

Abstract
Multiple mutations in different subunits of the tethering complex Conserved Oligomeric Golgi (COG) have been identified as a cause for Congenital Disorders of Glycosylation (CDG) in humans. Yet, the mechanisms by which COG mutations induce the pleiotropic CDG defects have not been fully defined. By detailed analysis of Cog8 deficiency in either HeLa cells or CDG-derived fibroblasts, we show that Cog8 is required for the assembly of both the COG complex and the Golgi Stx5-GS28-Ykt6-GS15 and Stx6-Stx16-Vti1a-VAMP4 SNARE complexes. The assembly of these SNARE complexes is also impaired in cells derived from a Cog7-deficient CDG patient. Likewise, the integrity of the COG complex is also impaired in Cog1-, Cog4-and Cog6-depleted cells. Significantly, deficiency of Cog1, Cog4, Cog6 or Cog8 distinctly influences the production of COG subcomplexes and their Golgi targeting. These results shed light on the structural organization of the COG complex and its subcellular localization, and suggest that its integrity is required for both tethering of transport vesicles to the Golgi apparatus and the assembly of Golgi SNARE complexes. We propose that these two key functions are generally and mechanistically impaired in COG-associated CDG patients, thereby exerting severe pleiotropic defects.
Keywords: CDG, COG complex, Golgi, SNARE, tethering
The Conserved Oligomeric Golgi (COG) is an evolutionarily conserved Golgi-associated tethering complex consisting of eight subunits (Cog1–Cog8) (1–7). Previous studies suggest that the complex can be organized into two functionally and structurally distinct subcomplexes: lobe A (Cog1–4) and lobe B (Cog5–8) (5,7–9). Although mutations in different COG subunits impair the integrity of the entire complex (10–13), only mutations in the first lobe severely affect cell growth in yeast (7). The Cog1–4 subunits are, therefore, considered as essential components of the complex. These observations suggest that mutations in different COG subunits might exert distinct cellular defects. Depletion of the Cog3 or Cog7 subunit in mammalian cells, for example, induces phenotypes that are similar in many aspects. Yet, Cog7-depleted cells exhibit unique characteristics (14).
Similarly, inherited mutations in the genes encoding the different COG subunits, which cause Congenital Disorders of Glycosylation (CDG) in humans, exert a relatively wide range of phenotypes. The phenotypic spectrum ranges from severe to mild disease and is characterized by pleiotropic glycosylation defects (15–20). A point mutation in the Cog7 gene, for example, was described in two infants, both of whom died within the first 3 months (21), whereas other COG mutations caused milder phenotypes (11–13,22). Nevertheless, in all cases, a mutation within one subunit destabilizes other COG subunits and alters their subcellular localization, thereby affecting the overall integrity and function of the COG complex. Yet, the connection between COG function, its complex integrity and CDG pathology remains largely unknown.
Previous studies in yeast and mammalian cells suggest that COG functions as a tethering factor for two distinct classes of vesicles: vesicles that recycle within the Golgi apparatus and vesicles that recycle to the Golgi from the endosomal compartments (4,23–26). The essential role of COG in intra-Golgi retrograde transport was demonstrated both in vitro and in intact cells, and was shown to affect the proper localization of Golgi glycosylation enzymes and consequently the Golgi glycosylation machinery and the global cellular glycosylation (11,14,21,27).
Our recent studies suggest that COG also functions in SNARE complex assembly. Specifically, it positively regulates the assembly of two major Golgi SNARE complexes: Syntaxin5 (Stx5)-GS28-Ykt6-GS15 (Stx5 complex) and Stx6-Stx16-Vti1a-VAMP4 (Stx6 complex), which regulate intra-Golgi and endosome-to-TGN retrograde transport, respectively (28,29). We showed that COG, via its different subunits, directly interacts with the Golgi SNAREs and their associated Sec1/Munc18 (SM) proteins, thereby promoting the assembly of fusogenic SNARE complex (24,30,31).
Consistently, intra-Golgi retrograde transport was also impaired in CDG-derived fibroblasts, and concomitantly the proper localization of Golgi glycosylation enzymes was affected (11,12,21,22,32). These observations suggest that CDG-associated COG mutations affect the tethering of Golgi-derived vesicles to the Golgi membranes. In addition, Golgi-to-ER retrograde transport was attenuated in most COG-associated CDG patients (11–13,32). Yet, it remains unclear whether the CDG-associated COG mutations also affect endosome-to-Golgi transport and the assembly of Golgi SNARE complexes.
Here, we show that CDG-associated COG mutations impair the assembly of Golgi SNARE complexes. We further show that the distribution of endogenous proteins that cycle between the endosome and the Golgi is impaired in COG8-mutated CDG-derived cells, suggesting that endosome-to-Golgi trafficking is also affected. Finally, we show that depletion of different COG subunits induces the assembly of different COG subcomplexes. We propose that the assembly of these subcomplexes and their Golgi targeting might distinctly influence the functions of COG in the tethering and SNARE complex assembly, and thereby could yield different cellular defects. This might explain why different COG mutations induce distinct phenotypes in yeast, mammals and CDG patients.
Results
Deficiency of the Cog8 subunit affects the Golgi morphology and the steady-state levels and distributions of the other COG subunits
We have previously shown that depletion of either the Cog4 or the Cog6 subunit affects the assembly of Golgi SNARE complexes and consequently attenuates endosome-to-TGN and intra-Golgi retrograde transport (24,30,31). However, little is known about the influence of the Cog8 subunit on either SNARE complex assembly or endosome-to-TGN trafficking. To define the role of the Cog8 subunit in mammalian cells, we utilized two cellular models: Cog8-deficient fibroblasts derived from a CDG patient (22) and Cog8-depleted HeLa cells that were established by a specific and efficient shRNA construct (Figure S1, Supporting Information). We first assessed the influence of Cog8 depletion on the organization of the Golgi complex by immunofluorescence (IF) analysis utilizing the Golgi markers p115 and Mannosidase II. As shown, depletion of Cog8 markedly affected the compact organization of the Golgi apparatus in HeLa cells (Figure 1A). Electron microscopy analysis further exposed a dilation of Golgi cisternae and accumulation of vesicles in the vicinity of the Golgi (Figure 1B), which possibly represent vesicles that failed to fuse with the Golgi membranes. These results are consistent with previous studies in which vesiculated Golgi membranes and swollen Golgi cisternae were observed in CDG-derived Cog8-deficient fibroblasts (12,13).
Figure 1.
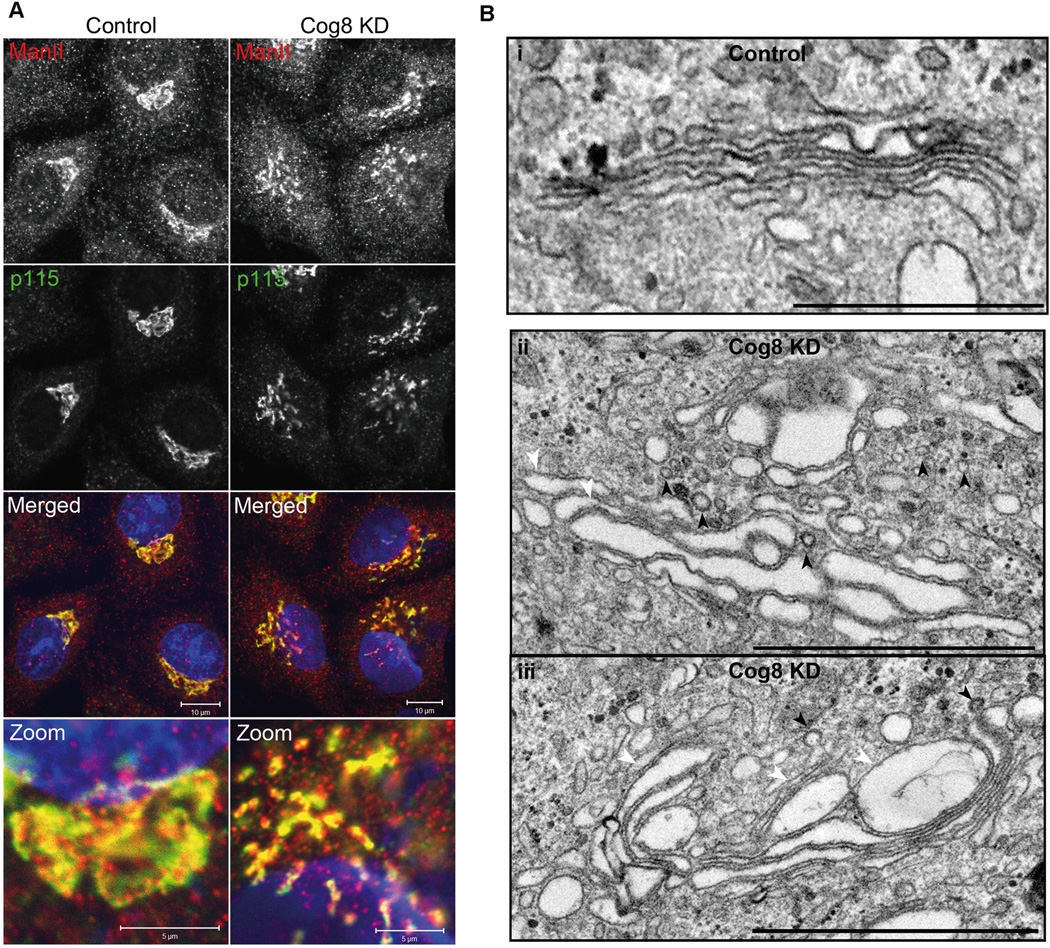
Downregulation of Cog8 by shRNA affects Golgi morphology in HeLa cells. A) IF analysis of Golgi morphology in Cog8-depleted HeLa cells. HeLa cells were transiently transfected with a pSUPER-puro (control) or Cog8 shRNA and 72 h later the cells were fixed and co-immunostained with anti-mannosidase II (ManII) (red) and anti-p115 antibodies (green). As shown, Cog8 depletion markedly affected the compact organization of the Golgi apparatus. Bars: 10 µm, zoom: 5 µm. B) Electron microscopy analysis of Golgi morphology in Cog8-depleted HeLa cells. Control and Cog8-depleted HeLa cells were fixed as described in Materials and Methods and analyzed by TEM. Shown are representative images. In control cells (i), the Golgi retained its characteristic structure consisting stacks of flattened cisternae. In Cog8-depleted cells (ii and iii), the Golgi cisternae were dilated and multiple vesicles (marked by arrowheads) were accumulated at the vicinity of the Golgi. Bars: 1 µm.
We next assessed the effect of Cog8 depletion on the steady-state levels of the different COG subunits using Western blot (WB) analysis. Knock down of Cog8 expression in HeLa cells significantly reduced (by ~80%) the level of Cog1 and also decreased the steady-state level of the Cog6 subunit (by ~30%) (Figure 2A). Similar effects were previously obtained in the CDG-derived Cog8-deficient fibroblasts consistent with the established interaction between the Cog1 and Cog8 subunits (8,9,11,12,22). Yet, a concomitant decrease in the steady-state levels of the Cog5 and Cog7 subunits was also observed in the patient’s cells used for this study (22). These differences could be due to the effect of chronic deficiency of Cog8 in the patient’s cells, which may destabilize additional COG subunits and eventually facilitate their degradation when compared with acute Cog8 deficiency in HeLa cells.
Figure 2.
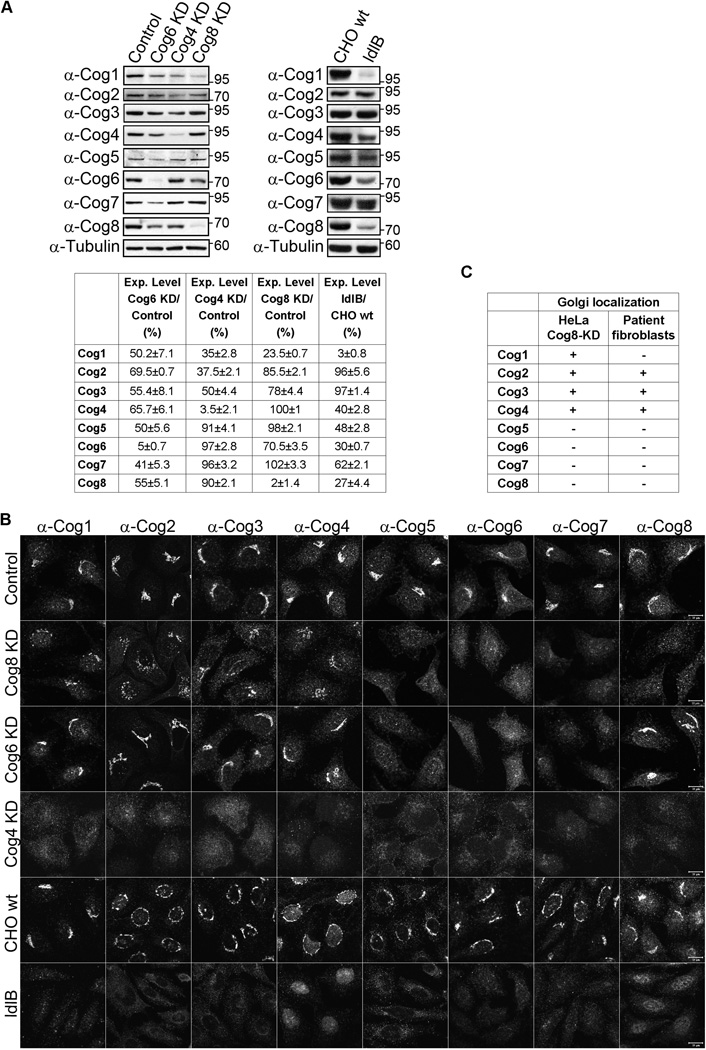
Deficiency of Cog8 affects the steady-state levels and distributions of the other COG subunits. A) Steady-state levels of COG subunits in HeLa cells or CHO cells deficient for different COG subunits. HeLa cells were transfected with either control, Cog4, Cog6 or Cog8 shRNA constructs. Twenty-four hours post-transfection, cells were incubated with 1 µg/mL puromycin for 72 h and subsequently total cell lysates were prepared. Cell lysates were also prepared for wild-type and Cog1-deficient CHO cells (ldlB). The steady-state levels of the eight COG subunits were determined by WB with the corresponding antibodies. The mean values ± SD of three independent experiments are shown. B) The steady-state distributions of the eight COG subunits in control, Cog8-, Cog6- and Cog4-depleted HeLa stable cell lines and in wild-type CHO cells and ldlB cells were determined by IF and confocal microscopy analysis. Shown are representative images. Bar 10 µm. C) The Golgi localization of the different COG subunits in Cog8-depleted HeLa cells as determined by this study is compared to their Golgi localization in Cog8-deficient fibroblasts obtained from a human CDG patient as previously determined by Kranz et al. (22).
We then examined the effect of Cog8 depletion on the Golgi localization of the different COG subunits using IF analysis. As shown, the Cog5, Cog6 and Cog7 subunits lost their characteristic Golgi localization in Cog8-depleted HeLa cells. The Cog1–4 subunits, however, were observed in the Golgi (Figure 2B), albeit a weak signal was detected for Cog1, possibly owing to its lower protein level. In CDG-derived Cog8-deficient fibroblasts, Cog1 was not detected in the Golgi, whereas the distributions of the other COG subunits were similar to those of Cog8-depleted HeLa cells (Figure 2C) (22), suggesting that deficiency of the Cog8 subunit similarly influences the distribution of the COG subunits in the two cell contexts. Interestingly, similar to the effects of Cog8 depletion in HeLa cells, depletion of the Cog6 subunit also impaired the Golgi localization of Cog5 and Cog7 subunits, whereas Cog1–4 and Cog8 were localized to the Golgi (Figure 2B). By contrast, deficiency of the Cog4 subunit in HeLa cells or the Cog1 subunit in CHO cells (ldlB) (1,5,10) caused the dispersal of all the COG subunits from the Golgi complex (Figure 2B).
These results suggest that depletion of subunits that belong to lobe B (Cog6 and Cog8) either in HeLa cells or CDG-derived cells affects the Golgi localization of lobe B but not the lobe A subunits, whereas depletion of subunits that belong to lobe A (Cog1 and Cog4) disrupts the Golgi localization of all the COG subunits.
Deficiency of the Cog8 subunit impairs the assembly of the COG complex in HeLa cells
The impaired colocalization between lobe A and lobe B subunits in Cog8-deficient cells (Figure 2B,C) suggests that the integrity of the whole COG complex was abrogated. Indeed, a defect in the formation of the whole COG complex in CDG-derived Cog8-deficient cells was previously demonstrated by glycerol gradient sedimentation analysis (12,13). We thus examined the assembly of the COG complex in Cog8-depleted HeLa cells utilizing co-immunoprecipitation (co-IP) assays. We immunoprecipitated (IP) one subunit of lobe A (Cog4) and one subunit of lobe B (Cog7) and examined their interactions with all the other COG subunits in control and Cog8-depleted HeLa cells by WB. As shown (Figure 3A), IP with anti-Cog4 antibody revealed that Cog4 interacts similarly with the Cog2 and the Cog3 subunits in both control and Cog8-depleted cells. Yet, the interactions of Cog4 with the Cog5 and Cog7 subunits were abolished in Cog8-depleted cells. Consistent with these results, IP with anti-Cog7 antibody showed that Cog7 interacts with Cog5, whereas its interactions with the Cog2, Cog3 or Cog4 subunits were abolished. Collectively, these results suggest that depletion of the Cog8 subunit results in the assembly of two separate subcomplexes: Cog2-Cog3-Cog4 and Cog5-Cog7. The interactions of Cog1 with either the lobe A or the lobe B subunits were significantly reduced possibly owing to its reduced steady-state level (Figure 2A). Similarly, Cog6 failed to interact with either lobe A or lobe B subunits, suggesting that Cog6 exists as an individual subunit in Cog8-depleted cells.
Figure 3.
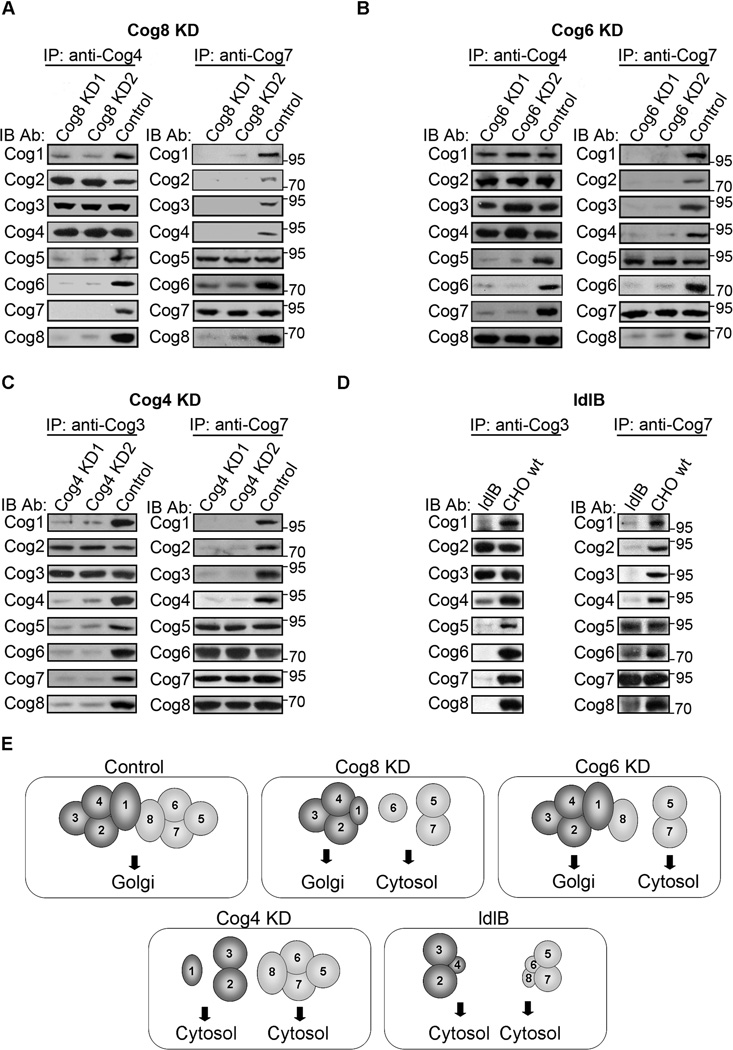
Deficiency of the Cog8 subunit impairs the assembly of the COG complex in mammalian cells. A–D) Analysis of COG complex assembly in HeLa and CHO cell lines deficient for different COG subunits. Control, Cog8-, Cog6- and Cog4-depleted (KD) HeLa stable cell lines (A–C) or wild-type CHO cells and ldlB cells (D) were solubilized and equal cell lysates were subjected to IP with the indicated antibodies. The presence of the different COG subunits in the immunocomplexes was determined by WB using the corresponding antibodies. For HeLa cells, two different clones of each stable cell line (KD1 and 2) were used in this analysis. E) A scheme describing the subunit composition of the subcomplexes formed in different COG mutants and their subcellular localization. The scheme summarizes the results of the WB, IF and co-IP analysis performed in control, Cog4-, Cog6- and Cog8-depleted HeLa cells as well as in Cog1-deficient CHO cells. A reduction in steady-state levels is represented by a smaller size of the relevant subunit.
To gain further insight into the assembly of the COG complex, we examined the influence of other lobe A and lobe B subunits (Cog1, Cog4 and Cog6) on COG complex assembly utilizing the same approach. In Cog6-depleted cells (Figure 3B), Cog4 interacts with Cog1, Cog2, Cog3 and Cog8 similar to the control cells. Yet, its interactions with Cog5 and Cog7 were abolished. Consistent with these results, Cog7 interacts with Cog5, whereas its interactions with all the other COG subunits were abolished. Collectively, these results suggest that depletion of the Cog6 subunit results in the assembly of Cog1-Cog2-Cog3-Cog4-Cog8 and Cog5-Cog7 separate subcomplexes. The influence of Cog4 depletion was different (Figure 3C); Cog3 interacts with Cog2 similar to the control cells, yet its interactions with all the other COG subunits were abolished. Cog7 interacts with Cog5, Cog6 and Cog8 similar to the control cells, but its interactions with Cog1, Cog2 and Cog3 were abolished. Collectively, these results suggest that depletion of the Cog4 subunit results in the assembly of Cog2-Cog3 and Cog5-Cog6-Cog7-Cog8 subcomplexes. It is worth mentioning that, in addition to the reduced steady-state levelofCog1 (by 65 ± 2.8%) in Cog4-depleted cells, its interactions with either the lobe A or the lobe B subunits were almost abolished (by 94 ± 3% and 99 ± 0.7%, respectively).
We further explored the effect of lobe A subunits on COG complex assembly by utilizing Cog1-deficient CHO cells (ldlB) previously established and analyzed by Krieger and coworkers (1,5,10). As shown in Figure 3D, in ldlB cells Cog3 interacts with Cog2 similar to the control cells, yet its interaction with the Cog4 subunit was substantially reduced possibly owing to the decreased steady-state level of Cog4 (Figure 2A). A similar decrease in Cog4 was previously shown by Oka et al. in ldlB cells (10). In addition, the interactions of Cog3 with the lobe B subunits and the interactions of Cog7 with the lobe A subunits were abolished. Yet, Cog7 interacts with Cog5, despite the decrease in their steady-state levels (Figure 2A), whereas its interactions with the Cog6 and Cog8 subunits were substantially reduced. Collectively, these results suggest that deficiency of Cog1 results in the assembly of Cog2-Cog3 and Cog5-Cog7 dimers with smaller pools of Cog2-Cog3-Cog4 and possibly Cog5Cog6-Cog7-Cog8 subcomplexes. Importantly, previous studies of COG complex assembly in ldlB cells utilizing gel filtration analysis suggested the existence of Cog2-Cog3Cog4 and Cog5-Cog6-Cog7-Cog8 subcomplexes based on the elution profiles of the different COG subunits (10). The complete overlap between the elution profiles of Cog2 and Cog3 when compared with their partial overlap with the elution profile of Cog4 (10) supports our results suggesting the dominance of a Cog2-Cog3 dimer in ldlB cells. Similarly, the extensive overlap between the elution profiles of Cog5 and Cog7 when compared with their limited overlap with the elution profiles of Cog6 and Cog8 (10) supports our results suggesting the dominance of a Cog5–Cog7 dimer in ldlB cells.
Overall, these results suggest that depletion of either the Cog8, Cog6, Cog4 or Cog1 subunits abolished the integrity of the entire COG complex and the connection between its two lobes. Nevertheless, depletion of each subunit distinctly influences the assembly of COG subcomplexes and their Golgi localization (Figure 3E).
Cog8 deficiency influences the assembly of the Stx5 and Stx6 Golgi SNARE complexes in both HeLa cells and CDG-derived fibroblasts
We have previously shown that the COG complex positively regulates the assembly of the Golgi Stx5 and Stx6 SNARE complexes. Our recent studies further suggest that multiple COG subunits including Cog4, Cog6 and Cog7 interact with multiple Golgi SNAREs and positively regulate SNARE complexes assembly (24,30,31). We therefore predicted that the proper assembly of the Golgi SNARE complexes would also be impaired in Cog8-depleted cells owing to the dispersal of the lobe B subunits from the Golgi. Accordingly, we assessed the effect of Cog8 deficiency on the assembly of the two SNARE complexes utilizing either Cog8-depleted HeLa cells or CDG-derived fibroblasts.
Localization studies of SNAREs that belong to the Stx5 SNARE complex revealed that the t-SNARE Stx5 was localized to the Golgi in both control and Cog8-deficient CDG-derived fibroblasts (Figure 4A). However, the Golgi localization of its interacting SNAREs, GS28 and GS15 was markedly reduced in the CDG patient’s cells as well as in Cog8-depleted HeLa cells (Figure 4A,B). WB analysis suggests that the steady-state levels of both GS28 and GS15 were markedly reduced in the patient’s cells (Figure 4C). By contrast, the protein level of GS15 was increased in Cog8-depleted HeLa cells. This increase in GS15 level may reflect an attempt to compensate for a tethering defect due to acute Cog8 depletion, as was previously proposed for other v-SNARE proteins (33–35). The differences in the steady-state levels of GS15 between the two mutant cell types could be related to the duration of Cog8 deficiency; acute and short-term deficiency (HeLa cells) may lead to the dispersal of GS15 from the Golgi due to tethering defects, whereas long-term deficiency (patient’s fibroblasts) may lead to its dispersal and eventually degradation.
Figure 4.
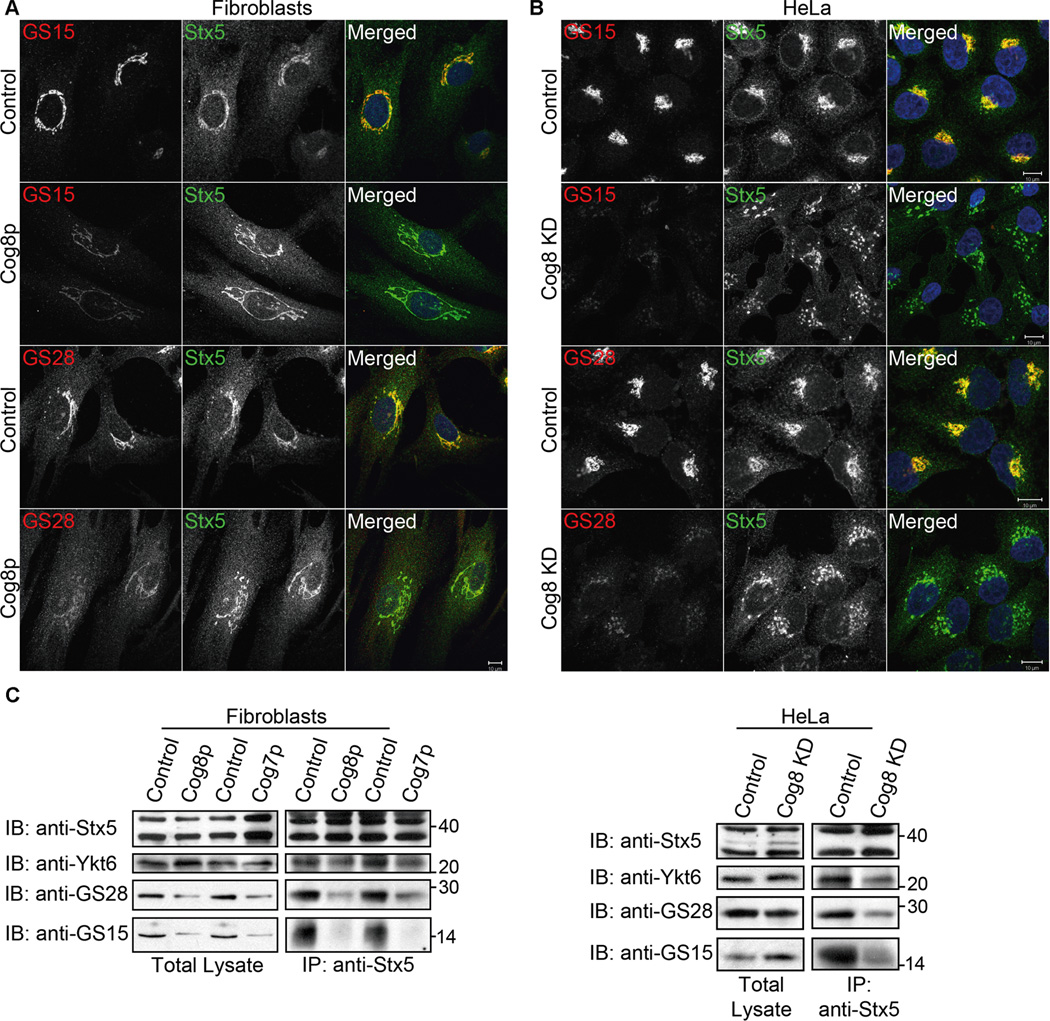
Deficiency of Cog8 affects the Golgi localization of GS28 and GS15 and their assembly into SNARE complex. A, B) The Golgi localization of Stx5 (green) and GS28 (red) or GS15 (red) was determined in Cog8-deficient fibroblasts (A) or Cog8-depleted HeLa cells (B) and the corresponding controls by co-immunostaining and confocal microscopy analysis. As shown, the Golgi localization of GS28 and GS15 was significantly reduced in both Cog8-deficient fibroblasts and HeLa cells. Bars: 10 µm. C) Cog8- and Cog7-deficient fibroblasts obtained from human patients (left panels) or Cog8-depleted HeLa cells (right panels) with their corresponding controls were pretreated with NEM, solubilized and subjected to IP with anti-Stx5 antibody. The presence of the indicated SNARE proteins in the immunocomplexes was determined by WB using the corresponding antibodies.
We next examined the influence of Cog8 deficiency on the assembly of the Stx5 complex. We treated the cells with n-ethylmaleimide (NEM) that inhibits disassembly of SNARE complexes, immunoprecipitated Stx5, and examined whether its interacting SNAREs are present in the same immunocomplex using WB analysis. As shown in Figure 4C, the interactions of Stx5 with either the v-SNARE GS15 or the t-SNAREs Ykt6 and GS28 were significantly reduced in both Cog8-deficient cell types. Importantly, the assembly of the Stx5 SNARE complex was also compromised in CDG-derived Cog7deficient fibroblasts (Figure 4C), suggesting that impaired SNARE complex assembly is a common defect found in COG-associated CDGs.
We next examined the influence of Cog8 deficiency on the assembly of the Stx6 SNARE complex that regulates endosome-to-TGN retrograde trafficking (29). IF analysis revealed that the Golgi localization of the v-SNARE VAMP4 was reduced in Cog8-deficient CDG-derived fibroblasts, concomitant with an increase in its cytosolic staining (Figure 5A). WB analysis demonstrated a 70 ± 9% decrease in VAMP4 protein level in the patient’s cells (Figure 5C) consistent with its weak staining in the Golgi. The three t-SNAREs, Stx6, Stx16 and Vti1a, however, were localized to the Golgi in the patient-derived fibroblasts, but could also be detected in punctate cytosolic structures (Figures 5A and S2A) that failed to colocalize with the early endosomal marker EEA1 (Figure S3). When compared with the CDG-derived cells, Cog8-deficient HeLa cells manifested stronger effects on the distribution of these SNARE proteins; VAMP4 was completely dispersed from the Golgi, although no decrease in its steady-state levels was observed (Figure 5C), and only a residual staining of the three t-SNAREs was detected at the Golgi (Figures 5B and S2B). Despite the differences in the steady-state distributions of the SNARE proteins, the assembly of the Stx6 SNARE complex was impaired in both Cog8-deficient cell types. As shown in Figure 5C, a significant reduction in the interaction between the t-SNARE Stx6 and the v-SNARE VAMP4 and between the t-SNAREs Stx6 and Stx16 was observed in both cell types. Importantly, the assembly of the Stx6 SNARE complex was also impaired in Cog7-deficient fibroblasts derived from CDG patient (Figure 5C) similar to the results obtained for the Stx5 complex (Figure 4C). Altogether, our results demonstrate that the Cog8 subunit is essential for the assembly of both Stx5 and Stx6 SNARE complexes, and that the assembly of these complexes is impaired in CDG patients carrying mutations in different COG genes.
Figure 5.
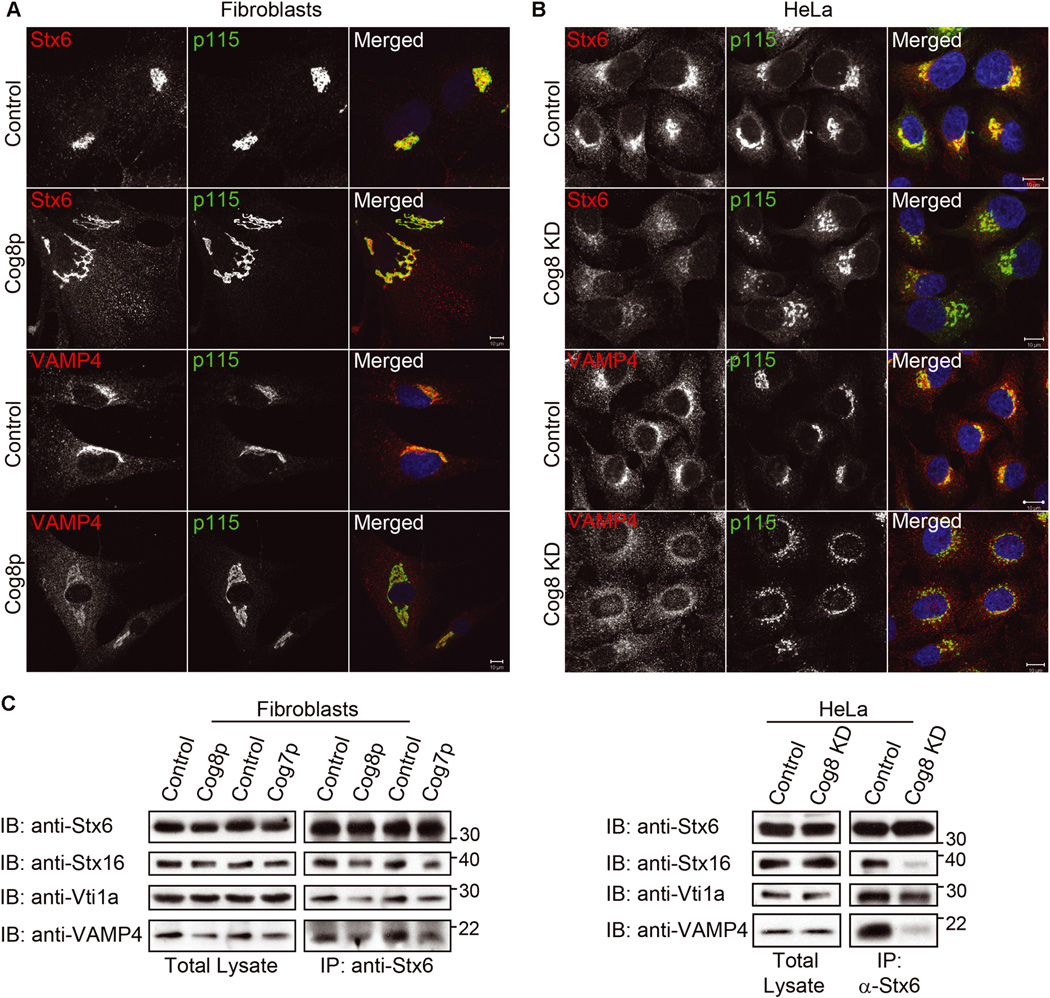
Deficiency of Cog8 affects the subcellular distribution of Stx6 and VAMP4 and impairs their assembly into SNARE complex. A) The Golgi localization of Stx6 or VAMP4 (red) in control or Cog8-deficient fibroblasts was determined by co-immunostaining with the Golgi marker p115 (green) and confocal microscopy analysis. In the patient’s cells, the Golgi localization of VAMP4 was reduced concomitantly with an increase in its cytosolic haze-like staining. In addition, a pool of Stx6 accumulated in punctate structures. Bars: 10 µm. B) HeLa cells were transiently transfected with a control or Cog8 shRNA construct, and 72 h later the cells were fixed and double immunostained with the indicated antibodies. The localization of Stx6 and VAMP4 (red) to the Golgi membranes was determined by co-immunostaining with p115 (green). In Cog8-depleted HeLa cells, VAMP4 was completely dispersed from the Golgi, while Stx6 displayed a weak residual Golgi staining. Bars: 10 µm. C) Cog8- and Cog7-deficient fibroblasts obtained from human patients (left panels) or Cog8-depleted HeLa cells (right panels) with their corresponding controls were pretreated with NEM, solubilized and subjected to IP with anti-Stx6 antibody. The presence of the indicated SNARE proteins in the immunocomplexes was determined by WB using the corresponding antibodies.
Cog8 deficiency influences endosome-to-TGN retrograde transport in HeLa cells and CDG-derived fibroblasts
The effect of Cog8 deficiency on the assembly of Stx5 and Stx6 SNARE complexes suggests that depletion of Cog8 could also affect endosome-to-TGN retrograde transport. To explore this possibility, we first examined the influence of Cog8 depletion on the transport of TGN38 from the plasma membrane to the TGN in HeLa cells using an antibody uptake assay as we previously described (24). TGN38/46 is a TGN resident protein that cycles between the plasma membrane, endosomes and the TGN via the endosome-to-TGN retrograde transport pathway (36). In brief, control and Cog8-depleted HeLa cells were transiently transfected with HA-TGN38, incubated with anti-HA antibodies at 37°C for different time periods, washed, fixed and double immunostained for the TGN marker Golgin 97 and TGN38-HA. As shown in Figure 6A, depletion of the Cog8 subunit substantially inhibited the transport of TGN38-HA to the TGN in HeLa cells. TGN38-HA failed to reach the TGN of Cog8-depleted cells even 2 h following anti-HA uptake (Figure S4), but could be detected at the TGN in 65% of the control cells (n =200) already at 30min following anti-HA antibody uptake. These results suggest that depletion of the Cog8 subunit in HeLa cells markedly attenuates endosome-to-TGN retrograde transport. Consistent with these observations, we found that endogenous TGN38/46 was dispersed from the Golgi of Cog8-depleted HeLa cells (Figure 6B). Yet, it was detected in the Golgi of the patient-derived fibroblasts, but was also visualized in cytosolic haze-like staining that could represent endosome-derived transport intermediates that failed to tether and/or fuse with the TGN (Figure 6C). CI-MPR, which delivers mannose-6- phosphate-labeled lysosomal enzymes from the TGN to the endosomes and then recycles back to the TGN via the endosome-to-TGN retrograde transport, localized mainly to the Golgi in control HeLa cells. Yet, it could hardly be detected in the Golgi of Cog8-depleted HeLa cells (Figure 6B) as well as Cog8-deficient CDG-derived fibroblasts (Figure 6C). These results suggest that the deficiency of the Cog8 subunit in HeLa cells and CDG patients markedly and similarly influenced the recycling of the CI-MPR. Collectively, these results suggest that Cog8 deficiency in either HeLa cells or CDG-derived fibroblasts influences endosome-to-TGN retrograde transport.
Figure 6.
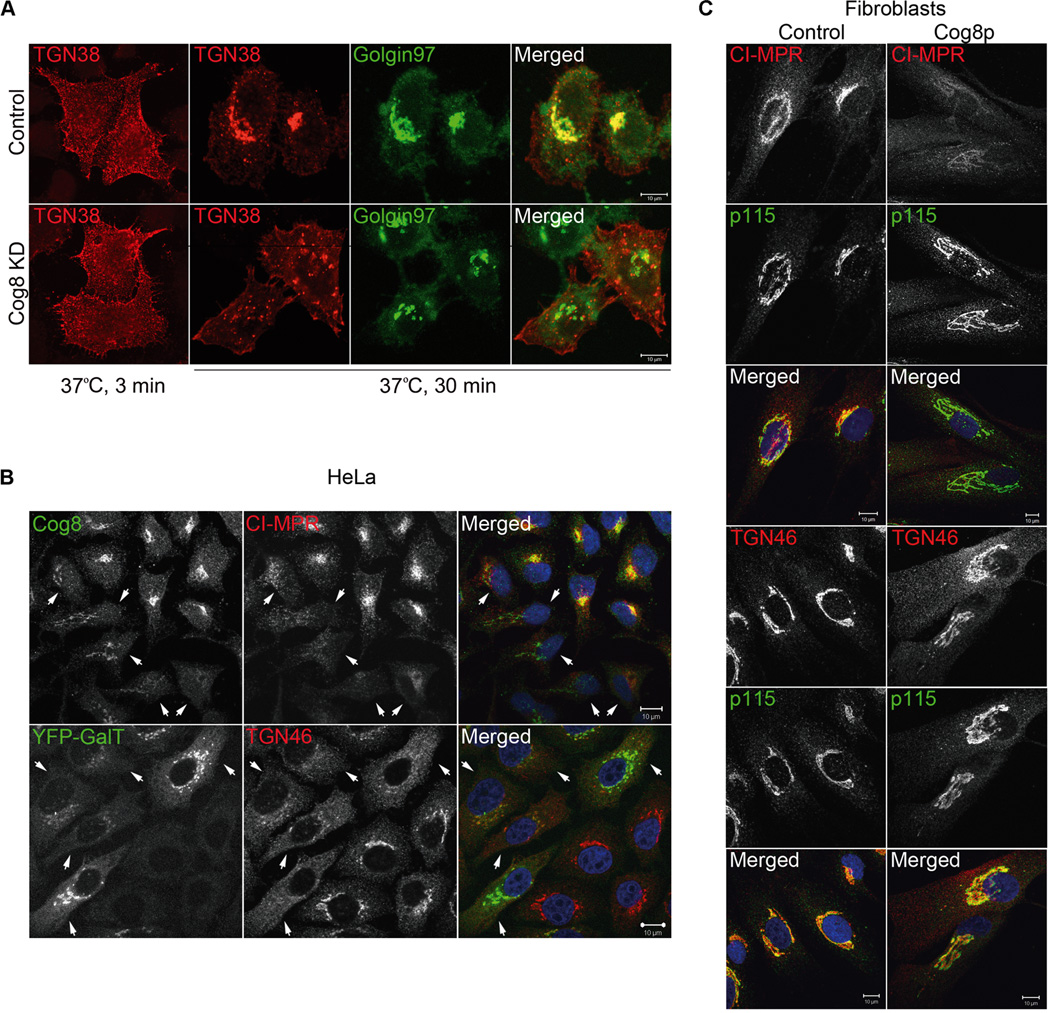
Attenuated endosome-to-TGN retrograde transport in Cog8-deficient cells. A) Endosome-to-TGN transport of HA-tagged TGN38 is attenuated in Cog8-depleted HeLa cells. Control and Cog8-depleted (KD) stable HeLa cell lines expressing TGN38-HA were incubated at 37°C with the anti-HA monoclonal antibody for various time periods. The colocalization of TGN38-HA (red) with the TGN marker Golgin 97 (green) was determined by immunostaining and confocal microscopy analysis. Shown are representative confocal images of control and Cog8-depleted cells at 3 and 30 min of antibody uptake. Bar: 10 µm. B, C) Deficiency of the Cog8 subunit impairs the Golgi localization of CI-MPR and TGN46. B) HeLa cells were transiently transfected with a Cog8 shRNA construct or cotransfected with the Cog8 shRNA construct together with a vector encoding YFP-Gal-T (5:1 molar ratio). Seventy-two hours later the cells were fixed and immunostained with the indicated antibodies. The Cog8-depleted cells (marked by arrowheads) were identified by immunostaining with anti-Cog8 antibody (upper panels) or by the YFP-Gal-T expression (lower panels). As shown, both CI-MPR and TGN46 were completely dispersed from the Golgi in Cog8-depleted HeLa cells. Bars: 10 µm. C) Control and Cog8-deficient fibroblasts obtained from a human patient (Cog8p) were fixed and co-immunostained for CI-MPR or TGN46 (red) and the Golgi marker p115 (green). As shown, the Golgi localization of CI-MPR was abolished and TGN46 was partially dispersed to the cytosol in the patient’s cells. Bars: 10 µm.
Discussion
Here, we showed for the first time that CDG-associated COG mutations impair the assembly of the Golgi Stx5 and Stx6 SNARE complexes in the patient-derived cells (Figures 4 and 5). This is an important observation that emphasizes the dual roles of the COG complex in tethering of transport vesicles to the Golgi membranes and in the assembly of Golgi SNARE complexes. We further show that deficiency of different COG subunits disrupts the integrity of the entire COG complex, but distinctly influences the assembly of COG subcomplexes and their Golgi localization (Figures 2 and 3).
Currently, the mechanism by which the COG is targeted to the Golgi complex is not known, as none of the COG subunits contain a Golgi-targeting motif. Nevertheless, our studies suggest that the Golgi localization of the COG complex is dependent on lobe A subunit(s), as the entire lobe B or its subcomplexes (which were observed in Cog4- or Cog1/Cog6/Cog8-depleted cells, respectively) failed to localize to the Golgi (Figures 2B and 3E). Both Cog1 and Cog4 were previously proposed to confer the Golgi localization of the complex (10,37). However, in CDG-derived Cog8-deficient fibroblasts, Cog2, Cog3 and Cog4, but not Cog1, were detected in the Golgi (Figure 2C) (22), suggesting that Cog1 is not essential for their Golgi localization. On the other hand, the Cog2–Cog3 dimer that was observed in Cog4-depleted cells (Figure 3C) failed to localize to the Golgi, suggesting that Cog4 is essential for the Golgi targeting of the entire complex. Yet, it is unclear how Cog4 mediates the Golgi targeting of the complex; it could be that its multiple interactions with Golgi SNAREs play a role in the Golgi targeting (30,31,38). Another possibility is that its interactions with the Cog2 and Cog3 subunits create a surface that mediates their association with the Golgi membranes. Further experiments should clarify this point.
The detailed analysis of the COG subcomplexes obtained in the different COG-depleted HeLa cells (Figure 3) supports the overall architecture of the complex, which was previously proposed by Ungar et al. and supported by additional studies. Accordingly, the Cog2-3-4 and the Cog5-6-7 trimers are linked via a Cog1–Cog8 dimer (8,11–13). Our results further suggest that the Cog8 subunit is essential for the assembly of the lobe B subcomplex, as depletion of Cog8 abolished the interaction of Cog6 with the Cog5–Cog7 dimer (Figure 3A). Yet, the entire lobe B (Cog5–Cog8) was assembled in Cog4-depleted cells (Figure 3C).
The integrity of the COG complex is not only essential for its Golgi targeting but also for its function in both vesicular tethering and SNARE complex assembly. The mislocalization of Golgi SNAREs, which was observed in both CDG-derived fibroblasts and Cog8-depleted HeLa cells (Figures 4 and 5), likely results from a defect in the tethering of vesicles to the Golgi membranes. This could explain the multiple vesicles accumulated in the vicinity of the Golgi in the EM analysis of Cog8-depleted HeLa cells (Figure 1B), and also the multiple punctate structures containing Golgi SNAREs or TGN proteins, which were observed in CDG-derived fibroblasts and failed to localize to the Golgi, TGN or early endosomes (Figures 5A, 6C, S2A and S3). Vesiculated Golgi membranes were also observed in the EM analysis of an additional Cog8-deficient CDG patient (13).
As for SNARE complex assembly, we have recently identified multiple interactions between COG subunits and the Golgi SNAREs that belong to the Stx5 and Stx6 complexes. These interactions are mediated by subunits of the two lobes: Cog4, Cog6 and Cog7, suggesting that the integrity of the entire COG complex is required for Golgi SNARE complexes assembly (31). Indeed, the assembly of the Stx5 and Stx6 SNARE complexes was impaired in Cog4-, Cog6-and Cog8-deficient HeLa cells (Figures 4 and 5) (24,30,31).
Our previous results also suggest that the COG complex facilitates the formation of fusogenic Golgi SNARE complexes and concomitantly prevents the formation of nonfusogenic Golgi SNARE interactions (31). Hence, it could be that different COG subcomplexes would distinctly influence the assembly of fusogenic and non-fusogenic SNARE subcomplexes on the Golgi membranes. On the other hand, COG subcomplexes that are not localized to the Golgi apparatus might interact with specific vesicles, such as COPI vesicles as COG interacts directly with specific COPI components (γCOP) (4). The subunit composition of the COG subcomplexes would, therefore, affect the interaction with components of the transport machinery (SNAREs and coats), whereas the subcellular localization of the subcomplexes could spatially restrict their effect to the Golgi membranes or possibly to vesicles in the cytosol. Thus, both the subunits composition of the COG subcomplexes and their subcellular localization could exert distinct cellular effects. We propose that these differences might explain the multiple phenotypes of different COG mutations in yeast, mammalian cells and CDG patients.
We predict that deficiency of different COG subunits will affect the assembly of the entire complex, and therefore will exert similar phenotypic characteristics such as glycosylation defects. Yet, the severity of the phenotype will be dependent on the composition of the subcomplexes, their subcellular distribution and impact on SNARE complex formation and function. In addition, different point mutations in COG subunits might impair specific interactions while preserving others leading to variable phenotypes in different CDG patients.
Overall, these studies shed light on the underlying mechanisms that connect CDG pathology with the functions and the integrity of the COG complex.
Materials and Methods
Antibodies, reagents and chemicals
Polyclonal antibodies against Golgin 97, GS15, Stx6, Cog2, Cog3, Cog4, Cog6 and Cog7 were previously described (24,30,39–41). Polyclonal antibodies against Cog1 were a generous gift from Dr M. Krieger (MIT, Cambridge, MA, USA) and polyclonal antibodies against Cog5 and Cog8 were generously provided by D. Ungar (University of York, York, UK). Polyclonal antibodies against Stx16, Stx5 and GS28 were a generous gift from Dr B.L. Tang (National University of Singapore, Singapore, Singapore), Dr S. Somlo (Yale University, New Haven, CT, USA) and Dr Z. Elazar (Weizmann Institute of Science, Rehovot, Israel), respectively. Polyclonal anti-TGN46 antibody was kindly provided by Dr M. Fukuda (Burnham Institute, La Jolla, CA, USA). Polyclonal anti-VAMP4 antibody and monoclonal anti-Stx6 antibody were purchased from Synaptic Systems and BD Biosciences, respectively. Polyclonal anti-Vti1a antibody was purchased from Proteintech Group, Inc. Monoclonal anti-CI-MPR antibody and polyclonal anti-EEA1 antibody were purchased from Abcam. Monoclonal anti-α-tubulin antibody and polyclonal anti-Cog2 antibody (Cat. No. SAB42000584) were purchased from Sigma-Aldrich. Alexa Fluor 488 donkey anti-mouse and anti-rabbit IgGs were purchased from Invitrogen. Cy3 (cyanine 3)-conjugated goat anti-rabbit and goat anti-mouse IgGs were purchased from Jackson ImmunoResearch Laboratories, Inc. Protein Agarose beads were purchased from Repligen Corp and Hoechst 33342 was purchased from Sigma-Aldrich.
DNA constructs
The DNA constructs encoding the Myc-tagged Cog7 and Cog8 subunits have been described previously (2,42).
The mammalian pSUPER-puro vector was used for the expression of shRNAs. Two shRNAs have been used to knockdown the expression of Cog8: shRNA1 and shRNA2 corresponding to nucleotides 1512–1531 (5'-GACCTTGTTCCGTATTTAA-3') and 550–569 (5'-CCTGTGTCCGGAAC AGTTA-3') of the human Cog8 cDNA, respectively. Both constructs yielded similar phenotypes and shRNA1 has been chosen for further analysis shown in the figures in this article. The shRNA constructs targeting the human Cog4 and Cog6 were previously described (24,30).
Cell culture and transfection
Wild-type CHO cells and ldlB cells were generously provided by Dr M. Krieger (MIT) and grown in Ham’s F-12 media supplemented with 5% fetal bovine serum, 100 µg/mL penicillin, 100 µg/mL streptomycin and 2mM L-glutamine.
Control fibroblasts and fibroblasts obtained from CDG patients were grown in low-glucose DMEM containing sodium pyruvate and L-glutamine (Gibco, Cat. No. 31885-023) and supplemented with 10% fetal bovine serum, 100 µg/mL penicillin and 100 µg/mL streptomycin.
HEK293 and HeLa cells were grown in DMEM (Gibco Cat. No. 41965-039) supplemented with 10% fetal bovine serum, 100 µg/mL penicillin and 100 µg/mL streptomycin. The cells were transfected using either the calcium phosphate method or transfection reagent (FuGENE HD; Roche). Stable HeLa cell lines depleted of the Cog8 subunit were established using the pSUPER-puro vector encoding shRNA1 (nucleotides 1512–1531) as previously described for Cog4 (30). Stable Hela cell lines depleted of the Cog6 and Cog4 subunits and a control cell line harboring an empty pSUPER-puro vector have been previously described (24,30). These stable HeLa cell lines were used for the IF and IP assays described in Figures 2B, 3A–C, 4C and 5C as well as for the transport assay described in Figure 6A. Transient transfections with the Cog8, Cog6 and Cog4 shRNA constructs or the pSUPER-puro empty vector were performed for the IB and IF experiments described in Figures 2A, 4B, 5B and 6B. In brief, HeLa cells grown on coverslips or in 90-mm tissue culture dishes were transiently transfected with either the appropriate shRNA construct or the pSUPERpuro vector using FuGENE HD. Twenty-four hours after transfection, the cells were either incubated with regular medium for 72 h and then analyzed by immunofluorescence (Figures 4B, 5B and 6B) or were incubated with 1 µg/mL puromycin for 72 h and then analyzed by SDS-PAGE and WB (Figure 2A).
Immunofluorescence and electron microscopy
For IF analysis, HeLa cells or fibroblasts were grown on coverslips, washed with PBS and fixed in 1% paraformaldehyde in either PBS or KM buffer (10mM 2-(N-morpholino)ethanesulfonic acid, pH6.2, 10mM NaCl, 1.5 mM MgCl2 and 2.5% glycerol) for 20 min at room temperature. Fixed cells were then permeabilized with either 0.1% Triton X-100 or 0.03% saponin (VAMP4 staining) in PBS. Cells were then immunostained at room temperature and analyzed by a confocal laser-scanning microscope (LSM 510; Carl Zeiss) essentially as previously described (43).
Transmission electron microscopy (TEM) analysis was performed essentially as previously described (44).
Cell extracts and immunoprecipitation assays
For the co-IP assays described in Figure 3A–D, cell extracts were prepared by solubilizing stable Hela or CHO cell lines in lysis buffer A (0.5% Triton X-100, 20 mM HEPES, pH 7.5, 100 mM NaCl, 5 mM MgCl2, 1 mM phenylmethylsulfonyl fluoride, 10 µg/mL leupeptin and 10 µg/mL aprotinin) for 30 min on ice. Cell lysates were centrifuged at 15 000× g for 15min at 4°C. Subsequently, supernatants were incubated for 3 h at 4°C with either anti-Cog3, anti-Cog4 or anti-Cog7 antibodies prebound to Protein A-agarose beads. The beads were then washed three times with lysis buffer, boiled in SDS sample buffer and separated by SDS-PAGE. The different COG subunits were detected by WB using the indicated antibodies.
Immunoprecipitation of SNARE complexes (Figures 4C and 5C) was performed after treatment with NEM essentially as previously described (45). HeLa cells or fibroblasts were extracted in lysis buffer B (0.5% Triton X-100, 50 mM Tris pH 7.5, 150 mM NaCl, 5 mM EDTA and protease inhibitors) for 1 h on ice. Following centrifugation, supernatants were incubated for overnight at 4°C with anti-Stx5 or anti-Stx6 antibodies prebound to Protein A-agarose beads. The beads were then washed three times with lysis buffer, boiled in SDS sample buffer and separated by SDS-PAGE. The different SNARE proteins were detected by WB using the indicated antibodies.
Densitometric analysis of immunoblots was performed using the ImageJ software (National Institutes of Health).
Transport assays and quantitation Cog8-depleted cells at each time point. The results represent a mean of TGN38 antibody uptake assay was performed essentially as we previously two independent experiments. Error bars indicate SDs. described (24). For the quantitative analysis of this assay confocal images of arbitrary fields from two independent experiments were acquired for both control and Cog8-depleted HeLa cells (n = 200 cells for each time point) using the LSM 510 software. The images were scored visually, and colocalization between the TGN marker Golgin 97 (green) and TGN38 (red) in the Golgi region was displayed as yellow in the merged image (Figure 6A). Cells with a yellow signal in the Golgi were counted as positive. The data shown are mean values ± SD.
Supplementary Material
Figure S1: Downregulation of the Cog8 subunit by shRNA in HeLa cells. A) HeLa cells were transiently transfected with either the pSUPERpuro vector (control) or with pSUPER-puro vectors encoding two different shRNAs for the human Cog8, shRNA1 and shRNA2, as indicated in Materials and Methods. Twenty-four hours after transfection, the cells were incubated with 1 µg/mL puromycin for 72 h and then analyzed for Cog8 or Cog7 expression by WB with the corresponding antibodies. As shown, both shRNAs efficiently downregulated Cog8 expression, without affecting the steady-state levels of Cog7. B) Specific downregulation of Cog8 by shRNA1. HeLa cells were cotransfected with the Cog8 shRNA1 construct or the pSUPER-puro vector as a control together with an expression vector encoding either the Myc-tagged Cog6 or Cog8. The expression of the Myc-tagged COG subunits was determined by WB with the anti-Myc antibody. As shown, Cog8 shRNA efficiently and selectively depleted the expression of the Cog8 subunit.
{kind=link}
Figure S2: Deficiency of Cog8 affects the subcellular distribution of Stx16 and Vti1a. A) The Golgi localization of Stx16 or Vti1a (red) in control or Cog8-deficient fibroblasts was determined by co-immunostaining with the Golgi marker p115 (green) and confocal microscopy analysis. In the patient’s cells, the Golgi localization of Stx16 and Vti1a was accompanied by cytosolic haze-like staining, which could represent transport intermediates that failed to fuse with the Golgi. Bars: 10 µm. B) HeLa cells were transiently transfected with a control or Cog8 shRNA construct, and 72 h later the cells were fixed and double immunostained with the indicated antibodies. The localization of Stx16 and Vti1a (red) to the Golgi membranes was determined by co-immunostaining with p115 (green). In Cog8-depleted HeLa cells, Stx16 and Vti1a were significantly dispersed from the Golgi and only residual staining was detected at the Golgi. Bars: 10 µm.
{kind=link}
Figure S3: Stx6 is not localized to early endosomes in Cog8-deficient fibroblasts. Cog8-deficient fibroblasts obtained from a human patient were fixed and double immunostained with anti-Stx6 (red) and anti-EEA1 (green) antibodies. As shown, no significant colocalization between Stx6 and EEA1 was detected. Higher magnification is shown in the zoom. Bar: 10 µm, zoom: 5 µm.
{kind=link}
Figure S4: The Golgi localization of TGN38-HA at different time points of antibody uptake in control and Cog8-depleted HeLa cells. The colocalization (yellow) between TGN38-HA (red) and Golgin 97 (green) was used to monitor the Golgi localization of TGN38-HA following antibody uptake. The localization of TGN38-HA was analyzed for ~200 control or Cog8-depleted cells at each time point. The results represent a mean of two independent experiments. Error bars indicate SDs.
{kind=link}
Acknowledgments
S. L. is the incumbent of the Joyce and Ben B. Eisenberg Chair of Molecular Biology and Cancer Research. This work was supported by the Binational Science Foundation (BSF), Grant No. 2011404 and by the Georges Lustgarten Cancer Research Fund.
Footnotes
The authors declare that there are no conflicts of interest.
References
- 1.Chatterton JE, Hirsch D, Schwartz JJ, Bickel PE, Rosenberg RD, Lodish HF, Krieger M. Expression cloning of LDLB, a gene essential for normal Golgi function and assembly of the ldlCp complex. Proc Natl Acad Sci U S A. 1999;96:915–920. doi: 10.1073/pnas.96.3.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Loh E, Hong W. Sec34 is implicated in traffic from the endoplasmic reticulum to the Golgi and exists in a complex with GTC-90 and ldlBp. J Biol Chem. 2002;277:21955–21961. doi: 10.1074/jbc.M202326200. [DOI] [PubMed] [Google Scholar]
- 3.Podos SD, Reddy P, Ashkenas J, Krieger M. LDLC encodes a brefeldin A-sensitive, peripheral Golgi protein required for normal Golgi function. J Cell Biol. 1994;127:679–691. doi: 10.1083/jcb.127.3.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suvorova ES, Duden R, Lupashin VV. The Sec34/Sec35p complex, a Ypt1p effector required for retrograde intra-Golgi trafficking, interacts with Golgi SNAREs and COPI vesicle coat proteins. J Cell Biol. 2002;157:631–643. doi: 10.1083/jcb.200111081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ungar D, Oka T, Brittle EE, Vasile E, Lupashin VV, Chatterton JE, Heuser JE, Krieger M, Waters MG. Characterization of a mammalian Golgi-localized protein complex, COG, that is required for normal Golgi morphology and function. J Cell Biol. 2002;157:405–415. doi: 10.1083/jcb.200202016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.VanRheenen SM, Cao X, Sapperstein SK, Chiang EC, Lupashin VV, Barlowe C, Waters MG. Sec34p, a protein required for vesicle tethering to the yeast Golgi apparatus, is in a complex with Sec35p. J Cell Biol. 1999;147:729–742. doi: 10.1083/jcb.147.4.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Whyte JR, Munro S. The Sec34/35 Golgi transport complex is related to the exocyst, defining a family of complexes involved in multiple steps of membrane traffic. Dev Cell. 2001;1:527–537. doi: 10.1016/s1534-5807(01)00063-6. [DOI] [PubMed] [Google Scholar]
- 8.Ungar D, Oka T, Vasile E, Krieger M, Hughson FM. Subunit architecture of the conserved oligomeric Golgi complex. J Biol Chem. 2005;280:32729–32735. doi: 10.1074/jbc.M504590200. [DOI] [PubMed] [Google Scholar]
- 9.Fotso P, Koryakina Y, Pavliv O, Tsiomenko AB, Lupashin VV. Cog1p plays a central role in the organization of the yeast conserved oligomeric Golgi complex. J Biol Chem. 2005;280:27613–27623. doi: 10.1074/jbc.M504597200. [DOI] [PubMed] [Google Scholar]
- 10.Oka T, Vasile E, Penman M, Novina CD, Dykxhoorn DM, Ungar D, Hughson FM, Krieger M. Genetic analysis of the subunit organization and function of the conserved oligomeric golgi (COG) complex: studies of COG5-and COG7-deficient mammalian cells. J Biol Chem. 2005;280:32736–32745. doi: 10.1074/jbc.M505558200. [DOI] [PubMed] [Google Scholar]
- 11.Foulquier F, Vasile E, Schollen E, Callewaert N, Raemaekers T, Quelhas D, Jaeken J, Mills P, Winchester B, Krieger M, Annaert W, Matthijs G. Conserved oligomeric Golgi complex subunit 1 deficiency reveals a previously uncharacterized congenital disorder of glycosylation type II. Proc Natl Acad Sci U S A. 2006;103:3764–3769. doi: 10.1073/pnas.0507685103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Foulquier F, Ungar D, Reynders E, Zeevaert R, Mills P, Garcia-Silva MT, Briones P, Winchester B, Morelle W, Krieger M, Annaert W, Matthijs G. A new inborn error of glycosylation due to a Cog8 deficiency reveals a critical role for the Cog1–Cog8 interaction in COG complex formation. Hum Mol Genet. 2007;16:717–730. doi: 10.1093/hmg/ddl476. [DOI] [PubMed] [Google Scholar]
- 13.Reynders E, Foulquier F, Leao Teles E, Quelhas D, Morelle W, Rabouille C, Annaert W, Matthijs G. Golgi function and dysfunction in the first COG4-deficient CDG type II patient. Hum Mol Genet. 2009;18:3244–3256. doi: 10.1093/hmg/ddp262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shestakova A, Zolov S, Lupashin V. COG complex-mediated recycling of Golgi glycosyltransferases is essential for normal protein glycosylation. Traffic. 2006;7:191–204. doi: 10.1111/j.1600-0854.2005.00376.x. [DOI] [PubMed] [Google Scholar]
- 15.Freeze HH, Ng BG. Golgi glycosylation and human inherited diseases. Cold Spring Harb Perspect Biol. 2011;3:a005371. doi: 10.1101/cshperspect.a005371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosnoblet C, Peanne R, Legrand D, Foulquier F. Glycosylation disorders of membrane trafficking. Glycoconj J. 2013;30:23–31. doi: 10.1007/s10719-012-9389-y. [DOI] [PubMed] [Google Scholar]
- 17.Foulquier F. COG defects, birth and rise! Biochim Biophys Acta. 2009;1792:896–902. doi: 10.1016/j.bbadis.2008.10.020. [DOI] [PubMed] [Google Scholar]
- 18.Zeevaert R, Foulquier F, Jaeken J, Matthijs G. Deficiencies in subunits of the Conserved Oligomeric Golgi (COG) complex define a novel group of Congenital Disorders of Glycosylation. Mol Genet Metab. 2008;93:15–21. doi: 10.1016/j.ymgme.2007.08.118. [DOI] [PubMed] [Google Scholar]
- 19.Reynders E, Foulquier F, Annaert W, Matthijs G. How Golgi glycosylation meets and needs trafficking: the case of the COG complex. Glycobiology. 2011;21:853–863. doi: 10.1093/glycob/cwq179. [DOI] [PubMed] [Google Scholar]
- 20.Miller VJ, Ungar D. Re’COG’nition at the Golgi. Traffic. 2012;13:891–897. doi: 10.1111/j.1600-0854.2012.01338.x. [DOI] [PubMed] [Google Scholar]
- 21.Wu X, Steet RA, Bohorov O, Bakker J, Newell J, Krieger M, Spaapen L, Kornfeld S, Freeze HH. Mutation of the COG complex subunit gene COG7 causes a lethal congenital disorder. Nat Med. 2004;10:518–523. doi: 10.1038/nm1041. [DOI] [PubMed] [Google Scholar]
- 22.Kranz C, Ng BG, Sun L, Sharma V, Eklund EA, Miura Y, Ungar D, Lupashin V, Winkel RD, Cipollo JF, Costello CE, Loh E, Hong W, Freeze HH. COG8 deficiency causes new congenital disorder of glycosylation type IIh. Hum Mol Genet. 2007;16:731–741. doi: 10.1093/hmg/ddm028. [DOI] [PubMed] [Google Scholar]
- 23.Ungar D, Oka T, Krieger M, Hughson FM. Retrograde transport on the COG railway. Trends Cell Biol. 2006;16:113–120. doi: 10.1016/j.tcb.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 24.Laufman O, Hong W, Lev S. The COG complex interacts directly with Syntaxin 6 and positively regulates endosome-to-TGN retrograde transport. J Cell Biol. 2011;194:459–472. doi: 10.1083/jcb.201102045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zolov SN, Lupashin VV. Cog3p depletion blocks vesicle-mediated Golgi retrograde trafficking in HeLa cells. J Cell Biol. 2005;168:747–759. doi: 10.1083/jcb.200412003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oka T, Ungar D, Hughson FM, Krieger M. The COG and COPI complexes interact to control the abundance of GEARs, a subset of Golgi integral membrane proteins. Mol Biol Cell. 2004;15:2423–2435. doi: 10.1091/mbc.E03-09-0699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peanne R, Legrand D, Duvet S, Mir AM, Matthijs G, Rohrer J, Foulquier F. Differential effects of lobe A and lobe B of the Conserved Oligomeric Golgi complex on the stability of {beta}1,4-galactosyltransferase 1 and {alpha}2,6-sialyltransferase 1. Glycobiology. 2011;21:864–876. doi: 10.1093/glycob/cwq176. [DOI] [PubMed] [Google Scholar]
- 28.Xu Y, Martin S, James DE, Hong W. GS15 forms a SNARE complex with syntaxin 5, GS28, and Ykt6 and is implicated in traffic in the early cisternae of the Golgi apparatus. Mol Biol Cell. 2002;13:3493–3507. doi: 10.1091/mbc.E02-01-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mallard F, Tang BL, Galli T, Tenza D, Saint-Pol A, Yue X, Antony C, Hong W, Goud B, Johannes L. Early/recycling endosomes-to-TGN transport involves two SNARE complexes and a Rab6 isoform. J Cell Biol. 2002;156:653–664. doi: 10.1083/jcb.200110081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laufman O, Kedan A, Hong W, Lev S. Direct interaction between the COG complex and the SM protein, Sly1, is required for Golgi SNARE pairing. EMBO J. 2009;28:2006–2017. doi: 10.1038/emboj.2009.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laufman O, Hong W, Lev S. The COG complex interacts with multiple Golgi SNAREs and enhances fusogenic SNARE complexes assembly. J Cell Sci. 2013 doi: 10.1242/jcs.122101. [DOI] [PubMed] [Google Scholar]
- 32.Steet R, Kornfeld S. COG-7-deficient human fibroblasts exhibit altered recycling of Golgi proteins. Mol Biol Cell. 2006;17:2312–2321. doi: 10.1091/mbc.E05-08-0822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pfeffer SR. Transport vesicle docking: SNAREs and associates. Annu Rev Cell Dev Biol. 1996;12:441–461. doi: 10.1146/annurev.cellbio.12.1.441. [DOI] [PubMed] [Google Scholar]
- 34.Sapperstein SK, Lupashin VV, Schmitt HD, Waters MG. Assembly of the ER to Golgi SNARE complex requires Uso1p. J Cell Biol. 1996;132:755–767. doi: 10.1083/jcb.132.5.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.VanRheenen SM, Cao X, Lupashin VV, Barlowe C, Waters MG. Sec35p, a novel peripheral membrane protein, is required for ER to Golgi vesicle docking. J Cell Biol. 1998;141:1107–1119. doi: 10.1083/jcb.141.5.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ghosh RN, Mallet WG, Soe TT, McGraw TE, Maxfield FR. An endocytosed TGN38 chimeric protein is delivered to the TGN after trafficking through the endocytic recycling compartment in CHO cells. J Cell Biol. 1998;142:923–936. doi: 10.1083/jcb.142.4.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Willett R, Kudlyk T, Pokrovskaya I, Schonherr R, Ungar D, Duden R, Lupashin V. COG complexes form spatial landmarks for distinct SNARE complexes. Nat Commun. 2013;4:1553. doi: 10.1038/ncomms2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shestakova A, Suvorova E, Pavliv O, Khaidakova G, Lupashin V. Interaction of the conserved oligomeric Golgi complex with t-SNARE Syntaxin5a/Sed5 enhances intra-Golgi SNARE complex stability. J Cell Biol. 2007;179:1179–1192. doi: 10.1083/jcb.200705145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu Y, Wong SH, Zhang T, Subramaniam VN, Hong W. GS15, a 15 kilodalton Golgi soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) homologous to rbet1. J Biol Chem. 1997;272:20162–20166. doi: 10.1074/jbc.272.32.20162. [DOI] [PubMed] [Google Scholar]
- 40.Zhang T, Hong W. Ykt6 forms a SNARE complex with syntaxin 5, GS28, and Bet1 and participates in a late stage in endoplasmic reticulum-Golgi transport. J Biol Chem. 2001;276:27480–27487. doi: 10.1074/jbc.M102786200. [DOI] [PubMed] [Google Scholar]
- 41.Lu L, Tai G, Hong W. Autoantigen Golgin-97, an effector of Arl1 GTPase, participates in traffic from the endosome to the trans-golgi network. Mol Biol Cell. 2004;15:4426–4443. doi: 10.1091/mbc.E03-12-0872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Loh E, Hong W. The binary interacting network of the conserved oligomeric Golgi tethering complex. J Biol Chem. 2004;279:24640–24648. doi: 10.1074/jbc.M400662200. [DOI] [PubMed] [Google Scholar]
- 43.Litvak V, Tian D, Carmon S, Lev S. Nir2, a human homolog of Drosophila melanogaster retinal degeneration B protein, is essential for cytokinesis. Mol Cell Biol. 2002;22:5064–5075. doi: 10.1128/MCB.22.14.5064-5075.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peretti D, Dahan N, Shimoni E, Hischberg K, Lev S. Coordinated lipid transfer between the endoplasmic reticulum and the Golgi complex requires the VAP proteins and is essential for Golgi-mediated transport. Mol Biol Cell. 2008;19:3871–3884. doi: 10.1091/mbc.E08-05-0498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perez-Victoria FJ, Bonifacino JS. Dual roles of the mammalian GARP complex in tethering and SNARE complex assembly at the trans-golgi network. Mol Cell Biol. 2009;29:5251–5263. doi: 10.1128/MCB.00495-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Downregulation of the Cog8 subunit by shRNA in HeLa cells. A) HeLa cells were transiently transfected with either the pSUPERpuro vector (control) or with pSUPER-puro vectors encoding two different shRNAs for the human Cog8, shRNA1 and shRNA2, as indicated in Materials and Methods. Twenty-four hours after transfection, the cells were incubated with 1 µg/mL puromycin for 72 h and then analyzed for Cog8 or Cog7 expression by WB with the corresponding antibodies. As shown, both shRNAs efficiently downregulated Cog8 expression, without affecting the steady-state levels of Cog7. B) Specific downregulation of Cog8 by shRNA1. HeLa cells were cotransfected with the Cog8 shRNA1 construct or the pSUPER-puro vector as a control together with an expression vector encoding either the Myc-tagged Cog6 or Cog8. The expression of the Myc-tagged COG subunits was determined by WB with the anti-Myc antibody. As shown, Cog8 shRNA efficiently and selectively depleted the expression of the Cog8 subunit.
Figure S2: Deficiency of Cog8 affects the subcellular distribution of Stx16 and Vti1a. A) The Golgi localization of Stx16 or Vti1a (red) in control or Cog8-deficient fibroblasts was determined by co-immunostaining with the Golgi marker p115 (green) and confocal microscopy analysis. In the patient’s cells, the Golgi localization of Stx16 and Vti1a was accompanied by cytosolic haze-like staining, which could represent transport intermediates that failed to fuse with the Golgi. Bars: 10 µm. B) HeLa cells were transiently transfected with a control or Cog8 shRNA construct, and 72 h later the cells were fixed and double immunostained with the indicated antibodies. The localization of Stx16 and Vti1a (red) to the Golgi membranes was determined by co-immunostaining with p115 (green). In Cog8-depleted HeLa cells, Stx16 and Vti1a were significantly dispersed from the Golgi and only residual staining was detected at the Golgi. Bars: 10 µm.
Figure S3: Stx6 is not localized to early endosomes in Cog8-deficient fibroblasts. Cog8-deficient fibroblasts obtained from a human patient were fixed and double immunostained with anti-Stx6 (red) and anti-EEA1 (green) antibodies. As shown, no significant colocalization between Stx6 and EEA1 was detected. Higher magnification is shown in the zoom. Bar: 10 µm, zoom: 5 µm.
Figure S4: The Golgi localization of TGN38-HA at different time points of antibody uptake in control and Cog8-depleted HeLa cells. The colocalization (yellow) between TGN38-HA (red) and Golgin 97 (green) was used to monitor the Golgi localization of TGN38-HA following antibody uptake. The localization of TGN38-HA was analyzed for ~200 control or Cog8-depleted cells at each time point. The results represent a mean of two independent experiments. Error bars indicate SDs.