

Abstract
Most diseases result in metabolic changes. In many cases, these changes play a causative role in disease progression. By identifying pathological metabolic changes, metabolomics can point to potential new sites for therapeutic intervention. Particularly promising enzymatic targets are those that carry increased flux in the disease state. Definitive assessment of flux requires the use of isotope tracers. Here we present techniques for finding new drug targets using metabolomics and isotope tracers. The utility of these methods is exemplified in the study of three different viral pathogens. For influenza A and herpes simplex virus, metabolomic analysis of infected versus mock-infected cells revealed dramatic concentration changes around the current antiviral target enzymes. Similar analysis of human-cytomegalovirus-infected cells, however, found the greatest changes in a region of metabolism unrelated to the current antiviral target. Instead, it pointed to the tricarboxylic acid (TCA) cycle and its efflux to feed fatty acid biosynthesis as a potential preferred target. Isotope tracer studies revealed that cytomegalovirus greatly increases flux through the key fatty acid metabolic enzyme acetyl-coenzyme A carboxylase. Inhibition of this enzyme blocks human cytomegalovirus replication. Examples where metabolomics has contributed to identification of anticancer drug targets are also discussed. Eventual proof of the value of metabolomics as a drug target discovery strategy will be successful clinical development of therapeutics hitting these new targets.
Metabolism was the first major biological process to be dissected into chemical reactions, and this knowledge provided the foundation for development of biochemistry into a thriving discipline. These initial “glory days” of metabolism were extraordinary fruitful. They gave rise to the metabolic network map and to important general concepts in biological regulation, such as feedback inhibition. Moreover, they led to practical inventions, such as the thymidylate synthase inhibitor 5-fluorouracil. This paragon of rational drug design, which remains among the most important weapons in the fight against cancer, differs from the naturally occurring metabolite uracil by only a single atom change (Fig. 1).
Figure 1.

Chemical structure of uracil and its anticancer analog, 5-fluorouracil.
With the discovery of the structure of DNA and the advent of molecular biology, however, metabolism slowly became viewed as passé. The resulting “dark ages” of metabolism research persisted for several decades. Remarkably, neither the metabolic disease epidemic, nor the successes of antimetabolites—including 3-hydroxy-3-methylglutaryl (HMG)-coenzyme A (CoA) reductase inhibitors (“statins”), the best-selling drugs of all time—were sufficient to rekindle broad interest in metabolism. Instead, the “omic” era of systems biology was required to launch a renaissance of metabolic investigation. Metabolism has proven to be uniquely amenable to predictive genome-scale quantitative modeling (Ibarra et al. 2002), and the metabolome, as measured by nuclear magnetic resonance (NMR) or mass spectrometry, has emerged as an important signature of biochemical activity (Fiehn 2002).
The initial utilization of metabolomic technology in the medical arena focused on biomarkers and diagnostics. Such efforts were grounded in the proven diagnostic value of certain metabolic measurements, for example, of serum glucose and cholesterol. Building on the importance of cholesterol as a marker of cardiovascular disease, metabolomics has been applied to look for signatures of heart disease. For example, multivariate statistical analysis of a proton NMR spectrum of serum metabolites was used to assess patients’ risk of coronary artery disease (Brindle et al. 2002). Typical of NMR-based metabolomics, individual compounds were not quantified; nevertheless, NMR signals associated with lipid fatty acid side chains and choline head groups were sufficient for accurate differentiation of normal individuals from those with triple vessel disease. In another study, plasma samples were taken from patients with inducible myocardial ischemia and targeted metabolites quantified using liquid chromatography–mass spectrometry (LC-MS; Sabatine et al. 2005). This allowed identification of specific metabolites that rose and fell in the pathological state. Citrate and other tricarboxylic acid (TCA) cycle metabolites were found to be depleted following exercise in patients with cardiac ischemia, consistent with prior evidence that ischemia reduces efflux of citrate from cardiac tissue (Panchal et al. 2001).
In the pharmaceutical industry, a major initial application of metabolomics was in toxicology. A consortium of pharmaceutical companies attempted to find biomarkers for drug-induced renal and hepatic toxicity (Lindon et al. 2003). Such efforts have so far failed to markedly alter the standard drug development pathway. Similarly, intensive efforts to find serum or urine hallmarks of cancer metabolism have yet to yield reliable biomarkers (Jentzmik et al. 2010). In contrast, there has recently been success in applying metabolomics to find early indicators of type II diabetes. Two groups have shown that elevations in branched chain and aromatic amino acids precede alterations in circulating glucose levels (Newgard et al. 2009; Wang et al. 2011). These seminal findings are now poised for translation into clinical prac-tice.
Interestingly, given the importance of metabolic inhibitors as therapeutics, there was not extensive early application of metabolomics for drug target identification or validation. This reflects in part the field’s initial development around proton NMR, which generates rich spectra from which quantitation of individual metabolites is often difficult owing to signal overlap. Such compound-specific quantitation is required to relate metabolomic data to metabolic maps and thereby infer enzymatic reactions that might constitute therapeutic targets. With the rise of MS-based metabolomics, this technical barrier was removed. Nevertheless, a biomarker-centric mentality was already embedded in the field. Slowly, this mentality is shifting, driven by the recognition that metabolic enzymes are preferred drug targets. Roughly 20% of currently prescribed, FDA-approved drugs target enzymes. As the case of 5-fluorouracil illustrates, often enzyme inhibitors are analogs of the enzyme’s naturally occurring substrates or products. When this is not the case, enzyme-active sites nevertheless are poised for inhibition with small-molecule drugs, and allosteric sites can be used for either enzyme activation or inhibition.
Here, we describe metabolomic technologies suitable for identifying enzymes that are potential targets for disease treatment. We focus on LC-MS and discuss methods for quantitating known metabolites and for identifying novel ones. We present methods for flux measurement, with an emphasis on enzymes that catalyze up-regulated fluxes as promising targets for disease treatment. Using examples from viral infections, we demonstrate the utility of these methods for drug target discovery. Similar examples from oncology are also discussed.
QUANTITATION OF KNOWN METABOLITES
Metabolomic assessment of a disease state typically begins with quantifying known metabolites. Although it is possible for metabolic activity to change without marked alterations in metabolite levels—for example, very small reactant and product concentration changes can markedly alter net flux through reactions near equilibrium—in most cases, concentration changes highlight impacted reactions. For example, nitrogen availability in microbes influences most strongly glutamine and α-ketoglutarate, metabolites that are directly involved in ammonia assimilation and that control nitrogen response regulatory proteins (Brauer et al. 2006; Yuan et al. 2006, 2009).
The most important technique for known metabolite quantitation is MS. Typically, a sample is separated chromatographically, either by gas chromatography (GC) or LC, with the column output directed to MS analysis. A targeted set of compounds, for which standards are available and thus chromatographic retention times can be experimentally confirmed, can be quantified using either technique. Both GC-MS and LC-MS can quantitate a substantial fraction of the metabolome, and the two together can measure more metabolites than either alone (Buscher et al. 2009). Because LC-MS can measure a broad spectrum of the metabolome without derivatization, it is particularly convenient.
LC separation is made more complex by the physical chemical diversity of the metabolome. LC-MS analysis of pharmaceuticals typically uses reversed-phase chromatography, that is, a hydrophobic column. Reversed-phase methods provide good separation power and reproducibility; however, many cellular metabolites are very hydrophilic and do not retain in reversed phase. One alternative is hydrophilic interaction chromatography (Bajad et al. 2006; Pesek et al. 2009). Another is addition of an ion-pairing agent to increase reversed-phase retention of polar metabolites. Our and several other laboratories use tributylamine as an ion-pairing agent to enhance separation of anionic compounds (Luo et al. 2007; Buescher et al. 2010; Lu et al. 2010). We currently generate most of our data using this approach. For amine-containing metabolites, including amino acids, reversed-phase retention and negative mode ionization can be further enhanced by carbobenzyloxy derivatization (Crutchfield et al. 2010).
In terms of MS, for known compound quantitation there are two basic choices. One is a triple quadrupole mass spectrometer operating in multiple reaction monitoring mode. This enables quantitation of a predetermined list of targeted compounds. Specificity is provided by two mass spectrometry steps (MS/MS). Alternatively, one can rely on a single step of high-resolution MS, for example, on a time-of-flight or stand-alone orbitrap instrument. In this case, specificity is provided by the mass-resolving power. Whereas a list of known compounds and their LC retention times is required for metabolite identification, the mass spectrometer operates in full-scan mode, detecting all incoming ions. Thus, the same data can also be used to find unexpected or novel metabolites. Figure 2 shows exemplary ion-specific chromatograms that result from such analysis.
Figure 2.

Representative raw chromatograms produced by LC-MS-based metabolomics. A metabolite extract was analyzed by LC–high-resolution MS in negative ion mode on a stand-alone orbitrap instrument. Each compound is identified by both its retention time and exact mass. (Reprinted, with permission, from Lu et al. 2010; © American Chemical Society.)
IDENTIFICATION OF UNEXPECTED OR NOVEL COMPOUNDS
Novel metabolites are particularly interesting from a drug discovery perspective. Enzymes and metabolites that are essential for survival and growth of common eukaryotic cell types have almost all been identified, for example, via knockout screens in yeast. In contrast, those that play a more peripheral role in normal physiology, or that exist only in special cell types, may remain to be discovered. Even when such metabolites have been reported in the literature, synthetic standards may not be available. Accumulation of such species in a disease state may point to the best enzymes to target therapeutically, because disease specificity may be greater than for better-known enzymes.
Either MS or NMR can find unexpected metabolites, with MS preferred for low-abundance compounds and NMR for those that lack ionizable groups. With either technique, the initial discovery is typically a peak whose abundance is altered in the diseased state. For peaks found by high-resolution LC-MS, the exact mass is immediately known. This mass yields a list of potential chemical formulae, a first step toward identification. Additional information can come from natural isotope abundances (Kind and Fiehn 2006), isotope-labeling studies (Hegeman et al. 2007; Clasquin et al. 2011), and MS/MS analysis. Together, this information is often sufficient to “guess” the structure. Correct identification is then confirmed by synthesis of the candidate compound and demonstration that the synthetic compound’s MS/MS spectrum and chromatographic retention time match those of the endogenous moiety.
FLUX MEASUREMENT
To understand comprehensively metabolic activity, in addition to metabolite concentrations, it is also essential to quantitate metabolic fluxes. Fluxes are the most direct metric of enzyme activity in living systems and, as such, are valuable for identifying enzymatic drug targets. Consider, for example, a compound whose concentration rises in a disease state. This may reflect impaired consumption or increased production. In the latter case of “high flux” targeting, the producing enzyme is a logical disease intervention.
Flux measurement in disease models has thus far been limited, primarily because fluxes are hard to measure. They are not physical entities that can be measured in an NMR machine or a mass spectrometer. Instead, they must be inferred based on nutrient uptake rates, waste output rates, and isotope tracer studies. For cultured cells, we have pioneered the approach of kinetic flux profiling for quantitating metabolic flux. Cells are switched from unlabeled media to otherwise identical media containing isotope-labeled nutrients. This results in labeling of downstream metabolites, with the labeling rates (as measured by LC-MS or NMR) directly proportional to flux through the metabolic pathway (Yuan et al. 2006; Munger et al. 2008). This approach provides information on metabolic flows, including in non-steady-state conditions such as transient nutrient deprivation or drug treatment (Yuan and Rabinowitz 2007; Kwon et al. 2008; Yuan et al. 2008). Different labeled substrates can trace diverse aspects of metabolism, for example, 13C-glutamine probes TCA cycle turning and efflux, whereas 15N-glutamine probes amination steps in nucleotide and amino acid synthesis.
Kinetic flux profiling is most useful in combination with other flux analysis methods. For example, glucose, glutamine, and oxygen uptake rates and associated waste excretion provide important constraints on intracellular metabolic activity. Steady-state isotope-labeling patterns, after feeding a mixture of labeled and unlabeled carbon, can be used to infer metabolic flux ratios (Sauer 2006). Such ratios are taken at points of metabolic convergence. For example, pyruvate is the point of convergence between glycolytic efflux via pyruvate kinase and TCA efflux via malic enzyme. The extent to which labeling of pyurvate resembles phosphoenolpyruvate versus malate reflects the relative fluxes by these two routes.
For systems close to steady state, all of these forms of data can be integrated within a flux-balanced metabolic model (Duarte et al. 2007; Yizhak et al. 2010). Global search algorithms are capable of computationally identifying the full range of fluxes consistent with the available data, given its confidence limits. Enzymes whose flux is altered in disease reflect potential drug targets.
Because many important diseases lack reliable cell culture models, an important challenge is development of methods for analyzing metabolic fluxes in vivo. In principle, both kinetic flux profiling and steady-state approaches can be applied, for example, after infusing a labeled nutrient into a rodent or human. So far, however, such studies have been used only to probe a few specific fluxes (Yang et al. 1996; Panchal et al. 2000; Beger et al. 2009; Fan et al. 2009; Castro-Perez et al. 2011), not for systems-level flux quantitation.
FINDING ANTIVIRAL TARGETS THROUGH METABOLOMICS
Viral infections are not canonically considered metabolic diseases. Nevertheless, viral replication involves substantial metabolic demands: synthesis of amino acids to build the viral proteome, nucleotides for the viral genome, carbohydrates for protein glycosylation, and energy to power all of these processes. For enveloped viruses, which include the majority of important human viral pathogens, components of the lipid envelope may also need to be synthesized. Consistent with viral replication involving substantial metabolic demands, nucleoside analog antimetabolites are the most important class of antiviral agents. Although it is nevertheless possible that viruses might be nothing more than metabolic leaches, syphoning off the metabolites that they need without overriding normal host cell activity, we hypothesized that viruses would actively perturb host cell metabolism.
To explore this possibility, we examined the metabolic response of cultured human fibroblasts to infection with influenza A (IVA), herpes simplex virus type-1 (HSV-1), and human cytomegalovirus (HCMV) (Munger et al. 2006, 2008; Vastag et al. 2011). The metabolome was sampled from mock and virally infected cells at various points following infection, with the time points tailored to the duration of each virus’ replication cycle. We observed substantial metabolome alterations induced by all three viruses (Fig. 3). The impacted metabolites, however, were largely distinct. Thus, the observed metabolic changes are virus specific and not a generic host cell stress response. In influenza-infected cells, acetylneuraminic acid exhibited the largest concentration increase, with few other metabolites showing large changes. In contrast, HSV-1 and HCMV infection resulted in broad-based metabolome changes, with HSV-1 infection particularly increasing levels of deoxypyrimidines such as dTTP and dTMP, whereas HCMV most strongly elevated concentrations of TCA cycle intermediates and acetylated amino acids (Munger et al. 2006, 2008; Chambers et al. 2010; Vastag et al. 2011).
Figure 3.

Metabolome remodeling during viral infections points to antiviral drug targets. Quiescent human fibroblasts were mock-infected or infected with influenza A virus (IVA), herpes simplex virus-1 (HSV-1), or human cytomegalovirus (HCMV). Relative metabolite levels in the infected and mock-infected cells were measured by LC-MS or a stand-alone orbitrap instrument and LC-MS/MS on a triple quadrupole instrument (QQQ). The resulting log-transformed fold changes in metabolite concentrations are shown in heat map format. LC-MS/MS measurements are designated by “QQQ” after the compound name. Each virus institutes a different metabolic program: IVA strongly increases acetylneuraminic acid, HSV-1 increases deoxypyrimidines, and HCMV increases metabolites of the TCA cycle and acetylated amino acids. The strongest metabolome changes induced by IVA and HSV-1 point to the preferred enzyme targets for treating these viruses; the changes induced by HCMV point to a new therapeutic opportunity. (Parts of this figure are adapted from Vastag et al. 2011.)
IVA encodes in its genome the enzyme neuraminidase, which catalyzes the cleavage of acetylneuraminic acid from glycoproteins. Oseltamivir, the leading current treatment for IVA infection, inhibits the viral neuraminidase. Thus, the metabolome response to IVA points to the preferred target for influenza treatment. Similarly, HSV-1 encodes in its genome an active thymidine kinase, which produces the deoxypyrimidines that are strongly elevated in HSV-1 infection. Acyclovir, the leading current treatment for HSV-1, is a nucleotide analog activated by the viral thymidine kinase. Thus, the metabolome response to HSV-1 highlights a key enzyme involved in HSV-1 treatment.
Like HSV-1, HCMV is currently treated primarily with a nucleoside analog, ganciclovir. The metabolic analysis of HCMV-infected cells, however, did not point primarily to deoxypyrimidines. This is consistent with HCMV’s lack of a functional thymidine kinase and with the poor therapeutic index of ganciclovir for treatment of HCMV, relative to acyclovir for treatment of HSV-1. Instead of pointing to deoxypyrimidine metabolism, metabolite concentration changes in HCMV infection point to two-carbon metabolism as a potential target for antiviral treatment. For example, citrate, which carries two-carbon units from the mitochondrion to the cytosol, is highly elevated, as are acetylated amino acids.
IDENTIFICATION OF N-ACETYL-ASPARTATE AS THE MOST STRONGLY UP-REGULATED METABOLITE IN HCMV INFECTION
Whereas acetylated amino acids are known to exist in humans, with N-acetyl-aspartate highly abundant in vertebrate brain, their route of synthesis remains unclear and they are not found on standard metabolic maps. Accordingly, they were not included on our lists of targeted metabolites and would not have been detected by our targeted methods. We were able, however, to identify the changes in these compounds through untargeted analysis, with N-acetyl-aspartate the most powerfully up-regulated metabolite in HCMV-infected cultured fibroblasts (Figs. 3 and 4A).
Figure 4.
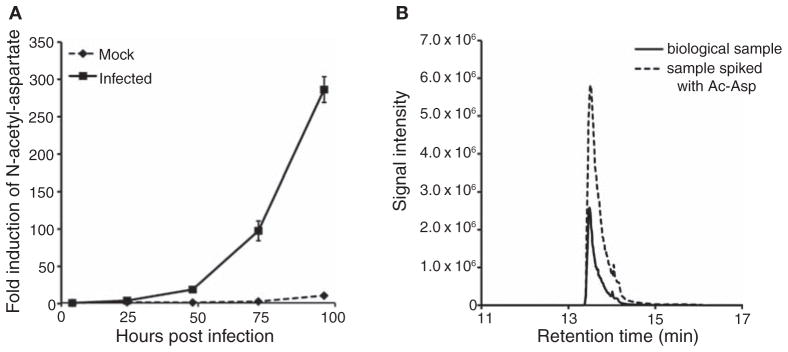
N-Acetyl-aspartate is the most strongly up-regulated metabolite in HCMV infection. (A) Time course of N-acetyl-aspartate accumulation. (B) Chromatogram showing overlapping LC-MS signal for biological sample and N-acetyl-aspartate standard.
The goal of identifying metabolites with concentration differences between sample groups carries a threefold challenge. First, peaks with significant differences between control and experimental sample groups must be identified. Second, the chemical formula of the source metabolite must be deduced. Finally, the structure of the metabolite has to be elucidated and confirmed. There are numerous free software packages that can assist with the first step. Some perform fully automated data processing and identification of peaks with significant changes between control and experimental samples (Katz et al. 2004; Baran et al. 2006; Katajamaa et al. 2006; Smith et al. 2006; Melamud et al. 2010). Complications arise in the second step, however, owing to the formation of fragments and adducts of the compound of interest during ionization. In addition, the analyst must deal with peaks that correspond to various natural isotope-containing forms of the compound. These two factors result in a typical compound producing up to 20 distinct peaks during LC-MS analysis. No current publicly available software package effectively selects from these the molecular ion peak; analyst expertise is still required. Because adducts and fragments are generated postchromatographically, in the ion source, they appear in the same spectrum as the parent peak, facilitating their pairing with the relevant molecular ion. Similarly, isotope-labeled forms of the parent metabolite hardly separate chromatographically and thus also appear in the same mass spectrum. Moreover, the intensity of the molecular ion, fragmentation, and adduct peaks correlate across biological samples. The primary task of the analyst is accordingly to figure out the isotope labeling, fragmentation, or adduct that accounts for each of these peaks. Once the molecular ion is defined, the relative intensity of the natural isotope-labeled peaks can be used to narrow down possible candidate formulas. Application of the rules developed by Kind et al. for filtering molecular formulas by accurate MS may yield only a single formula (Kind and Fiehn 2007); when multiple formulae are found, isotope-labeling experiments can be used to definitely determine the formula (Hegeman et al. 2007). Even with the molecular formula in hand, structure elucidation can be difficult. Data from high-resolution LC-MS experiments contains limited direct information about a compound’s structure. The retention time, however, can suggest the presence of certain functional groups, and coelution with a particular metabolite class may provide information about the nature of the unknown (e.g., charge, size, functional groups). In addition, if the metabolite fragments in the ionization source, the presence of some common fragments can be interpreted structurally. More preferably, the metabolite can be fragmented using tandem mass spectrometry (MS/MS). Once candidate structures are proposed, the presence or absence of particular functional groups can be confirmed by selective derivatization reactions. The resulting short list of preferred candidate structures can then be purchased or synthesized and their mass spectrum and retention times compared with the unknown.
N-Acetyl-aspartate was identified based on its in-source fragmentation pattern, with the products of decarboxylation, dehydration, and loss of the aspartate side chain detected as the most abundant fragments. In addition, it eluted a few seconds before a chemically related metabolite, N-acetyl-glutamate. Finally, its labeling by U-13C-glucose reflected the incorporation of two-carbon units from acetyl-CoA, whereas in U-13C-glutamine-fed cells, a four-carbon unit from aspartate was incorporated. To confirm the identity of the unknown, an authenticated N-acetyl-L-aspartate standard was purchased from Sigma-Aldrich. The standard coeluted with the unknown peak when added to biological samples (Fig. 4B). An analogous approach was applied for the identification of other metabolites from virally infected cells, including acetyl-neuraminic acid, carboxyethylcysteine, and glutarate. Whereas up-regulation of acetyl-aspartate is consistent with HCMV’s increasing the production of two-carbon units, which we show directly by flux analysis below, the reactions that produce acetyl-aspartate in HCMV-infected cells remain unknown, as does its biological function. Efforts to solve these mysteries are ongoing and may reveal additional antiviral targets.
PROBING TCA CYCLE FLUXES WITH ISOTOPE-LABELED GLUCOSE AND GLUTAMINE
Although concentration measurements are informative, they do not reveal the changes in influx and efflux that are responsible for the perturbation of metabolite levels. Feeding isotopic tracers, such as 13C-labeled glucose, provides an elegant way of tracing material flow through pathways. The rate of incorporation of a labeled nutrient into downstream metabolites can be analyzed to determine fluxes. Furthermore, metabolite-labeling patterns can indicate the biosynthetic source of a metabolite and the pathway used for its synthesis. For instance, citrate labeling during HSV-1 and HCMV infection of cultured human fibroblasts indicates that the viruses induce two different TCA cycle influx pathways. HCMV-infected fibroblasts produce a significant amount of citrate with two labeled carbon atoms (13C2-citrate; Fig. 5A), whereas their HSV-1-infected counterparts generate citrate with three labeled carbons (13C3-citrate; Fig. 5B). This implies that HCMV induces the flux of glycolytic carbon into the TCA cycle via pyruvate dehydrogenase and citrate synthase, whereas HSV-1 infection up-regulates anaplerotic TCA cycle influx via pyruvate carboxylase. These flux changes indicate that the two viruses manipulate TCA cycle flux to different ends: Whereas HCMV up-regulates the catalytic use of the pathway, taking two-carbon units from glycolysis to fatty acid biosynthesis, HSV-1 uses the TCA cycle to deliver glycolytic carbon through aspartate to pyrimidine nucleotide synthesis (Vastag et al. 2011). Moreover, the absence of 13C2-citrate in HSV-1-infected cells indicates that, unlike HCMV, HSV-1 does not induce fatty acid biosynthesis from glucose. These observations highlight entirely different sets of enzymes as potential targets for the treatment of these two herpesvirus infections.
Figure 5.

HCMV and HSV-1 induce divergent modes of TCA cycle activity. Infected fibroblasts were switched into media containing uniformly 13C-labeled glucose at t = 0 and citrate labeling was measured by LC-MS. (A) HCMV induces rapid formation of doubly labeled citrate, indicating entry of two-carbon units into the TCA cycle via pyruvate dehydrogenase. (B) HSV-1 induces rapid formation of triply labeled citrate, indicating entry of three-carbon units into the TCA cycle via pyruvate carboxylase. (Adapted from Vastag et al. 2011.)
SYSTEMS-LEVEL FLUX QUANTITATION IN HCMV-INFECTED CELLS
Observation of metabolite-labeling patterns, such as the enhanced entry of two-carbon units from glucose into citrate in HCMV-infected cells, provides immediate insight into metabolic network activity. Additional insights can be gained by integrating labeling patterns with other forms of information, such as metabolite uptake and excretion rates, to enable broad-based quantitation of metabolic fluxes. Such quantitation often requires iterative rounds of experiments and computation. For example, in attempting to measure metabolic flux changes induced by HCMV, we initially relied on 13C-glucose-labeling data. Whereas this results in some interesting observations (Fig. 5), some key fluxes, such as the efflux from the TCA cycle toward lipids, could not be resolved. We accordingly conducted additional experiments using 13C-labeled glutamine, which eventually enabled reliable resolution of these fluxes.
The ultimate results are presented as the color-coded metabolic map shown in Figure 6. The map is analogous to a standard textbook metabolic pathway diagram but with added information: absolute metabolite concentrations (font sizes), absolute fluxes (arrow sizes), and HCMV-induced changes in both (font and arrow colors). This map reveals that HCMV broadly increases central metabolic activity, including in glycolysis, nucleotide metabolism, and most strongly in the TCA cycle and its efflux to feed lipid metabolism. The finding that HCMV increases glucose-driven lipid synthesis was confirmed by measuring lipid labeling from radioactive glucose (Munger et al. 2008).
Figure 6.

HCMV hijacks host cell metabolism, directing carbon toward fatty acids. Metabolic map shows concentration and flux changes induced by HCMV infection. Font sizes represent metabolite pool sizes in uninfected cells, arrow widths show net fluxes in uninfected cells, and the color scale indicates the fold change in concentrations and fluxes induced by HCMV. Metabolites in gray were not measured. Note that the strongest flux changes are in the TCA cycle and its efflux toward fatty acids (Munger et al. 2008).
The observation of increased TCA efflux toward lipids was particularly intriguing from a drug discovery perspective, because a variety of inhibitors of lipid metabolism are currently used in clinical practice. Some of these are used to treat chronic diseases such as atherosclerosis and are quite safe. Given that our flux evidence particularly implicated the committed enzyme of fatty acid synthesis and elongation, acetyl-CoA carboxylase (ACC), we examined whether its inhibition would impair HCMV replication. We found that inhibition of ACC with 5-tetradecyloxy-2-furoic acid (TOFA) prevents efficient HCMV replication, with a >1000-fold decrement in infectious viral yield at TOFA concentrations that are known to be safe for long-term treatment of rodents (Fig. 7). Thus, our integrated metabolite and flux analysis of viral infections yielded a novel target for the treatment of HCMV.
Figure 7.

The acetyl-CoA carboxylase inhibitor tetradecyloxy-2-furoic acid causes a >1000-fold inhibition of HCMV replication. (A) Viral titers. (B) Viability of uninfected quiescent fibroblasts. (Based on data from Vastag et al. 2011.)
EXPANDING THE RANGE OF NOVEL ANTIVIRAL TARGETS
Our discovery of ACC as an anti-HCMV target exemplifies the power of systems biology: By looking broadly at all of metabolism, we were guided by the data toward lipid metabolism, an area previously unassociated with HCMV replication. The recognition that HCMV elevates and depends on ACC activity raises two fundamental questions: What does ACC do for the virus? And how does HCMV increase the activity of ACC and other host metabolic enzymes?
To investigate the role of ACC in viral replication, we measured the lipid and fatty acid composition of uninfected and HCMV-infected cells as well as of HCMV virions. We found that HCMV virions are highly enriched in phosphatidylethanolamines, with their phospholipid head group composition resembling that of synaptic vesicles. This led to the identification of the synaptic vesicle-like protein SNAP23 as required for HCMV budding (Liu et al. 2011). SNAP23 is involved in many cellular processes and its inhibition would probably be poorly tolerated; nevertheless, its identification proves the concept that lipidomic studies can also lead to the discovery of host proteins required for viral replication.
In experiments focusing specifically on fatty acids, we found that HCMV increases the concentrations of cellular lipids containing certain rare fatty acids and that these species contribute to formation of the viral envelope. Building from these observations, we have identified a set of additional lipid metabolic enzymes that are required for HCMV infection. Some of these enzymes are substantially downstream from ACC and represent more specific antiviral targets. Efforts to validate these targets are ongoing.
Investigation into how HCMV alters metabolism has also given rise to additional potential therapeutic targets. HCMV infection modulates many cellular signaling pathways (DeMeritt et al. 2004; Evers et al. 2004; Kudchodkar et al. 2006; Moorman et al. 2008; Yurochko 2008), with activation of calmodulin-dependent kinase kinase (CaMKK) required for HCMV to up-regulate glycolysis (McArdle et al. 2011). Interestingly, inhibition of CaMKK blocks viral replication. The likely pathway by which HCMV activates CaMKK involves the viral protein pUL37 × 1 causing increased cytosolic calcium (Sharon-Friling et al. 2006); virus-induced alterations in calcium homeostasis may also play a role in activation of pyruvate dehydrogenase and thus acetyl-CoA production.
Acetyl-CoA is the key substrate for ACC. HCMV infection increases expression of ACC-1 mRNA through activation of the sterol-regulatory-element-binding protein type 2 (SREBP-2) transcription factor (Spencer et al. 2011). SREBP-2 activation requires mTOR signaling (Spencer et al. 2011), which is maintained during HCMV infection by inhibition of the tuberous sclerosis protein complex (TSC1/2) (Moorman et al. 2008). Interestingly, TSC-2 interacts with viral protein pUL38 (Moorman et al. 2008), suggesting that this viral protein may be critical to the HCMV-induced triggering of fatty acid synthesis. Notably, inhibitors of mTOR also block HCMV replication, providing a second example of a metabolic regulatory kinase that is required by HCMV (Kudchodkar et al. 2004; Moorman et al. 2008; Clippinger et al. 2011). Although inhibition of mTOR is immunosuppressive, and thus inappropriate for treatment of most viral infections, HCMV infection is common in organ transplant recipients who require immunosuppressive therapy. Indeed, rapamycin protects against reactivation of HCMV in patients who have undergone allogeneic hematopoietic stem cell transplantation (Marty et al. 2007). Thus, mTOR inhibitors appear to have clinical potential in this patient subset as dual immunosuppressive/anti-HCMV agents.
METABOLOMICS AND ANTICANCER TARGET IDENTIFICATION
The prognosis for many cancers remains dismal. Although kinase inhibitors can decrease oncogene signaling and thereby tumor growth, in most cases they are insufficient to lead to long-term survival. Accordingly, there is great interest in metabolic enzyme inhibition as a complementary approach. Metabolomics is playing a central role in efforts to find and validate metabolic targets for cancer therapy.
One approach to target identification involves measuring the metabolic contents of tumor samples of varying grade or stage. The aim is to find metabolites whose levels correlate with cancer aggressiveness. The enzymes producing these metabolites then can be evaluated as potential therapeutic targets. This approach has been successfully applied to prostate cancer, where examination of 42 tissue samples, ranging from normal prostate to metastatic cancer, revealed six metabolites that are up-regulated in associated with disease progression: uracil, kynurenine, glycerol-3-phosphate, leucine, proline, and N-methyl-glycine (sarcosine) (Sreekumar et al. 2009). Follow-up studies have provided evidence that sarcosine contributes, perhaps through a signaling mechanism, to prostate cancer cell invasiveness. Knockdown of the enzyme that produces sarcosine, glycine-N-methyl-transferase, impaired prostate cancer cell invasiveness, suggesting its potential as a therapeutic target (Sreekumar et al. 2009).
A closely related approach involves analyzing a battery of cancer cell lines of differing aggressiveness. This approach revealed that aggressive cancer cell lines tend to have lower levels of monoacylglycerols and higher levels of free fatty acids (Nomura et al. 2010, 2011). These lipidomic changes are caused in part by higher activity of the enzyme monoacylglycerol lipase in aggressive cancer cells. Inhibition of monoacylglycerol lipase reverts the aggressive cancer cells’ lipidome to a state similar to that of less aggressive cells. Furthermore, it impairs tumor growth both in vitro and in vivo. Tumor growth can be restored, however, by the feeding of free fatty acids or a high-fat diet; thus, depending on the ability to control patients’ diets, monoacylglycerol lipase could be an anti-cancer target.
Metabolomics is also being used to expedite the translation of genomic observations into validated drug targets. A striking finding from sequencing of brain cancers was that almost half harbor point mutations at arginine 132 within the active site of the TCA cycle enzyme isocitrate hydrogenase-1 (IDH1) (Yan et al. 2009). Identical active site mutations within IDH1 or IDH2 were then also found in about 20% of acute myeloid leukemias (Mardis et al. 2009). The mutant IDH is a genetically dominant oncogene: A single copy of the mutant enzyme causes cancer. Metabolomic analysis of cells expressing mutant IDH revealed marked accumulation of a noncanonical metabolite, 2-hydroxyglutarate (Fig. 8). Biochemical analysis demonstrated that the mutant enzyme, although catalytically inactive at converting isocitrate into α-ketoglutarate, rapidly catalyzes NADPH-dependent reduction of α-ketoglutarate to the “oncometabolite” 2-hydroxyglutarate (Dang et al. 2009; Zhao et al. 2009; Ward et al. 2010).
Figure 8.

Mutant isocitrate dehydrogenase-1 (IDH1) produces the oncometabolite 2-hydroglutarate (2HG). (A) LC–high-resolution MS analysis of glioblastoma cells expressing wild-type IDH1 or the IDH1 R132H mutant. Expression of the mutant enzyme strongly increases three coeluting ion peaks, all of which form during electrospray ionization of 2HG: Peak A is [2HG-H]−, peak B is [2HG-H3O]−, and peak C is [2HG + Na − 2H]−. (B) Analysis of TCA cycle intermediate concentrations from glioblastoma tumor specimens with wild-type or mutant IDH1. (Adapted from Dang et al. 2009.)
Recent studies suggest that 2-hydroxyglutarate causes cancer by competing with α-ketoglutarate for the active site of ketoglutarate-dependent dioxygenases, including enzymes involved in DNA and histone modification. The evidence is most compelling for acute myeloid leukemia, where the α-ketoglutarate-dependent dioxygenase TET2, which functions as a cytosine demethylase, is a major tumor suppressor (Xu et al. 2011). Almost all acute myeloid leukemias have either an IDH mutation leading to 2-hydroxyglutarate production or homozygous deletion of TET2 (Figueroa et al. 2010). Thus, acute myeloid leukemia requires loss of TET2 activity, which can be achieved either through gene deletion or through an oncometabolite-producing enzyme mutation. Because the active site of mutant IDH is chemically unique, it provides a selective target for cancer therapy, which is being actively pursued in the pharmaceutical industry.
FUTURE OPPORTUNITIES
The opportunity to apply metabolomics to drug target discovery is as broad as the scope of diseases where metabolic changes play a causative role. This spans from infectious disease—where the basic metabolic pathways of two of the most important global pathogens, malaria and tuberculosis, have recently by augmented by metabolomic studies (Marrero et al. 2010; Olszewski et al. 2010)—to intrinsic pathologies of the human brain (Holmes et al. 2006; Kaddurah-Daouk 2006). Gary Siuzdak and colleagues have conducted remarkable studies in multiple sclerosis and neuropathic pain models that have not only identified characteristic metabolic changes but have also produced therapeutic leads. These successes, which hopefully will be published soon, have led Siuzdak to the coin the term “therapeutic metabolomics.” This term effectively highlights the transformation of metabolomics from a biomarker discovery tool to a central methodology for understanding and attacking pathophysiology. Ultimately, however, metabolomics per se will not be therapeutic, it will only yield drug targets. The next great hurdle of the current metabolic renaissance will be finding compounds that selectively hit these targets and proving their value in the clinic.
Acknowledgments
This work was supported by grants from the National Institutes of Health (CA82396 to T.S.; AI68678 and CA147961 to J.D.R.), Stand Up To Cancer, and the Beckman Foundation.
References
- Bajad SU, Lu W, Kimball EH, Yuan J, Peterson C, Rabinowitz JD. Separation and quantitation of water soluble cellular metabolites by hydrophilic interaction chromatography–tandem mass spectrometry. J Chromatogr A. 2006;1125:76–88. doi: 10.1016/j.chroma.2006.05.019. [DOI] [PubMed] [Google Scholar]
- Baran R, Kochi H, Saito N, Suematsu M, Soga T, Nishioka T, Robert M, Tomita M. MathDAMP: A package for differential analysis of metabolite profiles. BMC Bioinformatics. 2006;7:530. doi: 10.1186/1471-2105-7-530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beger RD, Hansen DK, Schnackenberg LK, Cross BM, Fatollahi JJ, Lagunero FT, Sarnyai Z, Boros LG. Single valproic acid treatment inhibits glycogen and RNA ribose turnover while disrupting glucose-derived cholesterol synthesis in liver as revealed by the [U-C(6)]-D-glucose tracer in mice. Metabolomics. 2009;5:336–345. doi: 10.1007/s11306-009-0159-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brauer MJ, Yuan J, Bennett BD, Lu W, Kimball E, Botstein D, Rabinowitz JD. Conservation of the metabolomic response to starvation across two divergent microbes. Proc Natl Acad Sci. 2006;103:19302–19307. doi: 10.1073/pnas.0609508103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brindle JT, Antti H, Holmes E, Tranter G, Nicholson JK, Bethell HW, Clarke S, Schofield PM, McKilligin E, Mosedale DE, Grainger DJ. Rapid and noninvasive diagnosis of the presence and severity of coronary heart disease using 1H-NMR-based metabonomics. Nat Med. 2002;8:1439–1444. doi: 10.1038/nm1202-802. [DOI] [PubMed] [Google Scholar]
- Buescher JM, Czernik D, Ewald JC, Sauer U, Zamboni N. Cross-platform comparison of methods for quantitative metabolomics of primary metabolism. Anal Chem. 2009;81:2135–2143. doi: 10.1021/ac8022857. [DOI] [PubMed] [Google Scholar]
- Buescher JM, Moco S, Sauer U, Zamboni N. Ultrahigh performance liquid chromatography–tandem mass spectrometry method for fast and robust quantification of anionic and aromatic metabolites. Anal Chem. 2010;82:4403–4412. doi: 10.1021/ac100101d. [DOI] [PubMed] [Google Scholar]
- Castro-Perez J, Previs SF, McLaren DG, Shah V, Herath K, Bhat G, Johns DG, Wang SP, Mitnaul L, Jensen K, et al. In vivo D2O labeling to quantify static and dynamic changes in cholesterol and cholesterol esters by high resolution LC/MS. J Lipid Res. 2011;52:159–169. doi: 10.1194/jlr.D009787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers JW, Maguire TG, Alwine JC. Glutamine metabolism is essential for human cytomegalovirus infection. J Virol. 2010;84:1867–1873. doi: 10.1128/JVI.02123-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clasquin MF, Melamud E, Singer A, Gooding JR, Xu X, Dong A, Cui H, Campagna SR, Savchenko A, Yakunin AF, et al. Riboneogenesis in yeast. Cell. 2011;145:969–980. doi: 10.1016/j.cell.2011.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clippinger AJ, Maguire TG, Alwine JC. The changing role of mTOR kinase in the maintenance of protein synthesis during human cytomegalovirus infection. J Virol. 2011;85:3930–3939. doi: 10.1128/JVI.01913-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crutchfield CA, Lu W, Melamud E, Rabinowitz JD. Mass spectrometry-based metabolomics of yeast. Methods Enzymol. 2010;470:393–426. doi: 10.1016/S0076-6879(10)70016-1. [DOI] [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMeritt IB, Milford LE, Yurochko AD. Activation of the NF-κB pathway in human cytomegalovirus-infected cells is necessary for efficient transactivation of the major immediate-early promoter. J Virol. 2004;78:4498–4507. doi: 10.1128/JVI.78.9.4498-4507.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte NC, Becker SA, Jamshidi N, Thiele I, Mo ML, Vo TD, Srivas R, Palsson BØ. Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proc Natl Acad Sci. 2007;104:1777–1782. doi: 10.1073/pnas.0610772104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evers DL, Wang X, Huang ES. Cellular stress and signal transduction responses to human cytomegalovirus infection. Microbes Infect. 2004;6:1084–1093. doi: 10.1016/j.micinf.2004.05.026. [DOI] [PubMed] [Google Scholar]
- Fan TW, Lane AN, Higashi RM, Farag MA, Gao H, Bousamra M, Miller DM. Altered regulation of metabolic pathways in human lung cancer discerned by 13C stable isotope-resolved metabolomics (SIRM) Mol Cancer. 2009;8:41. doi: 10.1186/1476-4598-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiehn O. Metabolomics—The link between genotypes and phenotypes. Plant Mol Biol. 2002;48:155–171. [PubMed] [Google Scholar]
- Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegeman AD, Schulte CF, Cui Q, Lewis IA, Huttlin EL, Eghbalnia H, Harms AC, Ulrich EL, Markley JL, Sussman MR. Stable isotope assisted assignment of elemental compositions for metabolomics. Anal Chem. 2007;79:6912–6921. doi: 10.1021/ac070346t. [DOI] [PubMed] [Google Scholar]
- Holmes E, Tsang TM, Huang JT, Leweke FM, Koethe D, Gerth CW, Nolden BM, Gross S, Schreiber D, Nicholson JK, Bahn S. Metabolic profiling of CSF: Evidence that early intervention may impact on disease progression and outcome in schizophrenia. PLoS Med. 2006;3:e327. doi: 10.1371/journal.pmed.0030327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibarra RU, Edwards JS, Palsson BO. Escherichia coli K-12 undergoes adaptive evolution to achieve in silico predicted optimal growth. Nature. 2002;420:186–189. doi: 10.1038/nature01149. [DOI] [PubMed] [Google Scholar]
- Jentzmik F, Stephan C, Miller K, Schrader M, Erbersdobler A, Kristiansen G, Lein M, Jung K. Sarcosine in urine after digital rectal examination fails as a marker in prostate cancer detection and identification of aggressive tumours. Eur Urol. 2010;58:12–18. doi: 10.1016/j.eururo.2010.01.035. discussion 20–21. [DOI] [PubMed] [Google Scholar]
- Kaddurah-Daouk R. Metabolic profiling of patients with schizophrenia. PLoS Med. 2006;3:e363. doi: 10.1371/journal.pmed.0030363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katajamaa M, Miettinen J, Oresic M. MZmine: Toolbox for processing and visualization of mass spectrometry based molecular profile data. Bioinformatics. 2006;22:634–636. doi: 10.1093/bioinformatics/btk039. [DOI] [PubMed] [Google Scholar]
- Katz JE, Dumlao DS, Clarke S, Hau J. A new technique (COMSPARI) to facilitate the identification of minor compounds in complex mixtures by GC/MS and LC/MS: Tools for the visualization of matched datasets. J Am Soc Mass Spectrom. 2004;15:580–584. doi: 10.1016/j.jasms.2003.12.011. [DOI] [PubMed] [Google Scholar]
- Kind T, Fiehn O. Metabolomic database annotations via query of elemental compositions: Mass accuracy is insufficient even at less than 1 ppm. BMC Bioinformatics. 2006;7:234. doi: 10.1186/1471-2105-7-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kind T, Fiehn O. Seven Golden Rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry. BMC Bioinformatics. 2007;8:105. doi: 10.1186/1471-2105-8-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudchodkar SB, Yu Y, Maguire TG, Alwine JC. Human cytomegalovirus infection induces rapamycin-insensitive phosphorylation of downstream effectors of mTOR kinase. J Virol. 2004;78:11030–11039. doi: 10.1128/JVI.78.20.11030-11039.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudchodkar SB, Yu Y, Maguire TG, Alwine JC. Human cytomegalovirus infection alters the substrate specificities and rapamycin sensitivities of raptor- and rictor-containing complexes. Proc Natl Acad Sci. 2006;103:14182–14187. doi: 10.1073/pnas.0605825103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon YK, Lu W, Melamud E, Khanam N, Bognar A, Rabinowitz JD. A domino effect in antifolate drug action in Escherichia coli. Nat Chem Biol. 2008;4:602–608. doi: 10.1038/nchembio.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindon JC, Nicholson JK, Holmes E, Antti H, Bollard ME, Keun H, Beckonert O, Ebbels TM, Reily MD, Robertson D, et al. Contemporary issues in toxicology the role of metabonomics in toxicology and its evaluation by the COMET project. Toxicol Appl Pharmacol. 2003;187:137–146. doi: 10.1016/s0041-008x(02)00079-0. [DOI] [PubMed] [Google Scholar]
- Liu ST, Sharon-Friling R, Ivanova P, Milne SB, Myers DS, Rabinowitz JD, Brown HA, Shenk T. Synaptic vesicle-like lipidome of human cytomegalovirus virions reveals a role for SNARE machinery in virion egress. Proc Natl Acad Sci. 2011;108:12869–12874. doi: 10.1073/pnas.1109796108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Clasquin MF, Melamud E, Amador-Noguez D, Caudy AA, Rabinowitz JD. Metabolomic analysis via reversed-phase ion-pairing liquid chromatography coupled to a stand alone orbitrap mass spectrometer. Anal Chem. 2010;82:3212–3221. doi: 10.1021/ac902837x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo B, Groenke K, Takors R, Wandrey C, Oldiges M. Simultaneous determination of multiple intracellular metabolites in glycolysis, pentose phosphate pathway and tricarboxylic acid cycle by liquid chromatography-mass spectrometry. J Chromatogr A. 2007;1147:153–164. doi: 10.1016/j.chroma.2007.02.034. [DOI] [PubMed] [Google Scholar]
- Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. New Engl J Med. 2009;361:1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrero J, Rhee KY, Schnappinger D, Pethe K, Ehrt S. Gluconeogenic carbon flow of tricarboxylic acid cycle intermediates is critical for Mycobacterium tuberculosis to establish and maintain infection. Proc Natl Acad Sci. 2010;107:9819–9824. doi: 10.1073/pnas.1000715107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty FM, Bryar J, Browne SK, Schwarzberg T, Ho VT, Bassett IV, Koreth J, Alyea EP, Soiffer RJ, Cutler CS, et al. Sirolimus-based graft-versus-host disease prophylaxis protects against cytomegalovirus reactivation after allogeneic hematopoietic stem cell transplantation: A cohort analysis. Blood. 2007;110:490–500. doi: 10.1182/blood-2007-01-069294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArdle J, Schafer XL, Munger J. Inhibition of calmodulin-dependent kinase kinase blocks human cytomegalovirus-induced glycolytic activation and severely attenuates production of viral progeny. J Virol. 2011;85:705–714. doi: 10.1128/JVI.01557-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melamud E, Vastag L, Rabinowitz JD. Metabolomic analysis and visualization engine for LC-MS data. Anal Chem. 2010;82:9818–9826. doi: 10.1021/ac1021166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorman NJ, Cristea IM, Terhune SS, Rout MP, Chait BT, Shenk T. Human cytomegalovirus protein UL38 inhibits host cell stress responses by antagonizing the tuberous sclerosis protein complex. Cell Host Microbe. 2008;3:253–262. doi: 10.1016/j.chom.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munger J, Bajad SU, Coller HA, Shenk T, Rabinowitz JD. Dynamics of the cellular metabolome during human cytome-galovirus infection. PLoS Pathog. 2006;2:e132. doi: 10.1371/journal.ppat.0020132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munger J, Bennett BD, Parikh A, Feng XJ, McArdle J, Rabitz HA, Shenk T, Rabinowitz JD. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat Biotechnol. 2008;26:1179–1186. doi: 10.1038/nbt.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF, Haqq AM, Shah SH, Arlotto M, Slentz CA, et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009;9:311–326. doi: 10.1016/j.cmet.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura DK, Long JZ, Niessen S, Hoover HS, Ng SW, Cravatt BF. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell. 2010;140:49–61. doi: 10.1016/j.cell.2009.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura DK, Lombardi DP, Chang JW, Niessen S, Ward AM, Long JZ, Hoover HH, Cravatt BF. Monoacylglycerol lipase exerts dual control over endocannabinoid and fatty acid pathways to support prostate cancer. Chem Biol. 2011;18:846–856. doi: 10.1016/j.chembiol.2011.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olszewski KL, Mather MW, Morrisey JM, Garcia BA, Vaidya AB, Rabinowitz JD, Llinas M. Branched tricarboxylic acid metabolism in Plasmodium falciparum. Nature. 2010;466:774–778. doi: 10.1038/nature09301. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Panchal AR, Comte B, Huang H, Kerwin T, Darvish A, des Rosiers C, Brunengraber H, Stanley WC. Partitioning of pyruvate between oxidation and anaplerosis in swine hearts. Am J Physiol Heart Circ Physiol. 2000;279:H2390–H2398. doi: 10.1152/ajpheart.2000.279.5.H2390. [DOI] [PubMed] [Google Scholar]
- Panchal AR, Comte B, Huang H, Dudar B, Roth B, Chandler M, Des Rosiers C, Brunengraber H, Stanley WC. Acute hibernation decreases myocardial pyruvate carboxylation and citrate release. Am J Physiol Heart Circ Physiol. 2001;281:H1613–H1620. doi: 10.1152/ajpheart.2001.281.4.H1613. [DOI] [PubMed] [Google Scholar]
- Pesek JJ, Matyska MT, Loo JA, Fischer SM, Sana TR. Analysis of hydrophilic metabolites in physiological fluids by HPLC-MS using a silica hydride-based stationary phase. J Sep Sci. 2009;32:2200–2208. doi: 10.1002/jssc.200900270. [DOI] [PubMed] [Google Scholar]
- Sabatine MS, Liu E, Morrow DA, Heller E, McCarroll R, Wiegand R, Berriz GF, Roth FP, Gerszten RE. Metabolomic identification of novel biomarkers of myocardial ischemia. Circulation. 2005;112:3868–3875. doi: 10.1161/CIRCULATIONAHA.105.569137. [DOI] [PubMed] [Google Scholar]
- Sauer U. Metabolic networks in motion: 13C-based flux analysis. Mol Syst Biol. 2006;2:62. doi: 10.1038/msb4100109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon-Friling R, Goodhouse J, Colberg-Poley AM, Shenk T. Human cytomegalovirus pUL37 × 1 induces the release of endoplasmic reticulum calcium stores. Proc Natl Acad Sci. 2006;103:19117–19122. doi: 10.1073/pnas.0609353103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CA, Want EJ, O’Maille G, Abagyan R, Siuzdak G. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal Chem. 2006;78:779–787. doi: 10.1021/ac051437y. [DOI] [PubMed] [Google Scholar]
- Spencer CM, Schafer XL, Moorman NJ, Munger J. Human cytomegalovirus induces the activity and expression of acetyl-coenzyme A carboxylase, a fatty acid biosynthetic enzyme whose inhibition attenuates viral replication. J Virol. 2011;85:5814–5824. doi: 10.1128/JVI.02630-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreekumar A, Poisson LM, Rajendiran TM, Khan AP, Cao Q, Yu J, Laxman B, Mehra R, Lonigro RJ, Li Y, et al. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature. 2009;457:910–914. doi: 10.1038/nature07762. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- Vastag L, Koyuncu E, Grady SL, Shenk TE, Rabinowitz JD. Divergent effects of human cytomegalovirus and herpes simplex virus-1 on cellular metabolism. PLoS Pathog. 2011;7:e1002124. doi: 10.1371/journal.ppat.1002124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang TJ, Larson MG, Vasan RS, Cheng S, Rhee EP, McCabe E, Lewis GD, Fox CS, Jacques PF, Fernandez C, et al. Metabolite profiles and the risk of developing diabetes. Nat Med. 2011;17:448–453. doi: 10.1038/nm.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting α-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225–234. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Xiao MT, Liu LX, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, et al. IDH1 and IDH2 mutations in gliomas. New Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Previs SF, Fernandez CA, Dugelay S, Soloviev MV, Hazey JW, Agarwal KC, Levine WC, David F, Rinaldo P, et al. Noninvasive probing of citric acid cycle intermediates in primate liver with phenylacetylglutamine. Am J Physiol. 1996;270:E882–E889. doi: 10.1152/ajpendo.1996.270.5.E882. [DOI] [PubMed] [Google Scholar]
- Yizhak K, Benyamini T, Liebermeister W, Ruppin E, Shlomi T. Integrating quantitative proteomics and metabolomics with a genome-scale metabolic network model. Bioinformatics. 2010;26:i255–i260. doi: 10.1093/bioinformatics/btq183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Rabinowitz JD. Differentiating metabolites formed from de novo synthesis versus macromolecule decomposition. J Am Chem Soc. 2007;129:9294–9295. doi: 10.1021/ja072997c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Fowler WU, Kimball E, Lu W, Rabinowitz JD. Kinetic flux profiling of nitrogen assimilation in Escherichia coli. Nat Chem Biol. 2006;2:529–530. doi: 10.1038/nchembio816. [DOI] [PubMed] [Google Scholar]
- Yuan J, Bennett BD, Rabinowitz JD. Kinetic flux profiling for quantitation of cellular metabolic fluxes. Nat Protoc. 2008;3:1328–1340. doi: 10.1038/nprot.2008.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Doucette CD, Fowler WU, Feng XJ, Piazza M, Rabitz HA, Wingreen NS, Rabinowitz JD. Metabolomics-driven quantitative analysis of ammonia assimilation in E. coli. Mol Syst Biol. 2009;5:302. doi: 10.1038/msb.2009.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurochko AD. Human cytomegalovirus modulation of signal transduction. Curr Top Microbiol Immunol. 2008;325:205–220. doi: 10.1007/978-3-540-77349-8_12. [DOI] [PubMed] [Google Scholar]
- Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, Yu W, Li Z, Gong L, Peng Y, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1α. Science. 2009;324:261–265. doi: 10.1126/science.1170944. [DOI] [PMC free article] [PubMed] [Google Scholar]