neurons, we first confirmed that kinesin5/Eg5 is expressed in neurons, as previously reported (Takemura et al., 1996; Baas 1998). Immunocytochemistry revealed Eg5/kinesin 5 localized in cell bodies, processes and growth cones of primary mouse E18 cortical neurons (Fig. 3A). We also confirmed that primary neurons take up FITC-labeled Aβ42 (Fig 3B) as has been previously demonstrated (Saavedra et al., 2007; Hu et al., 2009). Furthermore, kinesin 5 activity can be recovered by immunoprecipitation from the primary neuronal cultures, the first demonstration of kinesin 5 biochemical activity in mature neurons (Fig. 3C). More importantly, when the neurons were incubated with Aβ, neuronal kinesin 5 was inhibited in a dose-dependent manner (Fig. 3C), yet the treatment is not significantly toxic to neurons in such a short time. This result indicates that Aβ is taken up into primary neurons, where it must be at least partly enter the cytoplasm where it can bind to and inhibit the Eg5/kinesin 5 enzyme. Previously we demonstrated that Eg5/kinesin5 activity is also inhibited to undetectable levels in the brains of APP mice expressing high levels of the Aβ peptide (Borysov et al., 2010).
Figure 3.

Active Eg5/kinesin5 is present in primary mouse brain neurons. Immunocytochemistry confirms the presence of Eg5/kinesin 5 in cell bodies and processes of E18 neurons (A). FITC-labeled Aβ peptide is taken up into both the cell bodies and processes of primary neurons (B). Active Eg5 can be recovered from extracts of primary neuron cultures by immunoprecipitation but not from cells pre-treated with Aβ, indicating that Aβ taken up by neurons enters the cytoplasm where it can inhibit Eg5/kinesin 5 activity (C). To determine whether Aβ or monastrol reduced cell viability, a live/dead assay was performed after 48 hours of incubation (D). Neither 1 μM Aβsc (P=0.693) nor Aβ42 (p=0.419) significantly reduced viability, while 100 μM monastrol reduced viability by only 5% (p=0.007). In contrast, treatment of primary neurons with Aβ or monastrol severely disrupted the microtubule network shown by immunocytochemistry of α-tubulin (E).
Because Eg5/Kinesin 5 is engaged in the ATP-dependent sliding of microtubules with respect to each other, it is reasonable to expect that the structure and function of the microtubule network would be disrupted by inhibition of kinesin 5. We first established whether Aβ or monastrol treatment significantly reduces cell viability, and found that 1μM Aβ does not reduce cell viability over 48 hours, while 100μM monastol reduced viability by only 5% (Fig 3C). However, treatment of primary cortical neurons with Aβ or monastrol leads to a profound disruption of the microtubule transport network (green; Fig. 3D).
3.4 Kinesin 5 inhibition in primary neurons reduces cell surface localization of NMDA receptors
Immunocytochemical staining and confocal microscopy showed that, like H4 cells, primary neurons exhibit a significantly decreased level of surface NMDA/NR1 receptor subunits after inhibition of Eg5 by Aβ or monastrol (Fig. 4A). Interestingly, permeabilized neurons also showed a significant reduction in total NMDA/NR1 after Aβ or monastrol treatment (Fig 4B). Cross section confocal microscopy images show that in Aβ42 and monastrol treated neurons, the NMDA/NR1 receptors (green) are mainly inside the cells and are missing from the cell surface 4C–E, while the receptors are located both inside the cell and on the surface of the control cells (Fig.4F–H). Videos of three-dimensional reconstructed models of the confocal images showing the normal localization of the NMDA/NR1 receptor subunits in primary neurons treated with scrambled Aβ peptide and their mis-localization in Aβ42-treated neurons are shown in Movies 1 and 2.
Figure 4.

Treatment of primary neurons with Aβ or monastrol reduces cell surface localization of NMDA and p75 receptors. Confocal immuno-microscopy showed fewer cell surface NMDA/NR1 receptor subunits on non-permeabilized primary mouse E18 neurons after exposure to Aβ 1–40, Aβ 1–42, or monastrol (A; also see S1), with some reduction also seen in permeabilized cells (B). Cross section confocal microscopy images show that the NMDA/NR1 receptors (green) are located both inside the cell and on the surface of the control cells (C–E), while the receptors are mainly inside the cells and are missing from the cell surface in Aβ42 and monastrol treated neurons (F–H).
Top views of NMDA/NR1 receptor subunits with confocal fluorescence microscopy also revealed receptor mis-localization in primary neurons treated with Aβ42 (Fig.5A–D). The images reveal dispersed NMDA/NR1 receptor (red) in the cell body and in the processes after treatment with scrambled Aβ peptide (Fig.5A,B) for 24hr, while only aggregated, mainly peri-nuclear receptors were found after treatment with Aβ42 (Fig.5C,D).
Figure 5.
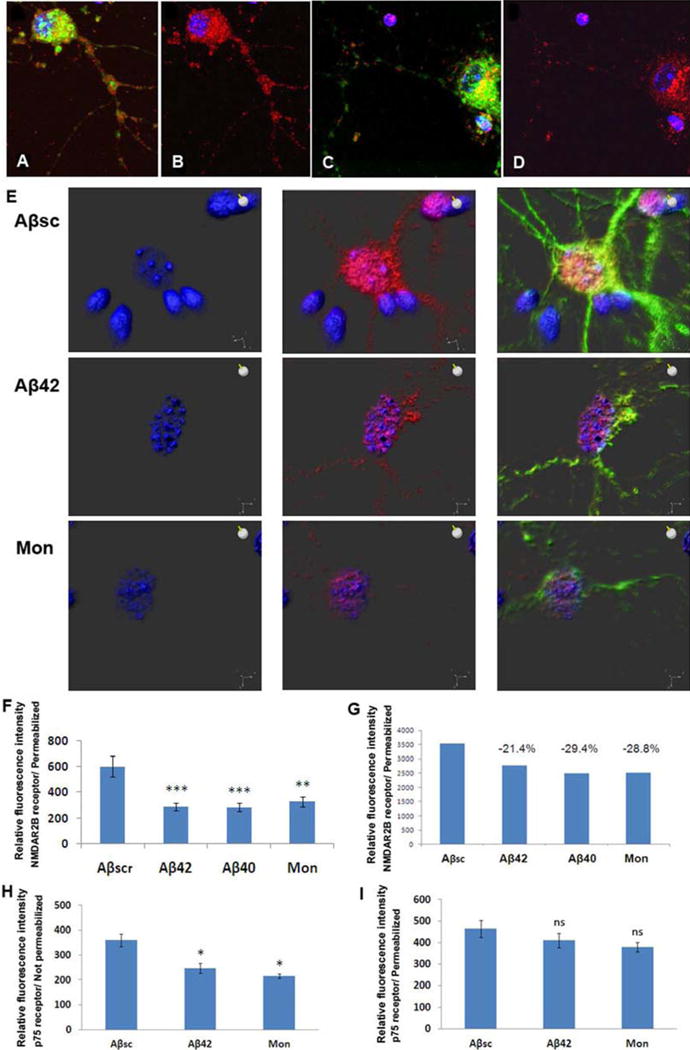
Top views of NMDA/NR1 receptor subunits with confocal immunoflourscence microscopy also revealed receptor mis-localization in primary neurons treated with Aβ42 (A–D). NMDA/NR1 receptor (red) is localized both in the cell body and in the processes after treatment with scrambled Aβ peptide (A,B) for 24hr, while only aggregated, mainly peri-nuclear receptors were found after treatment with Aβ42 (C,D). Similarly, total NMDAR2B receptor subunits (red) observed in permeabilized cell bodies and processes of primary neurons treated with scrambled Aβ42 peptide were reduced and became perinuclear after treatment of the neurons with Aβ or monastrol (E). Quantitation of NMDAR2B by confocal microscopy (F) and flow cytometry (G) confirmed a reduction in receptors induced by Aβ or monastrol. Finally, p75 receptors were significantly reduced in non-permeabilized (H) but not in permeabilized primary neurons (I) after 48h treatment with Aβ42 or monastrol.
Reduced levels of NMDAR2B receptor subunits were also observed by top view confocal imaging in permeabilized mouse primary neurons after treatment with Aβ42 or monastrol, compared to Aβsc (Fig. 5E). However, the level of WGA remained the same in each treatment groups (not shown). Videos illustrating the normal localization of NMDAR2B receptors in neurons treated with scrambled Aβ peptide and the changed receptor localization to a peri-nuclear remnant in cells treated with Aβ or monastrol are shown in Movies 3–5. Quantitation of NMDAR2B by confocal microscopy (Fig 5F) and flow cytometry (Fig. 5G) of permeabilized neurons confirmed a reduction in receptors induced by Aβ or monastrol. Similarly, p75 receptors were significantly reduced in non-permeabilized (Fig. 5H), while total p75 is not significantly reduced in permeabilized primary neurons (Fig. 5I) after 48h treatment with Aβ42 or monastrol.
3.5. Inhibition of Eg5 by monastrol causes functional defects in neuroplasticity
Glutamate activation of NMDA receptors leads to entry of Ca++ ions and the activation of pathways leading to long-term changes in synaptic sensitivity (Gladding and Raymond 2011). Both Aβ and monastrol treatment significantly reduced the stimulatory effect of glutamate on NMDA-dependent Ca++ entry into primary neurons (Fig. 6A).
Figure 6.

Glutamate-induced Ca++ entry and LTP/neuroplasticity are reduced by Aβ or monastrol; Eg5 activity is absent in APP/PS AD mice. Glutamate induced Ca++ entry into primary neurons was reduced by treatment with Aβ or monastrol by quantitative flourescence microscopic detection of Fura-2 (A). Hippocampal slices from non-transgenic mice were incubated in the presence of monastrol or buffer alone for 30 minutes. Subsequently, the slices were subjected to a high frequency (2 trains, 100pulses, 100Hz; HFS) stimulation, and their LTP responses were measured and found to be completely inhibited by monastol (B). Immunoprecipitation of Eg5 from extracts of brain showed recoverable activity in normal mice but no activity in APP/PS mice (C).
Glutamate activation of NMDA receptors is also necessary for Long Term Potentiation (LTP), perhaps the closest cellular correlate to neuroplasticity related to learning and memory, and it is considered significant for the understanding of Alzheimer’s disease that Aβ inhibits LTP (Cullen et al., 1997; Walsh et al., 2002; Wang et al., 2002; Townsend et al., 2006; Smith et al., 2009). Hippocampal slices from non-transgenic mice were incubated in the presence of 100μM monastrol or buffer alone for 30 minutes. Subsequently, the slices were subjected to a high frequency (2 trains, 100 pulses, 100 Hz; HFS) stimulation, and their LTP responses were measured. As shown in Fig. 6B, Eg5/kinesin 5 inhibition by monastrol significantly blocks LTP.
3.6. PS/APP mouse brain lacks Eg5/kinesin 5 activity
To test the ability of Aβ to inhibit Eg5/kinesin 5 in vivo, we assessed the activity of endogenous kinesin 5 in the brains of the PS1/APP transgenic mouse model of AD compared to control non-transgenic mice. Brain extracts were prepared, and Eg5/kinesin 5 was immunoprecipitated and subjected to the in vitro MT-dependent ATPase assay (Borysov et al., 2011). Consistent with the observed Aβ-mediated inhibition of Eg5/kinesin 5 in cultured neurons (Fig. 3A), kinesin 5 activity immunoprecipitated from brain extracts was almost completely absent from PS/APP compared to non-transgenic mice brains (Fig. 6C).
4. Discussion
The data indicate that Aβ reduces cell surface availability and function of neurotrophin and neurotransmitter receptors through inhibition of MT motors, specifically Eg5/kinesin 5. Consequently, cells are rendered less able to respond to their environment, which provides a potential mechanism for poor neuronal function and viability in AD and possibly Down syndrome. These data are completely consistent with the recent discovery, published during review of this paper, that Eg5 is required for the transport of certain proteins from the trans Golgi network to the cell surface (Wakana et al., 2013) and with our previous finding that Aβ inhibits the transport of the LDL receptor to the cell surface (Abisambra et al., 2010). Aβ inhibition of Eg5/kinesin 5 may also explain several previous observations that localization and function of various neurotransmitter receptors and other membrane proteins, including the NMDA receptor and the APP and PS proteins themselves, are disrupted in AD (Li et al., 1997; Honda et al., 2000; Almeida et al. 2005; Snyder et al., 2005; Shah et al., 2009; Liu et al., 2010). The fact that some polymorphisms linked to KIF11/Eg5 are associated with increased AD risk and/or Aβ production (Feuk et al., 2005; Reitz et al., 2012) further strengthens this hypothesis.
Our finding that Aβ applied externally to cells can enter cells and inhibit intracellular kinesin5/Eg5 adds to a growing body of evidence that intracellular Aβ is toxic and that cells take up Aβ into multiple cellular compartments where the peptide can affect various aspects of physiology. For example, intracellular Aβ can be detected inside neurons of 5xFAD mice by a new anti-Aβ antibody that does not detect APP (Youmans et al., 2012; compare with Winton et al., 2011). Furthermore, intra-neuronal Aβ correlates with toxicity in 5xFAD mice (Eimar and Vassar, 2013) and in PC12 cells and primary neurons, which take up Aβ through binding of the HH domain (Poduslo et al., 2010). Such Aβ uptake does not require the apoE cofactor needed for other aspects of Aβ function, such as polymerization (Saavedra et al., 2007; Potter and Wisniewski, 2012). Furthermore, similar to our finding of microtubule dysfunction by Aβ, Tang et al., (2012) found that Aβ oligomers added to cells impair the transport kinetics of synaptic cargoes, consistent with interference with the microtubule system. Finally, Yoon and colleagues (2009) found that intracellular Aβ interacts with superoxide dismutase (SOD) and inhibits its activity, also indicating that intracellular Aβ must reside, at least partly, in the cytpoplasmic domain where it can access soluble enzymes such as SOD and kinesin5/Eg5.
In addition to affecting neuroplasticity, the reduction in cell surface receptors induced by exposure to Aβ (and mimicked by monastrol) may also contribute directly to the neuronal cell loss in AD. For example, a prominent aspect of neurodegeneration in AD patients affects the cholinergic neurons of the basal forebrain (CBF), which have been shown to be critical for learning and memory and are the intended beneficiary of anti-cholinesterase therapies (Mufson et al., 2008). CBF neurons express both the Trk and p75NTR receptors throughout life, and are selectively lost in the absence of NGF whose optimal neurotrophic activity requires both Trk and p75 NTR (Hefti et al., 1984; Ruberti et al. 2000; Bui et al., 2002). Indeed, cholinergic neurons can be rescued from glutamate excitotoxicity or oxidative stress by exposure to NGF, NGF infusion increases cognition in both AD mice and human AD patients, and non-peptide ligands of p75NTR inhibit the neurotoxic and synaptotoxic effects of Aβ (Hefti et al., 1984; Ruberti et al., 2000; Bui et al., 2002; Tuszynski et al., 2005; Yang, 2008). Conversely, knocking out the NGF gene or blocking NGF activity by endogenous expression of anti-NGF antibody in normal mice replicates much of the cognitive decline and some of the pathology of AD transgenic mice (Ruberti et al., 2000; Capsoni et al., 2000). Interestingly, p75NTR is a major target of the γ-secretase enzyme, and its C-terminal fragment promotes expression, of, for example cyclin E (Jung et al., 2003; Parkhurst et al., 2010), while NGF deprivation also induces APP cleavage and the production of Aβ, indicating the activation of a positive feedback loop (Matrone et al., 2008; Parkhurst et al., 2010). In contrast, Aβ induces a long-term increase in expression of p75NTR, which we interpret as a compensatory response to poor localization, as we previously found for the LDL receptor in cells treated with Aβ (Chakravarthy et al., 2010; Abisambra et al., 2010). Finally, inflammation, which is an essential contributor to AD pathogenesis (Potter and Wisniewski, 2012), also increases p75NTR expression in many cells, although whether this reflects increased or decreased NGF sensitivity is less clear (Parkhurst et al., 2010). In sum, the reduction of cell surface p75NTR in CBF neurons by Aβ may lead directly to increased vulnerability of these cells in AD.
In addition to inhibiting receptor localization and function, Aβ-induced microtubule dysfunction leads to defective chromosome segregation during neurogenesis/mitosis and to 20–30% aneuploid/hyperploid neurons and other cells in familial and sporadic AD and in mouse and cell culture models thereof (Potter, 1991; Potter et al., 1995; Geller and Potter, 1999; Migliore et al., 1999; Boeras et al., 2008; Iourov et al. 2009; Granic et al., 2010; Arendt et al., 2010; Granic and Potter, 2011). The importance of chromosome mis-segregation in AD is indicated by the finding that the specific loss of aneuploid neurons during the transition from mild cognitive impairment to severe AD accounts for 90% of the neuronal cell death observed at autopsy (Arendt et al., 2010). The mechanism of AD-related chromosome mis-segregation became clear when Aβ was found to inhibit three specific microtubule motor kinesins that are essential for mitotic spindle structure and function (Borysov et al., 2011). That some of these motors also function in mature neurons (Baas 1998) and that Aβ disrupts the microtubule network in neurons and prevents the localization of the LDL receptor to the cell surface (Abisambra et al., 2010) prompted the current study of the effect of Aβ on the localization and function of neurotransmitter and neurotrophin receptors.
Together with previous results, these novel data suggest that protecting MT motors such as Eg5/kinesin 5 from inhibition by both extracellular and intracellular Aβ may constitute a promising approach to AD therapy with multiple benefits: 1) preventing Aβ from disrupting neurotrophin receptor localization and function required for neuron cell survival and neurite outgrowth, 2) preventing Aβ from disrupting neurotransmitter receptor localization and function required for neuroplasticity, and 3) preventing Aβ from disrupting chromosome segregation during neurogenesis and causing neuronal loss.
Supplementary Material
Movie 1. Video of three-dimensional reconstruction of the confocal images of primary neurons after 48 hr of Aβsc treatment showing normal localization of NMDA/NR1 receptors (green). Red: Cellmask.
Movie 2. Video of three-dimensional reconstruction of the confocal images of primary neurons after 48 hr of Aβ 42 treatment showing mis-localization of NMDA/NR1 receptors away from the cell surface. Red: Cellmask.
Movie 3. Video of successive two-dimensional top view confocal images of primary neurons after Aβsc treatment (48h) showing normal localization of NMDAR2B receptors (green) on the surface of a neuronal cell body and its processes.
Movie 4. Video of successive two-dimensional top view confocal images of primary neurons after Aβ42 treatment (48h) showing reduced levels and abnormal perinuclear localization of NMDAR2B receptors (green).
Movie 5 Video of successive two-dimensional top view confocal images of primary neurons after monastrol treatment (48h) showing reduced levels and abnormal perinuclear localization of NMDAR2B receptors (green).
Acknowledgments
We thank Dr. Moses Chao for a gift of p75 antibody, Dr. Byeong Cha of the USF Microscopy Core for help with the confocal imaging, Chris Katnik, Adam Behensky and Dr. Dominic D’Agostino for help with Ca++ imaging, and Michelle Norden, Adam Behansky and Chris Katnik for help with neuronal cultures. Work was supported by the USF Health Byrd Alzheimer’s Institute, the Eric Pfeiffer Chair for Research on Alzheimer’s Disease, the University of Colorado Denver, the Linda Crnic Institute for Down Syndrome, and the NIH (NS076291).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure Statement: The authors declare no conflict of interest.
References
- Abisambra JF, Fiorelli T, Padmanabhan J, Neame P, Wefes I, Potter H. LDLR expression and localization are altered in mouse and human cell culture models of Alzheimer’s disease. PLoS One. 2010;5:e8556. doi: 10.1371/journal.pone.0008556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida CG, Tampellini D, Takahashi RH, Greengard P, Lin MT, Snyder EM, Gouras GK. Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol Dis. 2005;20:187–198. doi: 10.1016/j.nbd.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Arendt T, Bruckner MK, Mosch B, Losche A. Selective cell death of hyperploid neurons in Alzheimer’s disease. Am J Pathol. 2010;177:15–20. doi: 10.2353/ajpath.2010.090955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baas PW. The role of motor proteins in establishing the microtubule arrays of axons and dendrites. J Chem Neuroanat. 1998;14:175–180. doi: 10.1016/s0891-0618(98)00012-x. [DOI] [PubMed] [Google Scholar]
- Boeras DI, Granic A, Padmanabhan J, Crespo NC, Rojiani AM, Potter H. Alzheimer’s presenilin 1 causes chromosome missegregation and aneuploidy. Neurobiol Aging. 2008;29:319–328. doi: 10.1016/j.neurobiolaging.2006.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borysov SI, Granic A, Padmanabhan J, Norden M, Walsczak C, Potter H. Alzheimer’s Aβ Disrupts Formation and Stability of the Mitotic Spindle and Directly Inhibits Mitotic/Neuronal Microtubule Motors. Cell Cycle. 2011;10:1397–1410. doi: 10.4161/cc.10.9.15478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui NT, König HG, Culmsee C, Bauerbach E, Poppe M, Krieglstein J, Prehn JH. p75 neurotrophin receptor is required for constitutive and NGF-induced survival signalling in PC12 cells and rat hippocampal neurones. J Neurochem. 2002;81:594–605. doi: 10.1046/j.1471-4159.2002.00841.x. [DOI] [PubMed] [Google Scholar]
- Capsoni S, Ugolini G, Comparini A, Ruberti F, Berardi N, Cattaneo A. Alzheimer-like neurodegeneration in aged antinerve growth factor transgenic mice. Proc Natl Acad Sci U S A. 2000;97:6826–6831. doi: 10.1073/pnas.97.12.6826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarthy B, Gaudet C, Ménard M, Atkinson T, Brown L, Laferla FM, Armato U, Whitfield J. Amyloid-beta peptides stimulate the expression of the p75(NTR) neurotrophin receptor in SHSY5Y human neuroblastoma cells and AD transgenic mice. J Alzheimers Dis. 2010;19:915–925. doi: 10.3233/JAD-2010-1288. [DOI] [PubMed] [Google Scholar]
- Cullen WK, Suh YH, Anwyl R, Rowan MJ. Block of LTP in rat hippocampus in vivo by beta-amyloid precursor protein fragments. Neuroreport. 1997;8:3213–3217. doi: 10.1097/00001756-199710200-00006. [DOI] [PubMed] [Google Scholar]
- Eimer WA, Vassar R. Neuron loss in the 5XFAD mouse model of Alzheimer’s disease correlates with intraneuronal Aβ42 accumulation and Caspase-3 activation. Mol Neurodegener. 2013;8:2. doi: 10.1186/1750-1326-8-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuk L, McCarthy S, Andersson B, Prince JA, Brookes AJ. Mutation screening of a haplotype block around the insulin degrading enzyme gene and association with Alzheimer’s disease. Am J Med Genet B Neuropsychiatr Genet. 2005;136B:69–71. doi: 10.1002/ajmg.b.30172. [DOI] [PubMed] [Google Scholar]
- Geller LN, Potter H. Chromosome missegregation and trisomy 21 mosaicism in Alzheimer’s disease. Neurobiol Dis. 1999;6:167–179. doi: 10.1006/nbdi.1999.0236. [DOI] [PubMed] [Google Scholar]
- Gladding CM, Raymond LA. Mechanisms underlying NMDA receptor synaptic/extrasynaptic distribution and function. Mol Cell Neurosci. 2011;48:308–320. doi: 10.1016/j.mcn.2011.05.001. [DOI] [PubMed] [Google Scholar]
- Granic A, Padmanabhan J, Norden M, Potter H. Alzheimer Abeta peptide induces chromosome mis-segregation and aneuploidy, including trisomy 21: requirement for tau and APP. Mol Biol Cell. 2010;21:511–520. doi: 10.1091/mbc.E09-10-0850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granic A, Potter H. Down syndrome model of Alzheimer’s disease: beyond trisomy 21 nondisjunction. In: Dey S, editor. Genetics and Etiology of Down Syndrome. InTech; Croatia: 2011. pp. 159–176. [Google Scholar]
- Hamano T, Mutoh T, Tabira T, Araki W, Kuriyama M, Mihara T, Yano S, Yamamoto H. Abnormal intracellular trafficking of high affinity nerve growth factor receptor, Trk, in stable transfectants expressing presenilin 1 protein. Brain Res Mol Brain Res. 2005;137:70–76. doi: 10.1016/j.molbrainres.2005.02.018. [DOI] [PubMed] [Google Scholar]
- Hardy J. The amyloid hypothesis for Alzheimer’s disease: a critical reappraisal. J Neurochem. 2009;110:1129–1134. doi: 10.1111/j.1471-4159.2009.06181.x. [DOI] [PubMed] [Google Scholar]
- Heald R, Walczak CE. Microtubule-based motor function in mitosis. Curr Opin Struct Biol. 1999;9:268–274. doi: 10.1016/s0959-440x(99)80037-2. [DOI] [PubMed] [Google Scholar]
- Hefti F, Dravid A, Hartikka J. Chronic intraventricular injections of nerve growth factor elevate hippocampal choline acetyltransferase activity in adult rats with partial septo-hippocampal lesions. Brain Res. 1984;293:305–309. doi: 10.1016/0006-8993(84)91237-x. [DOI] [PubMed] [Google Scholar]
- Honda T, Nihonmatsu N, Yasutake K, Ohtake A, Sato K, Tanaka S, Murayama O, Murayama M, Takashima A. Familial Alzheimer’s disease-associated mutations block translocation of full-length presenilin 1 to the nuclear envelope. Neurosci Res. 2000;37:101–111. doi: 10.1016/s0168-0102(00)00106-1. [DOI] [PubMed] [Google Scholar]
- Hsu TC, Satya-Prakash KL. Aneuploidy induction by mitotic arrestants in animal cell systems: possible mechanisms. Basic Life Sci. 1985;36:279–289. doi: 10.1007/978-1-4613-2127-9_18. [DOI] [PubMed] [Google Scholar]
- Hu X, Crick SL, Bu G, Frieden C, Pappu RV, Lee JM. Amyloid seeds formed by cellular uptake, concentration, and aggregation of the amyloid-beta peptide. Proc Natl Acad Sci U S A. 2009;106:20324–20329. doi: 10.1073/pnas.0911281106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber LJ, Chao MV. Mesenchymal and neuronal cell expression of the p75 neurotrophin receptor are distinguished during morphogenesis of transgenic animals. Developmental Biology. 1995;167:227–238. doi: 10.1006/dbio.1995.1019. [DOI] [PubMed] [Google Scholar]
- Iourov IY, Vorsanova SG, Liehr T, Yurov YB. Aneuploidy in the normal, Alzheimer’s disease and ataxia-telangiectasia brain: differential expression and pathological meaning. Neurobiol Dis. 2009;34:212–220. doi: 10.1016/j.nbd.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Jung KM, Tan S, Landman N, Petrova K, Murray S, Lewis R, Kim PK, Kim DS, Ryu SH, Chao MV, Kim TW. Regulated intramembrane proteolysis of the p75 neurotrophin receptor modulates its association with the TrkA receptor. J Biol Chem. 2003;278:42161–42169. doi: 10.1074/jbc.M306028200. [DOI] [PubMed] [Google Scholar]
- Kapoor TM, Mayer TU, Coughlin ML, Mitchison TJ. Probing spindle assembly mechanisms with monastrol, a small molecule inhibitor of the mitotic kinesin, Eg5. J Cell Biol. 2000;150:975–988. doi: 10.1083/jcb.150.5.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VM. Regulation of tau phosphorylation in Alzheimer’s disease. Ann NY Acad Sci. 1996;777:107–113. doi: 10.1111/j.1749-6632.1996.tb34408.x. [DOI] [PubMed] [Google Scholar]
- Lee VM, Trojanowski JQ. Progress from Alzheimer’s tangles to pathological tau points towards more effective therapies now. J Alzheimers Dis. 2006;9(3 Suppl):257–262. doi: 10.3233/jad-2006-9s328. [DOI] [PubMed] [Google Scholar]
- Li J, Xu M, Zhou H, Ma J, Potter H. Alzheimer presenilins in the nuclear membrane, interphase kinetochores, and centrosomes suggest a role in chromosome segregation. Cell. 1997;90:917–927. doi: 10.1016/s0092-8674(00)80356-6. [DOI] [PubMed] [Google Scholar]
- Liu SJ, Gasperini R, Foa L, Small DH. Amyloid-beta decreases cell-surface AMPA receptors by increasing intracellular calcium and phosphorylation of GluR2. J Alzheimers Dis. 2010;21:655–666. doi: 10.3233/JAD-2010-091654. [DOI] [PubMed] [Google Scholar]
- Liu YL, Peterson DA, Schubert D. Amyloid b peptide alters intracellular vesicle trafficking and cholesterol homeostasis. Proc Natl Acad Sci U S A. 2008;95:13266–13271. doi: 10.1073/pnas.95.22.13266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhang YW, Wang X, Zhang H, You X, Liao FF, Xu H. Intracellular trafficking of presenilin 1 is regulated by beta-amyloid precursor protein and phospholipase D1. J Biol Chem. 2009;284:12145–12152. doi: 10.1074/jbc.M808497200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailhes JB, Mastromatteo C, Fuseler JW. Transient exposure to the Eg5 kinesin inhibitor monastrol leads to syntelic orientation of chromosomes and aneuploidy in mouse oocytes. Mutat Res. 2004;559:153–167. doi: 10.1016/j.mrgentox.2004.01.001. [DOI] [PubMed] [Google Scholar]
- Mandelkow EM, Mandelkow E. Tau in Alzheimer’s disease. Trends Cell Biol. 1998;8:425–427. doi: 10.1016/s0962-8924(98)01368-3. [DOI] [PubMed] [Google Scholar]
- Matrone C, Ciotti MT, Mercanti D, Marolda R, Calissano P. NGF and BDNF signaling control amyloidogenic route and Aβ production in hippocampal neurons. Proc, Natl Acad Aci U S A. 2008;105:13139–13144. doi: 10.1073/pnas.0806133105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazumdar M, Sundareshan S, Misteli T. Human chromokinesin KIF4A functions in chromosome condensation and segregation. J Cell Biol. 2004;166:613–620. doi: 10.1083/jcb.200401142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliore L, Botto N, Scarpato R, Petrozzi L, Cipriani G, Bonuccelli U. Preferential occurrence of chromosome 21 malsegregation in peripheral blood lymphocytes of Alzheimer disease patients. Cytogenet Cell Genet. 1999;87:41–46. doi: 10.1159/000015389. [DOI] [PubMed] [Google Scholar]
- Mufson EJ, Counts SE, Perez SE, Ginsberg SD. Cholinergic system during the progression of Alzheimer’s disease: therapeutic implications. Expert Rev Neurother. 2008;8:1703–1718. doi: 10.1586/14737175.8.11.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan J, Levy M, Dickson DW, Potter H. Alpha1-antichymotrypsin, an inflammatory protein overexpressed in Alzheimer’s disease brain, induces tau phosphorylation in neurons. Brain. 2006;129:3020–3034. doi: 10.1093/brain/awl255. [DOI] [PubMed] [Google Scholar]
- Parkhurst CN, Zampieri N, Chao MV. Nuclear Localization of the p75 Neurotrophin Receptor Intracellular Domain. J Biol Chem. 2010;285:5361–5368. doi: 10.1074/jbc.M109.045054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pigino G, Pelsman A, Mori H, Busciglio J. Presenilin-1 mutations reduce cytoskeletal association, deregulate neurite growth, and potentiate neuronal dystrophy and tau phosphorylation. J Neurosci. 2001;21:834–842. doi: 10.1523/JNEUROSCI.21-03-00834.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poduslo JF, Gilles EJ, Ramakrishnan M, Howell KG, Wengenack TM, Curran GL, Kandimalla KK. HH domain of Alzheimer’s disease Abeta provides structural basis for neuronal binding in PC12 and mouse cortical/hippocampal neurons. PLoS One. 2010;5:e8813. doi: 10.1371/journal.pone.0008813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter H. Review and hypothesis: Alzheimer disease and Down syndrome--chromosome 21 nondisjunction may underlie both disorders. Am J Hum Genet. 1991;48:1192–1200. [PMC free article] [PubMed] [Google Scholar]
- Potter H, Ma J, Das S, Geller LN, Benjamin M, Kayyali US, Dressler D. Beyond beta-protein: New steps in the pathogenic pathway to Alzheimer’s disease. In: Mortimer JA, Iqbal K, Winblad B, Wisniewski HM, editors. Research Advances in Alzheimer’s Disease and Related Disorders. John Wiley and Sons Ltd.; New York: 1995. pp. 643–654. [Google Scholar]
- Potter H, Wisniewski T. Apolipoprotein E: Essential Catalyst of the Alzheimer Amyloid Cascade. Internat J Alz Dis. 2012;2012:489428. doi: 10.1155/2012/489428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A. Tau is essential to beta -amyloid-induced neurotoxicity. Proc Natl Acad Sci U S A. 2002;99:6364–6369. doi: 10.1073/pnas.092136199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitz C, Cheng R, Schupf N, Lee JH, Mehta PD, Rogaeva E, St George-Hyslop P, Mayeux R. Association between variants in IDE-KIF11-HHEX and plasma amyloid beta levels. Neurobiol Aging. 2012;33:199.e13–7. doi: 10.1016/j.neurobiolaging.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Ruberti F, Capsoni S, Comparini A, Di Daniel E, Franzot J, Gonfloni S, Rossi G, Berardi N, Cattaneo A. Phenotypic knockout of nerve growth factor in adult transgenic mice reveals severe deficits in basal forebrain cholinergic neurons, cell death in the spleen, and skeletal muscle dystrophy. J Neurosci. 2000;20:2589–2601. doi: 10.1523/JNEUROSCI.20-07-02589.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saavedra L, Mohamed A, Ma V, Kar S, de Chaves EP. Internalization of beta-amyloid peptide by primary neurons in the absence of apolipoprotein E. J Biol Chem. 2007;282:35722–35732. doi: 10.1074/jbc.M701823200. [DOI] [PubMed] [Google Scholar]
- Shah SB, Nolan R, Davis E, Stokin GB, Niesman I, Canto I, Glabe C, Goldstein LS. Examination of potential mechanisms of amyloid-induced defects in neuronal transport. Neurobiol Dis. 2009;346:11–25. doi: 10.1016/j.nbd.2009.05.016. [DOI] [PubMed] [Google Scholar]
- Smith JP, Lal V, Bowser D, Cappai R, Masters CL, Ciccotosto GD. Stimulus pattern dependence of the Alzheimer’s disease amyloid-β 42 peptide’s inhibition of long term potentiation in mouse hippocampal slices. Brain Research. 2009;1269:176–184. doi: 10.1016/j.brainres.2009.03.007. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Takemura R, Nakata T, Okada Y, Yamazaki H, Zhang Z, Hirokawa N. mRNA expression of KIF1A, KIF1B, KIF2, KIF3A, KIF3B, KIF4, KIF5, and cytoplasmic dynein during axonal regeneration. J Neurosci. 1996;16:31–35. doi: 10.1523/JNEUROSCI.16-01-00031.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Scott DA, Das U, Edland SD, Radomski K, Koo EH, Roy S. Early and selective impairments in axonal transport kinetics of synaptic cargoes induced by soluble amyloid β-protein oligomers. Traffic. 2012;13:681–693. doi: 10.1111/j.1600-0854.2012.01340.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tezapsidis N, Merz PA, Merz G, Hong H. Microtubular interactions of presenilin direct kinesis of Abeta peptide and its precursors. FASEB J. 2003;17:1322–1324. doi: 10.1096/fj.02-0980fje. [DOI] [PubMed] [Google Scholar]
- Tong L, Balazs R, Thornton PL, Cotman CW. Beta-amyloid peptide at sublethal concentrations downregulates brain-derived neurotrophic factor functions in cultured cortical neurons. J Neurosci. 2004;24:6799–6809. doi: 10.1523/JNEUROSCI.5463-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ. Effects of secreted oligomers of amyloid β-protein on hippocampal synaptic plasticity: a potent role for trimmers. J Physiol. 2006;572:477–492. doi: 10.1113/jphysiol.2005.103754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuszynski MH, Thal L, Pay M, Salmon DP, U HS, Bakay R, Patel P, Blesch A, Vahlsing HL, Ho G, Tong G, Potkin SG, Fallon J, Hansen L, Mufson EJ, Kordower JH, Gall C, Conner J. A Phase I clinical trial of nerve growth factor gene therapy for Alzheimer’s disease. Nat Med. 2005;11:551–555. doi: 10.1038/nm1239. [DOI] [PubMed] [Google Scholar]
- Wakana Y, Villeneuve J, van Galen J, Cruz-Garcia D, Tagaya M, Malhotra V. Kinesin-5/Eg5 is important for transport of CARTS from the trans-Golgi network to the cell surface. J Cell Biol. 2013;202:241–250. doi: 10.1083/jcb.201303163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walczak CE, Heald R. Mechanisms of mitotic spindle assembly and function. Int Rev Cytol. 2008;265:111–158. doi: 10.1016/S0074-7696(07)65003-7. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Wang Y, Greig NH, Yu QS, Mattson MP. Presenilin-1 mutation impairs cholinergic modulation of synaptic plasticity and suppresses NMDA currents in hippocampus slices. Neurobiol Aging. 2009;30:1061–1068. doi: 10.1016/j.neurobiolaging.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HW, Pasternak JF, Kuo H, Ristic H, Lambert MP, Chromy B, Viola KL, Klein WL, Stine WB, Krafft GA, Trommer BL. Soluble oligomers of β amyloid (1–42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res. 2002;924:133–140. doi: 10.1016/s0006-8993(01)03058-x. [DOI] [PubMed] [Google Scholar]
- Winton MJ, Lee EB, Sun E, Wong MM, Leight S, Zhang B, Trojanowski JQ, Lee VM. Intraneuronal APP, not free Aβ peptides in 3xTg-AD mice: implications for tau versus Aβ-mediated Alzheimer neurodegeneration. J Neurosci. 2011;31:7691–7699. doi: 10.1523/JNEUROSCI.6637-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Yang T. Small molecule, non-peptide p75 ligands inhibit Abeta-induced neurodegeneration and synaptic impairment. PLoS One. 2008;3:e3604. doi: 10.1371/journal.pone.0003604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon EJ, Park HJ, Kim GY, Cho HM, Choi JH, Park HY, Jang JY, Rhim HS, Kang SM. Intracellular amyloid beta interacts with SOD1 and impairs the enzymatic activity of SOD1: implications for the pathogenesis of amyotrophic lateral sclerosis. Exp Mol Med. 2009;41:611–617. doi: 10.3858/emm.2009.41.9.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youmans KL, Tai LM, Kanekiyo T, Stine WB, Jr, Michon SC, Nwabuisi-Heath E, Manelli AM, Fu Y, Riordan S, Eimer WA, Binder L, Bu G, Yu C, Hartley DM, LaDu MJ. Intraneuronal Aβ detection in 5xFAD mice by a new Aβ-specific antibody. Mol Neurodegener. 2012;7:8. doi: 10.1186/1750-1326-7-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie 1. Video of three-dimensional reconstruction of the confocal images of primary neurons after 48 hr of Aβsc treatment showing normal localization of NMDA/NR1 receptors (green). Red: Cellmask.
Movie 2. Video of three-dimensional reconstruction of the confocal images of primary neurons after 48 hr of Aβ 42 treatment showing mis-localization of NMDA/NR1 receptors away from the cell surface. Red: Cellmask.
Movie 3. Video of successive two-dimensional top view confocal images of primary neurons after Aβsc treatment (48h) showing normal localization of NMDAR2B receptors (green) on the surface of a neuronal cell body and its processes.
Movie 4. Video of successive two-dimensional top view confocal images of primary neurons after Aβ42 treatment (48h) showing reduced levels and abnormal perinuclear localization of NMDAR2B receptors (green).
Movie 5 Video of successive two-dimensional top view confocal images of primary neurons after monastrol treatment (48h) showing reduced levels and abnormal perinuclear localization of NMDAR2B receptors (green).