

SUMMARY
We investigated the in vivo relevance of the impact of sarA and saeRS on protease production using derivatives of the USA300 strain LAC. The results confirmed that mutation of saeRS or sarA reduces virulence in a bacteremia model to a comparable degree. However, while eliminating protease production restored virulence in the sarA mutant, it had little impact in the saeRS mutant. Additionally, constitutive activation of saeRS (saeRSC) enhanced the virulence of LAC and largely restored virulence in the isogenic sarA mutant. Based on these results, together with our analysis of the representative virulence factors alpha toxin, protein A (Spa), and extracellular nucleases, we propose a model in which the attenuation of saeRS mutants is defined primarily by decreased production of such factors, while constitutive activation of saeRS increases virulence, and reverses the attenuation of sarA mutants, because it results in both increased production and decreased protease-mediated degradation of these same factors. This regulatory balance was also apparent in a murine model of catheter-associated infection, with the results suggesting that the impact of saeRS on nuclease production plays an important role during the early stages of these infections that is partially offset by increased protease production in sarA mutants.
Keywords: Staphylococcus, protease, saeRS, sarA, bacteremia, biofilm
INTRODUCTION
Infections caused by Staphylococcus aureus are of extreme clinical significance based on both their frequency and severity (Otto, 2012). Moreover, the ability to treat these infections is increasingly compromised by reduced susceptibility of many strains, if not outright resistance, to currently available antibiotics including vancomycin (Holmes et al., 2012). Biofilm-associated infections are a specific concern because they do not respond adequately to antimicrobial therapy irrespective of the resistance status of the offending S. aureus strain (Romling and Balsalobre, 2012). One way to overcome these limitations would be to develop new antibiotics with efficacy against the most problematic resistant strains even in the context of a biofilm, but this has proven a difficult task, particularly at a time when pharmaceutical companies have de-emphasized antibiotic development (Boucher et al., 2009). An ancillary approach would be to develop methods to limit biofilm formation itself, thereby enhancing the therapeutic efficacy of both existing and any newly developed antibiotics.
Given the multifactorial nature of S. aureus biofilm formation (Archer et al., 2011, Laverty et al., 2013, Otto, 2008), we have placed a primary focus on targeting regulatory elements that modulate the production of critical virulence factors rather than the virulence factors themselves. A number of regulatory loci have been implicated in this regard, but to date few of these have been explored in the context of their therapeutic relevance in vivo. One exception is the staphylococcal accessory regulator (sarA), mutation of which limits biofilm formation in a murine model of catheter-associated infection to a degree that can be correlated with increased antibiotic susceptibility under both in vitro and in vivo conditions (Weiss et al., 2009, Weiss et al., 2009). Moreover, with the exception of the 8325-4 strain RN6390 and Newman, both of which can be explained by well-defined regulatory defects, the impact of sarA on biofilm formation is consistent in diverse clonal lineages of S. aureus including both methicillin-susceptible and methicillin-resistant strains (Beenken et al., 2003, Beenken et al., 2010, Mrak et al., 2012, Zielinska et al., 2011).
Mutation of sarA results in the increased production of extracellular proteases, and we have confirmed that this limits the accumulation of multiple virulence factors (Mrak et al., 2012, Tsang et al., 2008, Zielinska et al., 2011, Zielinska et al., 2012). This includes the fibronectin-binding protein FnbA, protein A (Spa), alpha toxin, and phenol-soluble modulins (PSMs) (Mrak et al., 2012, Zielinska et al., 2011), all of which have been implicated in various aspects of S. aureus pathogenesis including biofilm formation (Anderson et al., 2012, Caiazza et al., 2003, Cassat et al., 2013, Claro et al., 2013, Edwards et al., 2012, Lower et al., 2011, Kobayashi et al., 2011, Merino et al., 2009, O’Neill et al., 2008, Otto, 2010, Periasamy et al., 2012). A number of reports have also confirmed that proteases, including some derived from sources other than S. aureus, limit biofilm formation and promote dispersal from an established biofilm (Boles and Horswill, 2008, Chen et al., 2013, Mootz et al., 2013, Park et al., 2012, Sugimoto et al., 2013).
We also confirmed that eliminating the ability to produce extracellular proteases increases the virulence of sarA mutants in a murine bacteremia model (Zielinska et al., 2012). S. aureus proteases have been shown to degrade specific components of host defense systems including complement (Jusko et al., 2014, Kantyka et al., 2013, Zdzalik et al., 2012), but if these host proteins were the relevant in vivo targets, it would be anticipated that eliminating protease production would result in decreased rather than increased virulence and, conversely, that the increased production of proteases in sarA mutants would result in increased rather than decreased virulence owing to compromised host defenses. Thus, the more likely explanation is that the decreased accumulation of specific S. aureus virulence factors accounts for the attenuation of sarA mutants and, conversely, that restoration of these factors in protease-deficient sarA mutants accounts for this in vivo “phenotypic complementation” (Zielinska et al., 2012). Thus, these results suggest that inhibitors of sarA could potentially be used in conjunction with conventional antibiotics to enhance the efficacy of conventional antibiotics to overcome the therapeutic recalcitrance that characterizes biofilm-associated infections.
However, sarA is part of a large, highly interactive regulatory network, several components of which also modulate protease production (Oscarsson et al., 2006). This raises the possibility that differences in the functional status of other regulatory loci could impact sarA-defined in vivo phenotypes as well as therapeutic strategies targeting sarA. Among these is the saeRS two-component regulatory system, mutation of which also enhances protease production. Although this effect is modest by comparison to that associated with mutation of sarA (Mrak et al., 2012), the increased production of aureolysin in a LAC saeRS mutant was recently shown to have a dramatic impact on bone remodeling in osteomyelitis (Cassat et al., 2013), thus raising the possibility that saeRS would also be a viable target for therapeutic intervention.
Perhaps more importantly, the commonly-studied strain Newman has a point mutation in saeS that enhances phosphorylation of the SaeR response regulator (Mainiero et al., 2010, Schäfer et al., 2009), and we have confirmed that this “constitutive activation” limits protease activity even in an isogenic sarA mutant (Mrak et al., 2012, Zielinska et al., 2011). Indeed, Newman is one of the few S. aureus strains in which mutation of sarA has relatively little impact on biofilm formation (Beenken et al., 2003). Newman also has other defects of potential relevance in biofilm formation, most notably mutations that prevent anchoring of the fibronectin-binding proteins to the cell surface (Grundmeier et al., 2004), and this compromises the ability to address these issues in derivatives of Newman itself. Indeed, we confirmed that restoration of surface-anchored FnbA greatly enhances biofilm formation in Newman, but that the impact of mutating sarA on biofilm formation is still not evident unless the defect in saeS was repaired (Mrak et al., 2012). This suggests that the point mutation in saeS (L18P), or other changes that result in increased production and/or activity of SaeR, could compromise therapeutic strategies targeting sarA. However, these previous studies were limited to in vitro experiments, and it remains unclear whether the functional status of saeRS impacts sarA phenotypes in vivo. To avoid the limitation imposed by a lack of surface-anchored fibronectin-binding proteins in Newman, we chose to address this using the contemporary clinical isolate LAC.
RESULTS
To generate derivatives of LAC that differ with respect to the functional status of saeRS and sarA, we first mutated saePQRS (hereinafter referred to as saeRS) using the pKOR1 mutagenesis system (Bae and Schneewind, 2006, Mrak et al., 2012). This mutant was then complemented by chromosomal insertion of version of saeRS from Newman (Schäfer et al., 2009) as previously described (Mrak et al., 2012). The relevant mutation is a point mutation in saeS (L18P), which results in increased phosphorylation of SaeR, a phenotype that we refer to hereinafter as constitutive activation (saeRSC). We then introduced a sarA mutation the saeRS mutant and its saeRSC derivative using phage-mediated transduction from an existing LAC sarA mutant (Zielinska et al., 2012). Analysis of these mutants by qRT-PCR confirmed that levels of the saeR transcript were increased in the saeRSC derivative by comparison to the LAC parent strain in both the post-exponential (OD560 = 3.0) and stationary (overnight) growth phases (Fig. 1A). Western blot data examining the accumulation of SaeR itself was more difficult to interpret owing to greater variability between repetitive experiments, which we attribute to differences in the background observed in blots done with this antibody (Fig. 1B, lanes 3 and 6), thus making it difficult to achieve statistical significance. Nevertheless, examination of the collective results from these replicates clearly indicated that accumulation of SaeR during the stationary growth phase was higher in the saeRSC derivative than the WT parent strain (Fig. 1B, lanes 1 vs. 2). Additionally, the absence of the saeR transcript (Fig. 1A), and SaeR itself beyond this background (Fig. 1B), were also confirmed in the saeRS mutant.
Fig. 1. Phenotypic verification of sarA and saeRS mutants.
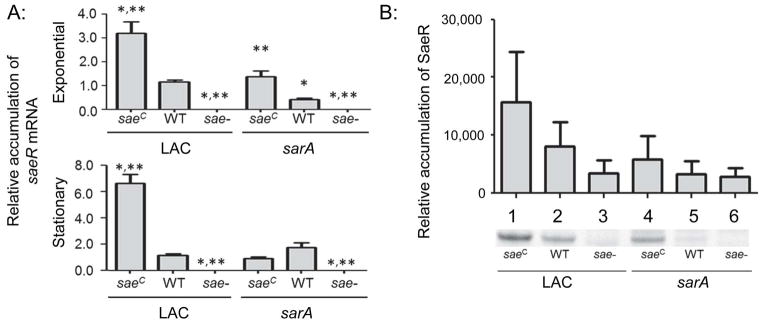
A: The abundance of saeR mRNA was assessed by qRT-PCR in the exponential and stationary growth phases. Strains on the left of each panel as indicated by the underline are strains derived from LAC itself, while strains on the right are derived from the isogenic LAC sarA mutant (e.g. the designation WT on the right indicates results observed with the LAC sarA mutant). Results shown represent the average ± standard deviation from two experiments, each of which was repeated in triplicate. Single asterisk indicates statistical significance by comparison to the LAC parent strain (WT). Double asterisks indicate significance by comparison to the sarA mutant. B: Western blots were performed in triplicate using cell lysates prepared from the same strains and rabbit polyclonal IgG targeting SaeR as primary antibody. Where necessary for legibility, the designation for the constitutively active saeRS derivative (saeRSC) and saeRS mutant were reduced to saeC and sae-respectively.
By comparison to the saeRSC derivative, accumulation of the saeR transcript was reduced in sarA and saeRSC/sarA mutants irrespective of growth phase, thus suggesting that sarA functions at some level upstream of saeRS. To the extent that sarA is upstream of the accessory gene regulator (agr) (Chien et al, 1998), while agr is upstream of saeRS (Novick and Jiang, 2003), one possible explanation for these results is that the impact of sarA on accumulation of the saeR transcript is due to reduced transcription via an indirect effect mediated through agr. However, accumulation of SaeR was higher in the saeRSC/sarA mutant by comparison to the isogenic sarA mutant (Fig. 1B, lanes 4 vs. 5) despite comparable levels of the saeR transcript. This suggests that SaeR accumulation is limited in sarA mutants owing to some post-transcriptional mechanism, the impact of which is limited by constitutive activation of saeRS. This is consistent with the observation that, while accumulation of SaeR in the saeRSC/sarA mutant was higher by comparison to the sarA mutant, it was lower in the saeRSC/sarA mutant by comparison to the isogenic saeRSC derivative itself (Fig. 1B, lanes 4 vs. 1).
One possible explanation for these results is that the relative level of extracellular proteases defines these phenotypes, and further analysis confirmed that overall protease production increases as the functional status of both saeRS and sarA decreases (Fig. 2A). However, these effects were not equivalent, with the impact of decreased sarA function playing the predominant role. Most importantly, protease activity was significantly increased saeRSC/sarA mutant by comparison to the isogenic saeRS mutant, but significantly decreased by comparison to the isogenic sarA mutant (Fig. 2A). This suggests that the increased accumulation of SaeR in the saeRSC mutant by comparison to the saeRSC/sarA mutant (Fig. 1B, lanes 1 vs. 4), and the increased accumulation of SaeR in the saeRSC/sarA mutant by comparison to its isogenic sarA mutant (Fig. 1B, lanes 4 vs. 5), could both be explained by the impact of these loci on the production of extracellular proteases.
Fig. 2. Protease production and its impact on accumulation of SarA.

A: Protease activity was assessed in the indicated strains using a FRET-based assay. The specific nature of each protease mutant (prot-) is described in the text. Single asterisk indicates statistical significance by comparison to the LAC parent strain (WT). Double asterisks indicate significance by comparison to the isogenic protease mutant. Triple asterisk indicates that the results observed with the sarA mutant generated in the constitutively active saeRS variant (saeRSC/sarA) were significantly different from those observed with both the sarA and saeRS/sarA mutants. Strain designations are the same as those defined for Fig. 1. B: Western blots were used to confirm mutation of sarA and assess accumulation of SarA as a function of saeRS and the production of extracellular proteases. Single asterisk indicates statistical significance by comparison to the corresponding saeRS derivative. The designation saeRS+ corresponds to LAC itself. The functional status of sarA, and the ability to produce proteases, are indicated by (+) and (−) signs below the blots.
To investigate this, we used derivatives of LAC and its sarA mutant that are unable to produce any of the 10 recognized extracellular proteases (Zielinska et al., 2012). We also generated a derivative of the saeRS and saeRSC mutants with additional mutations in the genes encoding aureolysin, SspA, SspB and ScpA. We did not mutate the spl operon in these strains for two reasons. First, both the spl mutation and the chromosomally inserted constitutively-active variant of saeRS are marked by erythromycin resistance, thus precluding transduction of one into the other. Second, and more importantly, mutation of sarA results in increased accumulation of all 10 extracellular proteases (Zielinska et al, 2012), while mutation of saeRS results in increased accumulation of aureolysin, SspA, SspB, and ScpA but decreased accumulation of the spl-encoded proteases (Cassat et al., 2013), thus suggesting that the functional status of the spl operon is unlikely to play a distinguishing role between these strains with respect to overall protease activity. This was confirmed by demonstrating that proteolytic activity was reduced to baseline levels in all protease mutants irrespective of the functional status of the spl operon (Fig. 2). Importantly, this was assessed using a casein-based protease assay, which is a known substrate of the spl-encoded proteases (Reed et al., 2001). Thus, we consider these protease-deficient derivatives equivalent in the context of the experiments we describe.
Western blot comparisons of whole cell lysates from these strains confirmed that the absence of SarA in all sarA mutants and that accumulation of SarA was unaffected by the functional status of saeRS (Fig. 2B), a result that would also be expected if saeRS is downstream of sarA. Accumulation of SaeR was higher in the saeRSC, sarA-positive derivatives than in the isogenic saeRSC/sarA mutants irrespective of the ability to produce extracellular proteases (Fig. 3A, lanes 1 and 2 vs. lanes 3 and 4), although as with our other SaeR western blots (Fig. 1B) these differences, while readily apparent, did not reach statistical significance. This sarA-dependent difference was not apparent in the saeS-repaired strains (saeRS+) (Fig. 3A, lanes 9 and 10 vs. lanes 11 and 12). Accumulation of SaeR in the saeRS+ sarA mutant (Fig. 3A, lane 11) appeared to be reduced by comparison to its protease-deficient derivative (Fig. 3A, lane 12), but this difference, while reproducible, did not reach statistical significance. These results demonstrate that, whatever the mechanism involved, the impact of mutating sarA on the accumulation of SaeR is limited by constitutive activation of saeRS. They also suggest that, while the increased production of extracellular proteases in sarA mutants has some impact on the accumulation of SaeR, the primary effect is mediated at the level of transcription and/or mRNA stability.
Fig. 3. Protease production and its impact on accumulation of SaeR and AgrA.

A: Western blots were used to assess the impact of saeRS, sarA, and protease production on the accumulation of SaeR. B. Western blots were used to assess the impact of saeRS, sarA, and protease production on the accumulation of AgrA. Single asterisk indicates statistical significance by comparison to the corresponding saeRS derivative. Double asterisks indicate significance by comparison to the isogenic sarA mutant. In both panels, the designation saeRS+ corresponds to LAC itself, with the functional status of sarA, and the ability to produce proteases, indicated by (+) and (−) signs below the blots.
Based on the current S. aureus regulatory paradigm indicating that sarA is upstream of agr while agr is upstream of saeRS, we also extended our studies to evaluate accumulation of AgrA as a function of the functional status of sarA and saeRS. In these experiments, mutation of saeRS was found to have no impact on the accumulation of AgrA (Fig. 3B, lanes 5 and 6 vs. 9 and 10), as would expected based on the linear regulatory paradigm outlined above. However, mutation of sarA resulted in decreased accumulation of AgrA, and in both the saeS mutant and its saeS-repaired derivative this could be correlated to a statistically significant degree with the increased production of extracellular proteases (Fig. 3B, lanes 7 vs. 8 and lanes 11 vs. 12). This is consistent with our previous proteomics comparisons in which the amount of AgrA observed in a LAC sarA mutant was reduced by comparison to the parent strain and partially restored in the protease-deficient sarA derivative (Zielinska et al., 2012). As with the impact of mutating sarA on accumulation of SaeR, these effects were also overcome by constitutive activation of saeRS (Fig. 3B, lanes 1–4).
Thus, one explanation for these results is that mutation of sarA results in reduced accumulation of AgrA owing to protease-mediated degradation, with this in turn resulting in reduced transcription of saeRS, both of which are largely overcome as might be expected by constitutive activation of saeRS. These results demonstrate that the impact of sarA on the accumulation of AgrA is not mediated entirely by its impact on the production of AgrA (Chien et al, 1998), but also indirectly by the increased production of extracellular proteases in sarA mutants (Fig. 3B, lane 12). This further emphasizes both the complexity of S. aureus regulatory circuits and the potential importance of sarA in repressing the production of extracellular proteases as a means of maintaining the integrity of these circuits.
To further examine the potential significance of these results, we also examined the accumulation alpha toxin and protein A (Spa) in these mutants. We chose these as representative virulence factors because their production and/or accumulation has been shown to be impacted by both sarA and saeRS (Mainiero et al., 2010, Mrak et al., 2012). The results confirmed that mutation of saeRS alone limits the production and/or accumulation of both of these virulence factors irrespective of the ability to produce extracellular proteases (Fig. 4A and 4B, lanes 5 and 6), with this effect being essentially absolute in the case of alpha toxin (Fig. 4A, lanes 5 and 6). They also demonstrated that mutation of sarA has the same phenotypic effect, but in this case the accumulation of both alpha toxin and Spa was restored in the protease-deficient sarA mutants (Fig. 4A and 4B, lanes 11 and 12). Accumulation of Spa, but not alpha toxin, was also restored in the protease-deficient saeRS/sarA mutant (Fig. 4A and 4B, lanes 7 and 8).
Fig. 4. Protease production and its impact on accumulation of alpha toxin and protein A (Spa).

A: Western blots were used to assess the impact of saeRS, sarA, and protease production on the accumulation of alpha toxin. B. Western blots were used to assess the impact of saeRS, sarA, and protease production on the accumulation of Spa. Single asterisk indicates statistical significance by comparison to the corresponding saeRS derivative. Double asterisks indicate significance by comparison to the isogenic sarA mutant. In both panels, the designation saeRS+ corresponds to LAC itself, with the functional status of sarA, and the ability to produce proteases, indicated by (+) and (−) signs below the blots.
Accumulation of alpha toxin was also restored in the saeRSC/sarA mutant irrespective of the ability to produce extracellular proteases (Fig. 4A, lanes 3 and 4), thus suggesting that the impact of saeRS on the alpha toxin phenotype is in fact mediated at the level of its production. Conversely, the impact of increased protease production in sarA mutants on the accumulation of Spa was evident irrespective of the functional status of saeRS (Fig. 4B, lanes 3 vs. 4, lanes 7 vs. 8, and lanes 11 vs. 12). We conclude that the impact of sarA on the degradation of Spa is phenotypically epistatic to that of saeRS on the production of Spa, but that the opposite is true with respect to alpha toxin.
While the results of the experiments discussed above are informative, they are also based on in vitro studies, and under such circumstances it would be anticipated that the impact of extracellular proteases would be magnified, perhaps to the point of biological insignificance, owing to the proximity of the proteases on their S. aureus targets imposed by the constrained environment of a test tube. Thus, to determine the in vivo relevance of these results, we used a murine bacteremia model to examine the relative impact of the functional status of saeRS and sarA. While a primary focus of our work is on the impact of these loci in biofilm formation, we chose to use this model first based on the logic that any such physical constraint would be minimized in the dynamic context of blood flow. The results confirmed that mutation of sarA or saeRS dramatically decreased the virulence of LAC as assessed by almost every in vivo parameter examined, the only exception being bacterial burdens in the kidney, which were reduced in the sarA mutant but not in the saeRS mutant (Figs. 5 and 6). Concomitant mutation of sarA and saeRS also appeared to have an additive effect, although in most cases this was difficult to assess owing to the impact of mutation of either locus alone.
Fig. 5. Survival in bacteremia as a function of the functional status of saeRS and sarA.
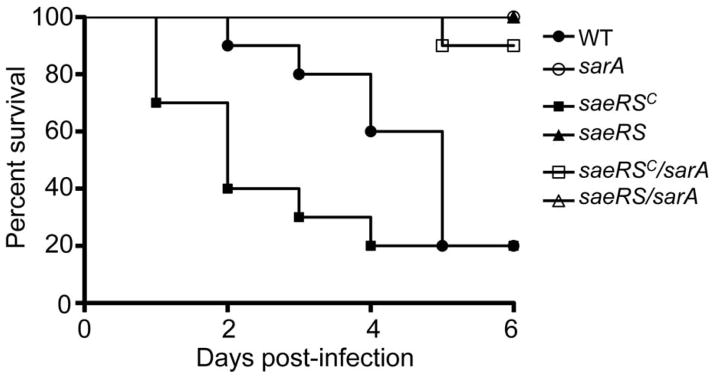
Results shown are Kaplan-Meirer survival curves of mice infected by tail vein injection of 5 × 107 cfu of the indicated strains. Results observed with all strains other than LAC and its saeRSC and saeRSC/sarA mutants overlapped at 100% survival. Results observed with all sarA mutants were statistically significant by comparison to the parent strain irrespective of the functional status of saeRS, but they were not significant by comparison to each other. The difference between the parent strain and the saeRSC derivative was also significant.
Fig. 6. Impact of saeRS vs. sarA on virulence in a bacteremia model.

Results are shown as colony forming units (cfu) per organ obtained from the indicated tissues 6 days after intravenous injection of the indicated S. aureus strains. As in Fig. 1, for simplicity in labeling, the designation WT in the group underlined on the right indicates results observed with the LAC sarA mutant. Boxes indicate the 25th and 75th percentiles for each group and define the interquartile range (IQR), with the (+) within each box indicating the mean and the horizontal line indicating the median. Vertical lines define the lowest and highest data points within 1.5 IQR of the lower and higher quartile respectively, with individual dots representing single data points outside this range. A single asterisk indicates significance by comparison to the parent strain, while double asterisks indicate significance by comparison to the isogenic sarA mutant.
Conversely, constitutive activation of saeRS reversed the attenuation of sarA mutants to a statistically significant degree in all tissues (Fig. 6). This was not evident as assessed based on survival curves, but this could be potentially be explained by the time frame of the experiments themselves (Fig. 5). Indeed, in this context, constitutive activation of saeRS increased the virulence even of LAC itself. These same general trends were evident when virulence was assessed based the ability to cause secondary bone and joint infection, although the only statistically significant difference as assessed based on overall histopathology scores was the difference between the results observed with the constitutively active variant of LAC and the sarA/saeRS double mutant (Fig. 7). Thus, when taken together, these results are consistent with the hypothesis the impact of sarA and saeRS on virulence in this model is mediated, at least in part, via different mechanisms. This was confirmed by demonstrating that eliminating protease production reversed the attenuation of sarA mutants in this model, but had little impact in saeRS mutants, with the only significant difference observed between the saeRS mutant and its protease-deficient derivative being bacterial burdens in the spleen (Fig. 8).
Fig. 7. Impact of saeRS and sarA on development of secondary musculoskeletal infections.

Results illustrate overall histopathological scores for each experimental group. Boxes indicate the 25th and 75th percentiles for each group and define the interquartile range (IQR), with the horizontal line indicating the median. Vertical lines define the lowest and highest data points within 1.5 IQR of the lower and higher quartile respectively, with individual dots representing single data points outside this range. The only statistically significant difference observed was that between the constitutively active saeRS derivative (saeRSC) and the saeRS/sarA mutant.
Fig. 8. Impact of protease production on phenotypes of saeRS and sarA mutants.

Upper left illustrates Kaplan-Meirer survival curves of mice infected by tail vein injection of 5 × 107 cfu of the indicated strains. Remaining panels illustrate colony counts obtained from the indicated tissues 6 days after intravenous injection of the indicated S. aureus strains. Boxes indicate the 25th and 75th percentiles for each group and define the interquartile range (IQR), with the horizontal line indicating the median. Vertical lines define the lowest and highest data points within 1.5 IQR of the lower and higher quartile respectively, with individual dots representing single data points outside this range. The asterisk indicates significance by comparison to the sarA mutant. Double asterisks indicate significance of the protease-deficient mutant by comparison to the isogenic protease-positive regulatory mutant.
Mutation of saeRS or sarA also limited the capacity of LAC to form a biofilm in vitro, and in this case the biofilm-deficient phenotype of both mutants was reversed to a comparable degree by eliminating their capacity produce extracellular proteases (Fig. 9A). This was also true in the saeRS/sarA and saeRSC/sarA mutants, although in the latter strain the impact of eliminating protease production had a reduced effect by comparison to all of these other strains. This is consistent with the fact that this strain also produced reduced amounts of extracellular proteases relative to these strains (Fig. 2A). Interestingly, unlike the bacteremia model in which mutation and constitutive activation of saeRS had opposite effects as would be expected, constitutive activation of saeRS also limited biofilm formation, albeit to a lesser degree than that observed with the isogenic saeRS and sarA mutants. Additionally, this phenotype was not altered in the protease-deficient saeRSC derivative (Fig. 9A). One possible explanation for this is the observation that extracellular nucleases were recently shown to be produced in reduced amounts in saeRS mutants (Olson et al., 2013). Although this previous report did not examine the impact of constitutive activation of saeRS on nuclease production, we confirmed that it is increased and that this is not impacted by eliminating the production of extracellular proteases (Fig. 9B). Thus, to the extent that nucleases limit biofilm formation in vitro (Beenken et al., 2012, Tsang et al., 2008), this could explain both the reduced capacity of the saeRSC mutants to form a biofilm and the reduced impact of eliminating the protease production in this strain.
Fig. 9. Impact of saeRS, and sarA on biofilm formation and nuclease activity in vitro.

Top: Biofilm formation was assessed using a microtiter plate biofilm. Single asterisk indicates statistical significance by comparison to the LAC parent strain (WT). Double asterisks indicate significance of protease-deficient derivatives by comparison to corresponding protease-producing strain. Note that the results observed with the protease-deficient saeRSC/sarA mutant were significant, while those observed with the protease-deficient saeRS, sarA, and saeRS/sarA mutants were not. Note also that eliminating protease production had a significant impact in the saeRS mutant and all sarA mutants irrespective of the functional status of saeRS, but had no impact in the saeRSC derivative. Bottom: Nuclease activity as determined using a FRET-based assay. Strain designations are the same as those indicated in Fig. 2. Single asterisk indicates statistical significance by comparison to the LAC parent strain (WT). Double asterisks indicate significance observed with protease-deficient derivatives by comparison to the same strain with the capacity to produce extracellular proteases.
We recently demonstrated that the impact of extracellular nucleases differs dramatically under in vitro vs. in vivo conditions (Beenken et al., 2012), and based on this we also examined the impact of the functional status of saeRS and sarA in vivo using a murine model of catheter-associated biofilm formation (Weiss et al., 2009). In LAC itself, mutation of saeRS resulted in a slight increase in the capacity to form a biofilm by comparison to its isogenic saeRSC variant (Fig. 10). These relative effects were not evident in the isogenic sarA mutants. In fact, unlike the results observed in our previous study (Zielinska et al., 2012), mutation of sarA in LAC had relatively little impact on biofilm formation. However, it did reverse the increase in biofilm formation observed in the isogenic saeRS mutant (Fig. 10). To some degree, these results must be interpreted with caution by comparison to our earlier report in that these experiments had to be stopped earlier (3 vs. 5 days) owing to the hypervirulence of the saeRSC derivative even by comparison to LAC itself (Fig. 5), which resulted in dramatic skin lesions and the rapid loss of subcutaneously implanted catheters (data not shown). Nevertheless, one potential interpretation of these results, particularly given the impact of the functional status of saeRS on nuclease production (Fig. 9B), is that nucleases are an important limiting factor in biofilm formation during the early stages of biofilm formation in vivo at a time when the impact of mutating sarA on protease production has not yet become fully evident. This is consistent with the observation that biofilm formation was increased, and nuclease production decreased, in the saeRS mutant, while the opposite phenotypes were observed in the saeRSC mutant (Fig. 9). Presumably, the impact of sarA on protease production would predominate relative to that of nucleases at later time points both because they would accumulate to higher levels and because the absence of nucleases would become detrimental has the host mounts a response (Berends et al., 2010). Nevertheless, the fact that mutation of sarA reversed the impact of mutating saeRS suggests that proteases are playing some role even at this early stage of biofilm formation in vivo.
Fig. 10. Impact of saeRS and sarA on biofilm formation in vivo.

Biofilm formation was assessed using a murine model of implant-associated biofilm formation (Weiss et al., 2009). Strain designations are the same as in Fig. 1. Boxes indicate the 25th and 75th percentiles for each group and define the interquartile range (IQR), with the horizontal line indicating the median. Vertical lines define the lowest and highest data points within 1.5 IQR of the lower and higher quartile respectively, with individual dots representing single data points outside this range. Numbers above and within the graph indicate statistical significance between the designated groups.
DISSCUSION
We previously demonstrated that mutation of sarA attenuates the virulence of the USA300 strain LAC in murine models of both bacteremia and implant-associated infection and that this was due in part to the increased production of extracellular proteases (Zielinska et al., 2012). We also demonstrated that expression of both saeRS and sarA is associated with reduced production of extracellular proteases (Mrak et al., 2012). Mutation of saeRS had a less significant impact on protease activity than that of sarA, but it was nevertheless correlated with a reduced capacity to form a biofilm at least under in vitro conditions. This suggests that saeRS and sarA may be viable therapeutic targets based on the common mechanistic theme of their ability to repress protease production, perhaps to the point that therapeutic efficacy could be maximized by developing and exploiting therapeutic strategies that target the intersection of these two regulatory pathways. At the same time, it also raises the possibility that constitutive activation of saeRS could repress protease production to an extent that could compromise therapeutic strategies targeting sarA. Addressing these issues under in vivo conditions was the primary motivation behind the experiments we report.
To these ends, we generated derivatives of the USA300 strain LAC that differ in the functional status of both saeRS and sarA. We also generated derivatives of each that were unable to produce the most relevant extracellular proteases. These strains were compared using both in vitro methods and in vivo models of bacteremia and catheter-associated biofilm formation. The results confirmed that both saeRS and sarA repress the overall production of extracellular proteases, but they also confirmed that saeRS plays a modest role in this regard by comparison to sarA (Mrak et al., 2012). Mutation of either locus severely attenuated the virulence of LAC in a bacteremia model, but the results we present suggest that this does not occur via the common mechanism of increased protease production. Specifically, eliminating protease production restored virulence in the sarA mutant, but not in the saeRS mutant. Rather, the reduced virulence of the saeRS mutant appeared to be a function of the reduced production of critical virulence factors, the specific examples we examined being alpha toxin, protein A, and extracellular nucleases. The possibility that the impact of sarA and saeRS is mediated via different mechanisms is consistent with the observation that concomitant mutation of sarA and saeRS appeared to have an additive effect. Although this was difficult to discern owing to the dramatic impact of mutating each locus alone, this nevertheless suggests an important regulatory balance between saeRS and sarA, with the first being defined primarily by the production of such virulence factors, and the second being defined primarily by their protease-mediated degradation.
One caveat in this regard is that we eliminated the production of all 10 extracellular proteases in the sarA mutant, but we did not eliminate production of the spl-encoded proteases in the LAC saeRS mutant or it’s constitutively active saeRSC variant. This leaves open the possibility that the failure to restore virulence in the saeRS mutant could be due to the continued production of the spl-encoded proteases. However, while mutation of saeRS or sarA results in an overall increase in protease production, the regulatory impact of these loci is not equivalent in that mutation of saeRS results in decreased accumulation of the spl-encoded proteases, while mutation of sarA has the opposite effect (Cassat et al., 2013, Zielinska et al., 2012). This is consistent with our demonstration that overall protease production was reduced to baseline levels in all protease-deficient mutants including those in which the spl operon was left intact. Thus, it seems unlikely that the production of spl-encoded proteases could account for the failure to restore virulence in the saeRS mutant by eliminating the production of aureolysin, SspA, SspB, and ScpA, all of which are produced in increased amounts in both sarA and saeRS mutants (Cassat et al., 2013).
We would also note that, while we did observe some differences in overall growth rate among these strains, they were minor with respect to both growth rate and overall yield (Fig. 11). Moreover, there was no correlation between these differences and relative virulence. For instance, the sarA and protease-deficient sarA mutants grew at comparable rates, but differed dramatically with respect to virulence. Similarly, the saeRSC derivative grew somewhat slower than the isogenic saeRS mutant, but exhibited greater virulence (Fig. 5). Thus, based on these collective results, we propose a model in which the attenuation of saeRS and sarA mutants occurs via different mechanisms, with the impact of saeRS being primarily transcriptional and that of sarA being primarily a function of the increased production of extracellular proteases (Fig. 12). However, these are clearly not the exclusive functions of either locus. For example, a recent report demonstrated that mutation of saeRS in LAC limits virulence in a murine osteomyelitis model owing in part to the aureolysin-mediated degradation of phenol-soluble modulins (PSMs) and its impact on osteoblast function and bone remodeling (Cassat et al., 2013), thus suggesting that the impact of saeRS as a repressor of protease production does in fact play an important role in this specific clinical context. Nevertheless, our results confirm that these different regulatory functions can be distinguished from each other in the context of bacteremia and, to a lesser extent, catheter-associated biofilm formation in vivo.
Fig. 11. Impact of saeRS and sarA on growth in vitro.
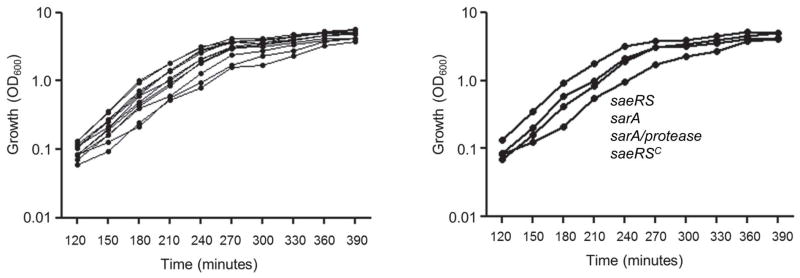
Growth was assessed in tryptic soy broth based on optical density (OD600) at the indicated time points. The panel on the left illustrates the results observed with all strains included in these studies and is intended only to illustrate that the differences in growth rate and overall yield were similar in all strains. The panel on the right breaks out the growth curves of the indicated strains because these were considered the most relevant in the overall context of this report. The strains are listed vertically to correspond to the order of the growth curves observed with each strain (i.e. the greatest difference observed among all strains was that between the saeRS mutant and its constitutively active variant).
Fig. 12. Model for the relative impact of saeRS and sarA in vivo.
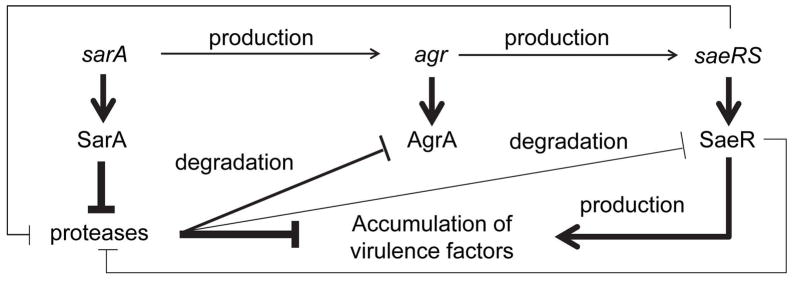
The model proposes that the primary phenotypic impact of sarA under in vivo conditions is due to the increased production of extracellular proteases and the impact of these proteases with respect degradation of critical targets, potentially even including intracellular regulatory proteins. In contrast, while the functional status of saeRS impacts protease production even in an isogenic sarA mutant, the impact of this is small by comparison to the impact of saeRS on the production of these same virulence factors. The end result is a balance between saeRS-mediated production and sarA-mediated degradation, the outcome of which can have a dramatic impact on clinical outcome depending on the context in which it is evaluated. The weight of connecting lines indicates the relative impact of each component of these proposed regulatory interactions.
Indeed, our results suggest that the regulatory balance between saeRS-mediated production and sarA-mediated degradation can be extended to a tissue-specific context. Specifically, mutation of saeRS limited bacterial burdens after systemic infection in the spleen and heart, but had no impact in the kidney. Although the combined effect of mutating sarA and saeRS was evident in all of these tissues, as well as the relative capacity to cause hematogenous bone and joint infection, it was less evident in the kidney than in the spleen and heart. Similarly, eliminating protease production in a LAC saeRS mutant increased virulence as assessed by colony counts in the spleen but had little impact in the heart or kidney. Such tissue-specific results have been reported in other contexts including the relative capacity to produce specific heme-dependent terminal oxidases (Hammer et al., 2013). In fact, to the extent that osteomyelitis is a biofilm-associated infection (Brady et al., 2008), this could explain why we observed relatively little impact of mutating saeRS in our murine catheter-model in that the impact of PSMs on osteoblast viability would presumably not be a factor in this model. Thus, these collective results suggest that saeRS and sarA could both be viable therapeutic targets owing to their impact on protease production. However, given that mutation of sarA has a much greater impact on protease production than mutation of saeRS (Mrak et al., 2012), it will be important to assess the relative impact of mutating each of these loci, both alone and in combination with each other, in the specific context of osteomyelitis. Given the results we report, it also remains imperative to determine the extent to which constitutive activation of saeRS impacts virulence, and whether this has a compromising effect in isogenic sarA mutants, in this clinical context.
Finally, while we previously demonstrated that the increased production of extracellular proteases limits the accumulation of a large number of both surface-associated and extracellular virulence factors in sarA mutants (Mrak et al., 2012, Zielinska et al., 2012), this is the first report we are aware of suggesting that it also limits accumulation of critical intracellular proteins including the response regulators SaeR and AgrA. Our experiments were done with cell lysates prepared from stationary phase cultures, which leaves open the possibility that this reflects the impact of extracellular proteases on intracellular proteins released into the extracellular environment owing to cell lysis during processing. These lysates were prepared from washed cells, which would presumably limit this possibility, but given our focus on S. aureus proteases it was not possible to include protease inhibitors during processing, thus it cannot be eliminated entirely. Nevertheless, these results at least raise the possibility that the increased production of extracellular proteases in sarA mutants, or more likely the dysregulation of the protease activation cascade (Shaw et al., 2004), may well impact the accumulation of intracellular as well as extracellular proteins, thus further complicating the regulatory balance between the saeRS-mediated production of virulence factors and the sarA-mediated protease degradation of these same virulence factors. At the same time, if this is ultimately proven to be true, it suggests that inhibitors of sarA could limit the positive regulatory functions of AgrA and SaeR, both of which make important contributions to the overall virulence of S. aureus.
EXPERIMENTAL PROCEDURES
Ethics Statement
All animal experiments were done in accordance with the policies of the Public Health Service (PHS) policy in the Care and Use of Laboratory Animals, the Animal Welfare Act, and the NIH Guide for the Care and Use of Laboratory Animals in an AAALAC (Association for Assessment and Accreditation of Laboratory Animal Care) internationally accredited facility. All animal procedures were reviewed and approved by the Institutional Animal Care and Use Committee of the University of Arkansas for Medical Sciences.
Bacterial strains and growth conditions
The S. aureus strains used in this study included a plasmid-cured, erythromycin-sensitive derivative of the USA300 strain LAC (Wörmann et al., 2011). To generate derivatives of this strain that differ with respect to the functional status of saeRS, we mutated saeRS using the pKOR1 mutagenesis system (Bae and Schneewind, 2006, Mrak et al., 2012) then complemented this strain by chromosomal insertion of the constitutively active version of saePQRS (saeRSC) from Newman (Luong and Lee et al., 2007, Mainiero et al., 2010, Schäfer et al., 2009). Isogenic sarA and agr mutants were generated by Φ11-mediated transduction from existing mutants (Zielinska et al., 2012, Blevins et al., 1999). Mutations in aur, scpAB, and sspABC were generated using the pKOR1 system, while mutation of the spl operon was done by Φ11-mediated transduction from existing mutants (Zielinska et al., 2012 and Wörmann et al., 2011).
Strains were maintained at −80°C in tryptic soy broth (TSB) containing 25% (vol/vol) glycerol. For each experiment, strains were retrieved from cold storage by plating on tryptic soy agar (TSA) with appropriate antibiotic selection. Antibiotics were used at the following concentrations: erythromycin (Erm; 5 μg per ml), tetracycline (Tet; 5 μg per ml), kanamycin (Kan; 50 μg per ml), and neomycin (Neo; 50 μg per ml), with kanamycin and neomycin being used together to avoid selection for spontaneous mutants. For phenotypic assays, strains were inoculated into TSB or biofilm media, as specified, at an initial optical density at 560 nm (OD560) of 0.05 and to the post-exponential (OD560 = 3.0) or stationary (16 hr) growth phases. All cultures employed for phenotypic assays were grown without antibiotic selection at 37°C with constant aeration and a medium-to-flask volume ratio of 0.40.
Production of extracellular proteases
Protease activity in standardized samples of conditioned medium from stationary phase cultures was assessed using the casein-based Protease Fluorescent Detection Kit (Sigma, St. Louis, MO) with a 1-hour incubation period (Zielinska et al., 2012).
Western Blotting
Accumulation of alpha-toxin and Spa were assessed using standardized samples of conditioned medium from stationary phase cultures. SaeR and AgrA production were assessed using whole cell lysates prepared from intact cells harvested from stationary phase cultures. Western blots were done in triplicate with different biological replicates using appropriate rabbit polyclonal IgG antibodies as previously described (Blevins et al., 1999, Mrak et al., 2012, Zielinska et al., 2012,). Blots were blocked with 0.5% skim milk containing 0.1 mg/ml human IgG (Sigma Chemical Co., St. Louis, MO).
Nuclease Activity
Nuclease activity was assessed using a fluorescence resonance energy transfer (FRET)-based assay (Beenken et al., 2012). Briefly, 25 μl sterilized supernatants from stationary phase cultures (16-hour) were mixed with an equal volume of FRET substrate diluted to 2 μM in buffer consisting of 20 mM Tris, pH 8.0, and 10 mM CaCl2. Results were assessed after 15 min at 30°C using a BioTek Synergy 2 microtiter plate reader (BioTek Instruments, Winooski, VT) with an excitation wavelength of 530 nm and an emission wavelength of 590 nm.
Transcriptional analysis
To assess the levels of saeR, total bacterial RNA was isolated using the Qiagen RNeasy Mini Kit. Quantitative, real-time RT-PCR was performed using primers and TaqMan probes corresponding to saeR as previously described (Mrak et al., 2012). Results were calibrated by comparison to those obtained with the same RNA samples with the 16S ribosomal RNA gene (Zielinska et al., 2011). Results are reported as relative units by comparison to the results observed with the parent strain, the value for which was set to 1.0.
Assessment of biofilm formation in vitro
Biofilm formation was assessed in vitro using a microtiter plate assay in which the wells were first coated with human plasma proteins and the medium (tryptic soy broth) was supplemented with both salt and glucose (Beenken et al., 2010).
Assessment of biofilm formation in vivo
Biofilm formation was assessed in vivo using a murine model of catheter-associated biofilm formation (Weiss et al., 2009). Briefly, uncoated catheters were implanted into each flank of NIH Swiss mice and inoculated by direct injection into the lumen of each catheter with 105 colony-forming units (cfu) of the test strain in a total volume of 100 μl of phosphate-buffered saline (PBS). Catheters were harvested after 3 days and processed for bacterial counts as previously described (Beenken et al., 2012). Because each mouse had two catheters implanted, and because previous experiments have confirmed the absence of cross-contamination between catheters in opposite flanks of the same mouse (Weiss et al., 2009), each catheter was treated as an independent data point (n = 10).
Bacteremia model
Groups of 10 five to eight week old female outbred NIH-Swiss mice (Harlan, Indianapolis, Ind.) were infected via tail vein injection with 5 × 107 cfu (Blevins et al., 2003) of each strain under study. Tissues were harvested from any mice that died or required euthanasia; otherwise, tissues were harvested after 6 days (Zielinska et al., 2012). Briefly, organs were removed aseptically and homogenized on ice. Dilutions of each homogenate were then plated on CHROMagar (Blevins et al., 2003) and the number of colony-forming units (cfu) per organ determined following overnight incubation at 37°C. Additionally, the left hind limb was removed and processed for histological analysis as previously described (Zielinska et al., 2012). All tissue sections were evaluated microscopically in a blinded fashion and scored for the degree of inflammation (0–3) based on the extent of acute inflammatory cells seen in the synovial space and bone. The sections were also scored for the absence (0) or presence (1) of Gram-positive cocci/abscess formation, articular cartilage erosion, cortical bone erosion and physis destruction. Statistical analysis was based on a total histopathological score generated for each animal based on these parameters.
Statistical analysis
Western blot data was logarithmically transformed and analyzed using analysis of variance (ANOVA) models with Tukey’s Multiple Comparison Test. Bacterial count data from harvested catheters were logarithmically transformed and analyzed using analysis of variance (ANOVA) models to evaluate the effect of each mutation. Pair-wise testing was performed using t-tests on the logarithmically transformed data. The significances of the ANOVA and t-test analyses were calculated using permutation tests. Wilcoxon rank-sum tests were used to analyze protease activity and histopathology data, while Kaplan-Meier methods were used to calculate survival distributions for lethality studies. Survival distributions were compared using log-rank tests. Statistical analyses were performed using R (version 2.7; The Foundation for Statistical Computing), SigmaPlot and GraphPad Prism 5.0. P-values ≤0.05 were considered to be statistically significant.
Acknowledgments
Research in the Smeltzer laboratory is supported by grants from the National Institute of Allergy and Infectious Disease (R01-AI069087, R56-AI074935, R56-AI093126) and the Department of Defense U.S. Army Medical Research and Materiel Command (W81XWH-10-1-0991, W81XWH-12-2-0020). AKZ was also supported by a pre-doctoral fellowship from the American Heart Associated (10PRE3220017). Support was also provided by the Translational Research Institute (UL1TR000039) through the NIH National Center for Research Resources and National Center for Advancing Translational Sciences, and by the core facilities supported by the Center for Microbial Pathogenesis and Host Inflammatory Responses (P20-GM103450). The content is the responsibility of the authors and does not necessarily represent the views of the NIH or Department of Defense. We thank Dr. Christiane Wolz for providing the constitutively active saeRS construct and Dr. Alex Horswill for providing protease mutants. We also thank Drs. Eric Skaar and James Cassat at Vanderbilt University Medical Center for careful review of our manuscript.
References
- Anderson MJ, Lin YC, Gillman AN, Parks PJ, Schlievert PM, Peterson ML. Alpha-toxin promotes Staphylococcus aureus mucosal biofilm formation. Front Cell Infect Microbiol. 2012;2:64. doi: 10.3389/fcimb.2012.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer NK, Mazaitis MJ, Costerton JW, Leid JG, Powers ME, Shirtliff ME. Staphylococcus aureus biofilms: properties, regulation, and roles in human disease. Virulence. 2011;2:445–459. doi: 10.4161/viru.2.5.17724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae T, Schneewind O. Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid. 2006;55:58–63. doi: 10.1016/j.plasmid.2005.05.005. [DOI] [PubMed] [Google Scholar]
- Beenken KE, Blevins JS, Smeltzer MS. Mutation of sarA in Staphylococcus aureus limits biofilm formation. Infect Immun. 2003;71:4206–4211. doi: 10.1128/IAI.71.7.4206-4211.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beenken KE, Mrak LN, Griffin LM, Zielinska AK, Shaw LN, Rice KC, Horswill AR, Bayles KW, Smeltzer MS. Epistatic relationships between sarA and agr in Staphylococcus aureus biofilm formation. PLoS One. 2010;5:e10790. doi: 10.1371/journal.pone.0010790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beenken KE, Spencer H, Griffin LM, Smeltzer MS. Impact of extracellular nuclease production on the biofilm phenotype of Staphylococcus aureus under in vitro and in vivo conditions. Infect Immun. 2012;80:1634–1638. doi: 10.1128/IAI.06134-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berends ET, Horswill AR, Haste NM, Monestier M, Nizet V, von Köckritz-Blickwede M. Nuclease expression by Staphylococcus aureus facilitates escape from neutrophil extracellular traps. J Innate Immun. 2010;2:576–86. doi: 10.1159/000319909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blevins JS, Gillaspy AF, Rechtin TM, Hurlburt BK, Smeltzer MS. The staphylococcal accessory regulator (sar) represses transcription of the Staphylococcus aureus collagen adhesion gene (cna) in an agr-independent manner. Mol Microbiol. 1999;33:317–326. doi: 10.1046/j.1365-2958.1999.01475.x. [DOI] [PubMed] [Google Scholar]
- Blevins JS, Elasri MO, Allmendinger SD, Beenken KE, Skinner RA, Thomas JR, Smeltzer MS. Role of sarA in the pathogenesis of Staphylococcus aureus musculoskeletal infection. Infect Immun. 2003;71:516–523. doi: 10.1128/IAI.71.1.516-523.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boles BR, Horswill AR. Agr-mediated dispersal of Staphylococcus aureus biofilms. PLoS Pathog. 2008;4:e1000052. doi: 10.1371/journal.ppat.1000052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis. 2009;48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- Brady RA, Leid JG, Calhoun JH, Costerton JW, Shirtliff ME. Osteomyelitis and the role of biofilms in chronic infection. FEMS Immunol Med Microbiol. 2008;52:13–22. doi: 10.1111/j.1574-695X.2007.00357.x. [DOI] [PubMed] [Google Scholar]
- Caiazza NC, O’Toole GA. Alpha-toxin is required for biofilm formation by Staphylococcus aureus. J Bacteriol. 2003;185:3214–3217. doi: 10.1128/JB.185.10.3214-3217.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassat JE, Hammer ND, Campbell JP, Benson MA, Perrien DS, Mrak LN, Smeltzer MS, Torres VJ, Skaar EP. A secreted bacterial protease tailors the Staphylococcus aureus virulence repertoire to modulate bone remodeling during osteomyelitis. Cell Host Microbe. 2013;13:759–772. doi: 10.1016/j.chom.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Krishnan V, Macon K, Manne K, Narayana SV, Schneewind O. Secreted proteases control autolysin-mediated biofilm growth of Staphylococcus aureus. J Biol Chem. 2013;288:29440–29452. doi: 10.1074/jbc.M113.502039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien Y, Manna AC, Projan SJ, Cheung AL. SarA level is a determinant of agr activation in Staphylococcus aureus. J Biol Chem. 1998;30(5):991–1001. doi: 10.1046/j.1365-2958.1998.01126.x. [DOI] [PubMed] [Google Scholar]
- Claro T, Widaa A, McDonnell C, Foster TJ, O’Brien FJ, Kerrigan SW. Staphylococcus aureus protein A binding to osteoblast tumour necrosis factor receptor 1 results in activation of nuclear factor kappa B and release of interleukin-6 in bone infection. Microbiology. 2013;159:147–54. doi: 10.1099/mic.0.063016-0. [DOI] [PubMed] [Google Scholar]
- Edwards AM, Bowden MG, Brown EL, Laabei M, Massey RC. Staphylococcus aureus extracellular adherence protein triggers TNFα release, promoting attachment to endothelial cells via protein A. PLos One. 2012;7:e43046. doi: 10.1371/journal.pone.0043046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundmeier M, Hussain M, Becker P, Heilman C, Peters G, Sinha B. Truncation of fibronectin-binding proteins in Staphylococcus aureus strain Newman leads to deficient adherence and host cell invasion due to loss of cell wall anchor function. Infect Immun. 2004;72:7155–7163. doi: 10.1128/IAI.72.12.7155-7163.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer ND, Reniere ML, Cassat JE, Zhang Y, Hirsch AO, Indriati Hood M, Skaar EP. Two heme-dependent terminal oxidases power Staphylococcus aureus organ-specific colonization of the vertebrate host. MBio. 2013;4:e00241–13. doi: 10.1128/mBio.00241-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes NE, Johnson PD, Howden BP. Relationship between vancomycin-resistant Staphylococcus aureus, vancomycin-intermediate S. aureus, high vancomycin MIC, and outcome in serious S. aureus infections. J Clin Microbiol. 2012;50:2548–2552. doi: 10.1128/JCM.00775-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jusko M, Potempa J, Kantyka T, Bielecka E, Miller HK, Kalinska M, Dubin G, Garred P, Shaw LN, Blom AM. Staphylococcal proteases aid in evasion of the human complement system. J Innate Immun. 2014;6:31–46. doi: 10.1159/000351458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantyka T, Pyrc K, Gruca M, Smagur J, Plaza K, Guzik K, Zeglen S, Ochman M, Potempa J. Staphylococcus aureus proteases degrade lung surfactant protein A potentially impairing innate immunity of the lung. J Innate Immun. 2013;5:251–260. doi: 10.1159/000345417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi SD, Malachowa N, Whitney AR, Braughton KR, Gardner DJ, Long D, Bubeck Wardenburg J, Schneewind O, Otto M, Deleo FR. Comparative analysis of USA300 virulence determinants in a rabbit model of skin and soft tissue infection. J Infect Dis. 2011;204:937–941. doi: 10.1093/infdis/jir441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laverty G, Gorman SP, Gilmore BF. Biomolecular mechanisms of staphylococcal biofilm formation. Future Microbiol. 2013;8:509–524. doi: 10.2217/fmb.13.7. [DOI] [PubMed] [Google Scholar]
- Lower SK, Lamlertthon S, Casillas-Ituarte NN, Lins RD, Yongsunthon R, et al. Polymorphisms in fibronectin binding protein A of Staphylococcus aureus are associated with infection of cardiovascular devices. Proc Natl Acad Sci USA. 2011;108:18372–18377. doi: 10.1073/pnas.1109071108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luong TT, Lee CY. Improved single-copy integration vectors for Staphylococcus aureus. J Microbiol Methods. 2007;70:186–90. doi: 10.1016/j.mimet.2007.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainiero M, Goerke C, Geiger T, Gonser C, Herbert S, Wolz C. Differential target gene activation by the Staphylococcus aureus two-component system saeRS. J Bacteriol. 2010;192:613–623. doi: 10.1128/JB.01242-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino N, Toledo-Arana A, Vergara-Irigaray M, Valle J, Solano C, Calvo E, Lopez JA, Foster TJ, Penadés JR, Lasa I. Protein A-mediated multicellular behavior in Staphylococcus aureus. J Bacteriol. 2009;191:832–843. doi: 10.1128/JB.01222-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootz JM, Malone CL, Shaw LN, Horswill AR. Staphopains modulate Staphylococcus aureus biofilm integrity. Infect Immun. 2013;81:3227–3238. doi: 10.1128/IAI.00377-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrak LN, Zielinska AK, Beenken KE, Mrak IN, Atwood DN. saeRS and sarA act synergistically to repress protease production and promote biofilm formation in Staphylococcus aureus. PLoS One. 2012;7:e38453. doi: 10.1371/journal.pone.0038453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick RP, Jiang D. The staphylococcal saeRS system coordinates environmental signals with agr quorum sensing. Microbiol. 2003;149:2709–17. doi: 10.1099/mic.0.26575-0. [DOI] [PubMed] [Google Scholar]
- Olson ME, Nygarrd TK, Ackermann L, Watkins RL, Zurek OW, Pallister KB, et al. Staphylococcus aureus nuclease is an SaeRS-dependent virulence factor. Infect Immun. 2013;81:1316–1324. doi: 10.1128/IAI.01242-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill E, Pozzi C, Houston P, Humphreys H, Robinson DA, Loughman A, Foster TJ, O’Gara JP. A novel Staphylococcus aureus biofilm phenotype mediated by the fibronectin-binding proteins, FnBPA and FnBPB. J Bacteriol. 2008;190:3835–3850. doi: 10.1128/JB.00167-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oscarsson J, Kanth A, Tegmark-Wisell K, Arvidson S. SarA is a repressor of hla (alpha-hemolysin) transcription in Staphylococcus aureus: its apparent role as an activator of hla in the prototype strain NCTC 8325 depends on reduced expression of sarS. J Bacteriol. 2006;188:8526–8533. doi: 10.1128/JB.00866-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto M. Basis of virulence in community-associated methicillin-resistant Staphylococcus aureus. Annu Rev Microbiol. 2010;64:143–162. doi: 10.1146/annurev.micro.112408.134309. [DOI] [PubMed] [Google Scholar]
- Otto M. MRSA virulence and spread. Cell Microbiol. 2012;10:1513–1521. doi: 10.1111/j.1462-5822.2012.01832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto M. Staphylococcal biofilms. Curr Top Microbiol Immunol. 2008;322:207–228. doi: 10.1007/978-3-540-75418-3_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Lee JH, Kim CJ, Lee JC, Cho MH, Lee J. Extracellular protease in Actinomycetes culture supernatants inhibits and detaches Staphylococcus aureus biofilm formation. Biotechnol Lett. 2012;34:655–661. doi: 10.1007/s10529-011-0825-z. [DOI] [PubMed] [Google Scholar]
- Periasamy S, Joo HS, Duong AC, Bach TH, Tan VY, Chatterjee SS, Cheung GY, Otto M. How Staphylococcus aureus biofilms develop their characteristic structure. PNAS (USA) 2012;109:1281–1286. doi: 10.1073/pnas.1115006109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed SB, Wesson CA, Liou LE, Trumble WR, Schlievert PM, Bohach GA, Bayles KW. Molecular characterization of a novel Staphylococcus aureus serine protease operon. Infect Immun. 2001;69:1521–1527. doi: 10.1128/IAI.69.3.1521-1527.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Römling U, Balsalobre C. Biofilm infections, their resilience to therapy and innovative treatment strategies. J Intern Med. 2012;272:541–561. doi: 10.1111/joim.12004. [DOI] [PubMed] [Google Scholar]
- Schäfer D, Lâm TT, Geiger T, Mainiero M, Engelmann S, Hussain M, Bosserhoff A, Frosch M, Bischoff M, Wolz C, Reidl J, Sinha BA. A point mutation in the sensor histidine kinase SaeS of Staphylococcus aureus strain Newman alters the response to biocide exposure. J Bacteriol. 2009;191:7306–7314. doi: 10.1128/JB.00630-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw L, Golonka E, Potempa J, Foster SJ. The role and regulation of the extracellular proteases of Staphylococcus aureus. Microbiology. 2004;150:217–28. doi: 10.1099/mic.0.26634-0. [DOI] [PubMed] [Google Scholar]
- Sugimoto S, Iwamoto T, Takada K, Okuda K, Tajima A, Iwase T, Mizunoe Y. Staphylococcus epidermidis Esp degrades specific proteins associated with Staphylococcus aureus biofilm formation and host-pathogen interaction. J Bacteriol. 2013;195:1645–1655. doi: 10.1128/JB.01672-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang LH, Cassat JE, Shaw LN, Beenken KE, Smeltzer MS. Factors contributing to the biofilm-deficient phenotype of Staphylococcus aureus sarA mutants. PLoS One. 2008;3:e3361. doi: 10.1371/journal.pone.0003361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss EC, Spencer HJ, Daily SJ, Weiss BD, Smeltzer MS. Impact of sarA on antibiotic susceptibility of Staphylococcus aureus in a catheter-associated in vitro model of biofilm formation. Antimicro. Agents Chemother. 2009;53:2475–2482. doi: 10.1128/AAC.01432-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss EC, Zielinska A, Beenken KE, Spencer HJ, Daily SJ, Smeltzer MS. Impact of sarA on daptomycin susceptibility of Staphylococcus aureus biofilms in vivo. Antimicrob Agents Chemother. 2009;53:4096–4102. doi: 10.1128/AAC.00484-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wörmann ME, Reichmann NT, Malone CL, Horswill AR, Gründling A. Proteolytic cleavage inactivates the Staphylococcus aureus lipoteichoic acid synthase. J Bacteriol. 2011;193:5279–5291. doi: 10.1128/JB.00369-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zdzalik M, Karim AY, Wolski K, Buda P, Wojcik K, Brueggemann S, Wojciechowski P, Eick S, Calander AM, Jonsson IM, Kubica M, Polakowska K, Miedzobrodzki J, Wladyka B, Potempa J, Dubin G. Prevalence of genes encoding extracellular proteases in Staphylococcus aureus - important targets triggering immune response in vivo. FEMS Immunol Med Microbiol. 2012;66:220–229. doi: 10.1111/j.1574-695X.2012.01005.x. [DOI] [PubMed] [Google Scholar]
- Zielinska AK, Beenken KE, Joo HS, Mrak LN, Griffin LM, Loung TT, Lee CY, Otto M, Shaw LN, Smeltzer MS. Defining the strain-dependent impact of the staphylococcal accessory regulator (sarA) on the alpha-toxin phenotype of Staphylococcus aureus. J Bacteriol. 2011;193:2948–2958. doi: 10.1128/JB.01517-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielinska AK, Beenken KE, Mrak LN, Spencer HJ, Post GR, Skinner RA, Tackett AJ, Horswill AR, Smeltzer MS. sarA-mediated repression of protease production plays a key role in the pathogenesis of Staphylococcus aureus USA300 isolates. Mol Microbiol. 2012;86:1183–1196. doi: 10.1111/mmi.12048. [DOI] [PMC free article] [PubMed] [Google Scholar]