

Abstract
Glucocorticoids are primary stress hormones necessary for life that regulate numerous physiological processes in an effort to maintain homeostasis. Synthetic derivatives of these hormones have been mainstays in the clinic for treating inflammatory diseases, autoimmune disorders, and hematological cancers. The physiological and pharmacological actions of glucocorticoids are mediated by the glucocorticoid receptor (GR), a member of the nuclear receptor superfamily of ligand-dependent transcription factors. Ligand-occupied GR induces or represses the transcription of thousands of genes by direct binding to DNA response elements and/or by physically associating with other transcription factors. The traditional view that glucocorticoids act through a single GR protein has changed dramatically with the discovery of a large cohort of receptor isoforms with unique expression, gene-regulatory, and functional profiles. These GR subtypes are derived from a single gene by alternative splicing and alternative translation initiation mechanisms. Post-translational modification of these GR isoforms further expands the diversity of glucocorticoid responses. Here, we discuss the origin and molecular properties of the GR isoforms and their contribution to the specificity and sensitivity of glucocorticoid signaling in healthy and diseased tissues.
INTRODUCTION
Glucocorticoids are hormones essential for life that are synthesized and released by the adrenal cortex in a circadian manner and in response to stress. The secretion of these hormones is controlled by the hypothalamic-pituitary-adrenal (HPA) axis (Figure 1). Internal and external signals trigger the hypothalamus to release corticotropin releasing hormone (CRH), which acts on the anterior pituitary to stimulate the synthesis and secretion of adrenocorticotropic hormone (ACTH). ACTH then acts on the adrenal cortex to stimulate the production and secretion of glucocorticoids. Acting on nearly every tissue and organ in the body, glucocorticoids function to maintain homeostasis both in response to normal diurnal changes in metabolism and in the face of stressful perturbations. Glucocorticoids regulate a plethora of physiological processes including intermediary metabolism, immune function, skeletal growth, cardiovascular function, reproduction, and cognition (1, 2). In a classic negative feedback loop, glucocorticoids also target the hypothalamus and anterior pituitary to inhibit the production and release of CRH and ACTH and thereby limit both the magnitude and duration of the glucocorticoid increase (Figure 1).
Figure 1.
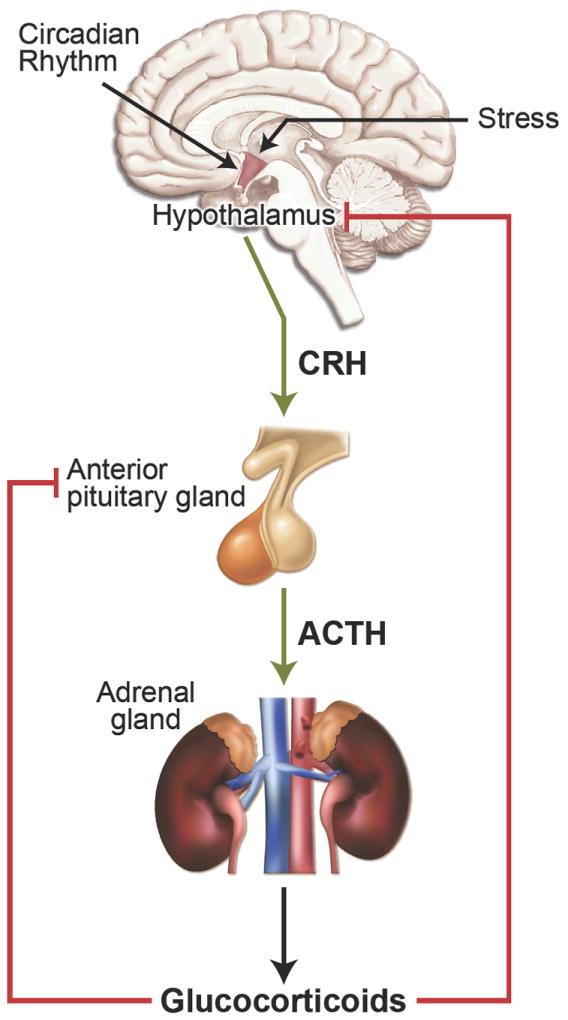
Regulation of glucocorticoid hormone secretion by the hypothalamic-pituitary-adrenal (HPA) axis.
Because of their powerful anti-inflammatory and immunosuppressive actions, glucocorticoids are one of the most widely prescribed drugs in the world today (3, 4). Synthetic glucocorticoids have been indispensable over the last half century for treating inflammatory and autoimmune diseases, such as asthma, allergy, sepsis, rheumatoid arthritis, ulcerative colitis, and multiple sclerosis. They are also commonly prescribed to prevent organ transplant rejection and to treat cancers of the lymphoid system such as leukemias, lymphomas, and myelomas. Unfortunately, the therapeutic benefits of glucocorticoids are limited by severe side effects that develop in patients chronically treated with these steroids (3, 5). Adverse effects include osteoporosis, skin atrophy, diabetes, abdominal obesity, glaucoma, growth retardation in children, and hypertension. In addition, patients on long term glucocorticoid therapy frequently develop tissue-specific glucocorticoid resistance. Understanding the factors at a molecular level that control the cellular response to glucocorticoids is a critical goal of current research as progress in this area will facilitate the development of novel glucocorticoids with improved benefit/risk ratios.
The physiological and pharmacological actions of glucocorticoids are mediated by the glucocorticoid receptor (GR, NR3C1), a member of the nuclear receptor superfamily of ligand-dependent transcription factors (6). Following glucocorticoid binding, GR induces or represses the transcription of target genes which can comprise up to 10-20% of the human genome (7-9). Consistent with the pleiotropic actions of glucocorticoids, GR is expressed in nearly every cell of the body and is necessary for life after birth (10). The cellular response to glucocorticoids is remarkably diverse, exhibiting profound variability in specificity and sensitivity (11, 12). For example, glucocorticoids induce the killing of thymocytes and osteoblasts but promote the survival of hepatocytes and cardiomyocytes. In addition, glucocorticoid sensitivity varies among individuals, within tissues of the same individual, and even within the same cell during different stages of the cell cycle (13, 14). While cell-type specific alterations in ligand bioavailability and cofactor expression can modulate the glucocorticoid response, recent studies have also identified a major role for an expanding array of GR isoforms (15, 16). GR is derived from a single gene; however, multiple GR proteins exist due to alternative splicing and alternative translation initiation. This large cohort of functionally distinct receptor subtypes are subject to various post-translational modifications that further regulate their signaling properties. Consequently, the cellular response to glucocorticoids is determined in large measure by the expressed complement and composite actions of the individual GR isoforms. In this review, we discuss the molecular heterogeneity of GR and its potential contribution to the regulation and dysregulation of glucocorticoid signaling.
GR SIGNALING
Classic GR Signaling Pathway
GR is a modular protein composed of three major domains: an N-terminal transactivation domain (NTD), a central DNA-binding domain (DBD), and a C-terminal ligand binding domain (LBD) (Figure 2) (17). The DBD and LBD are separated by a flexible region of the protein termed the hinge region. The DBD is the most conserved domain across the nuclear receptor family and contains two zinc finger motifs that recognize and bind target DNA sequences called glucocorticoid-responsive elements (GREs). The NTD houses a powerful transcriptional activation function (AF1) that interacts with coregulators and the basal transcription machinery and is the primary site for post-translational modifications (Figure 2). The LBD, consisting of 12 α-helices and four β-sheets, forms a hydrophobic pocket for binding glucocorticoids and also contains an activation function (AF2) that interacts with coregulators in a ligand-dependent manner (18). Two nuclear localization signals, NL1 and NL2, are located at the DBD/hinge region junction and within the LBD, respectively.
Figure 2.
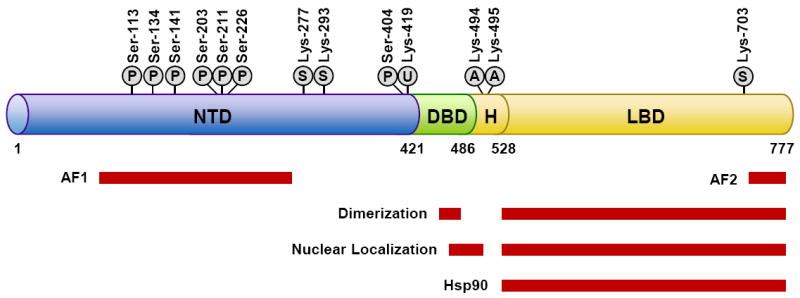
GR domain structure and sites of post-translational modification. Shown are the domains of GR and regions of the receptor involved in transactivation (AF1 and AF2), dimerization, nuclear localization, and hsp90 binding. Also depicted are the amino acid residues modified by phosphorylation (P), sumoylation (S), ubiquitination (U), and acetylation (A). Numbers are for human GR.
In the absence of hormone, GR resides predominantly in the cytoplasm of cells as part of a large multi-protein complex that includes chaperone proteins (hsp90, hsp70, and p23) and immunophilins of the FK506 family (FKBP51 and FKBP52) (Figure 3) (19, 20). These proteins maintain the receptor in a conformation that is transcriptionally inactive but favors high affinity ligand binding. Cortisol, the most abundant endogenous glucocorticoid in man, is transported in the blood predominantly bound to corticosteroid-binding globulin (CBG). CBG not only facilitates cortisol distribution but also plays a role in its release to tissues. CBG-free cortisol passively diffuses across the plasma membrane; however, its bioavailability within the cell is controlled by two enzymes working in an opposing manner (21). 11β-Hydroxysteroid dehydrogenase type 2 (11β-HSD2) oxidizes cortisol into the inactive metabolite cortisone; whereas 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) converts cortisone to cortisol (Figure 3). Changes in the level and/or activity of these enzymes can contribute to cellular differences in glucocorticoid sensitivity. In fact, inhibitors of 11β-HSD1 have been designed to limit the adverse metabolic side effects of elevated endogenous glucocorticoids (22). In contrast to cortisol, most synthetic glucocorticoids do not bind CBG and are not metabolized by 11β-HSD2. Upon binding glucocorticoids, GR undergoes a conformational change resulting in the dissociation of the associated proteins. This structural rearrangement exposes the two nuclear localization signals, and GR is rapidly translocated into the nucleus through nuclear pores.
Figure 3.
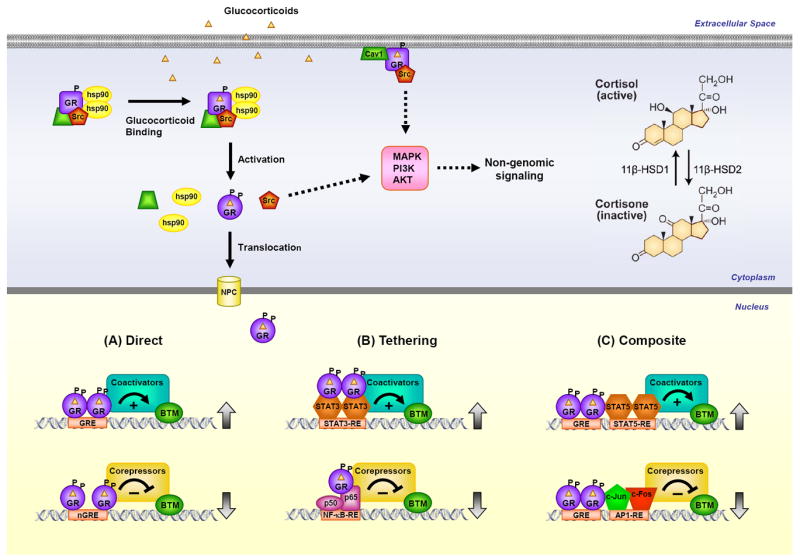
GR signaling pathways. Glucocorticoid-activated GR regulates gene expression in 3 primary ways: binding directly to DNA (A), tethering itself to other DNA-bound transcription factors (B), or binding directly to DNA and interacting with neighboring DNA-bound transcription factors (C). GR can also signal in a non-genomic manner through alterations in the activity of various kinases.
Once inside the nucleus, GR binds directly to GREs and regulates the expression of target genes (Figure 3) (23, 24). The consensus GRE sequence, GGAACAnnnTGTTCT, is an imperfect palindrome that is comprised of two 6-base pair half sites. GR binds this element as a homodimer, with each half site occupied by one receptor subunit. The three nucleotide spacing between the two half sites is strictly required for GR to dimerize on this element. The GRE has been shown to mediate the glucocorticoid-dependent induction of many genes and therefore is often referred to as an activating or positive GRE. However, recent genome-wide analyses have revealed that GR occupancy of the canonical GREs can also lead to the repression of target genes (25). These findings suggest a critical role for factors outside the GRE sequence, such as epigenetic regulators and chromatin context, in determining the polarity of the transcriptional response. A negative GRE (nGRE) has also been recently described that mediates glucocorticoid-dependent repression of specific genes (26). The consensus nGRE sequence, CTCC(n)0-2GGAGA, is palindromic but differs from the classic GRE in sequence, in having a variable spacer that ranges from 0 to 2 nucleotides, and in being occupied by two GR monomers that do not homodimerize (27). These nGREs are abundant throughout the genome, however more work is needed to clarify the extent this motif is utilized by GR for directly repressing target genes and whether this element can also be employed in the activation of gene expression.
Global GR recruitment assays indicate that only a small fraction of GREs are actually occupied by the receptor, and the specific sites of GR binding vary in a tissue-specific manner due to differences in chromatin accessibility and exposure of the GRE (28). These findings suggest that the widely varying effects of glucocorticoids on different tissues can be attributed in part to cell-type specific differences in the chromatin landscape that influence which GREs are accessible for GR binding. In addition, the concentration of glucocorticoids at which GR binds GREs and regulates gene expression varies throughout the genome (29). Some GREs are occupied by GR at very low concentrations of glucocorticoids (hypersensitive) whereas others require high doses of ligand for GR binding to occur. Both chromatin accessibility and other DNA binding proteins appear to govern the sensitivity of specific GREs. The identification of hypersensitive GREs suggests that low-dose glucocorticoid therapy may provide a novel treatment option for regulating specific sets of genes that avoids the deleterious side effects of high exogenous glucocorticoids.
Genome-wide analyses have also found that the majority of GR binding sites are located outside the promoter of glucocorticoid-responsive genes in intergenic or intragenic regions often far removed from the transcription start site (30). For example, glucocorticoid induction of βarrestin1 and repression of βarrestin2 occur via an intron1 GRE and intron11 nGRE, respectively (31). Both βarrestin isoforms play critical roles in the termination and transduction of GPCR signals. By altering the ratio of βarrestin1 and βarrestin2, glucocorticoids shift the balance of G protein and βarrestin-dependent signaling responses for a given GPCR (31). This redirection of the GPCR signaling profile may account for the superior clinical efficacy of glucocorticoid/β2 adrenergic receptor (β2AR) agonist combination therapies currently used for treating asthma and COPD. The β2AR is preferentially desensitized by βarrestin2. Therefore, the glucocorticoid-dependent reduction in βarrestin2 may counteract β2AR desensitization and allow agonist bound β2AR to promote a greater and/or more sustained relaxation response in airway smooth muscle cells. In addition, lower levels of βarrestin2 in lung epithelial cells may impair the pro-inflammatory effects of β2AR agonists that depend on the signaling function of βarrestin2. Another example of a GR binding site located a great distance from the transcription start site is the intragenic nGRE recently identified in exon 6 of the GR gene that mediates homologous down regulation of GR expression, a mechanism that may limit therapeutic responses to glucocorticoids (32). The location of GREs and nGREs at great distances from the transcription start site suggests that these response elements can loop to the promoter areas of target genes to regulate transcription.
The interaction of GR with DNA is highly dynamic, with GR cycling between bound and unbound states every few seconds (33). Once bound to the GRE, the receptor undergoes additional conformational changes that lead to the recruitment of coregulators and chromatin-remodeling complexes that modulate gene transcription rates by affecting the activity of RNA polymerase II (34-36). Cofactors that mediate transcriptional activation include steroid receptor coactivators (SRC1-3), the histone acetyltransferases CBP/p300, and the nuclear methylase coactivator-associated arginine methyltransferase (CARM1). NCoR and SMRT are established corepressors that are recruited to GR bound to nGREs. The specific cofactors assembled and their subsequent activity are dictated by both the nature of the glucocorticoid ligand and the specific GRE sequence bound by the receptor (37, 38).
GR can also regulate the transcription of target genes by physically interacting with other transcription factors (Figure 3). The association of GR with specific members of the STAT family, either apart from or in conjunction with GRE binding, has been shown to enhance the transcription of responsive genes (39). In contrast, the interaction of GR with the proinflammatory transcription factors, AP1 and NF-κB, antagonizes their activity and is considered to be a primary mechanism by which glucocorticoids suppress inflammation. GR directly binds the Jun subunit of AP1 and the p65 subunit of NF-κB and interferes with the transcriptional activation function of these two proteins (40, 41). For some genes, the repression is accomplished by GR tethering itself to these DNA-bound proteins without itself directly interacting with the DNA. For other genes, however, GR functions in a composite manner, binding directly to a GRE and physically associating with AP1 or NF-κB bound to a neighboring site on the DNA. GR-dependent recruitment of the GR interacting protein 1 (GRIP1), a transcriptional coregulator of the p160 family, is important for this inhibition (42). Interestingly, recent work has demonstrated that the anti-inflammatory actions of glucocorticoids differ in a sex-specific manner (43). Glucocorticoids regulate a greater number of inflammatory genes and elicit a greater anti-inflammatory response in male rats compared to female rats. Sex-specific expression profiles of coregulatory molecules may underlie the sexual dimorphism by modulating the repressive actions of GR on AP1 and NF-κB. The sex-specific differences in the anti-inflammatory actions of glucocorticoids not only provide a mechanistic basis for the predisposition of females to autoimmune diseases, such as rheumatoid arthritis and systemic lupus erythematous, but also suggest that therapeutic doses of glucocorticoids should be optimized according to the sex of the patient.
Non-Classical GR Signaling Pathway
While the principal effects of glucocorticoids are mediated by transcriptional responses that occur in minutes to hours, a growing body of evidence suggests that GR may also act via non-genomic mechanisms to elicit rapid cellular responses that occur within a few seconds to minutes and do not require changes in gene expression (Figure 3) (44, 45). Multiple mechanisms appear to be involved in these signaling events that ultimately impinge on the activity of various kinases, such as PI3K, AKT, and MAPKs. For example, the glucocorticoid-dependent release of accessory proteins associated with unliganded GR in the cytoplasm, such as the non-receptor tyrosine kinase c-Src, can mediate non-genomic actions of glucocorticoids. When liberated from the GR complex, c-Src activates multiple kinase cascades that lead to the phosphorylation of annexin 1, inhibition of cytosolic phospholipase A2 activity, and impaired release of arachidonic acid (46, 47). GR has also been reported to localize at the plasma membrane in caveolae via an interaction with caveolin1 (48). Glucocorticoid activation of this membrane associated GR regulates gap junction intercellular communication and neural progenitor cell proliferation by a mechanism that requires c-Src activity and rapid MAPK-dependent phosphorylation of connexin-43 (49). The existence of non-genomic signaling adds greater complexity and diversity to glucocorticoids and their biological actions, and raises the possibility that selective modulators of GR-dependent genomic or non-genomic pathways may be therapeutically advantageous.
GR Polymorphisms
The capacity of GR to function as a transcriptional activator or repressor is affected by several polymorphisms in the GR gene that alter the amino acid sequence of the encoded receptor. The ER22/23EK GR polymorphism occurs in ~3% of the population and results in an arginine to lysine change at position 23 within the NTD (50, 51). GR with the ER22/23EK polymorphism exhibits reduced transcriptional activity on both glucocorticoid responsive reporters and endogenous genes and has been associated with glucocorticoid insensitivity. Persons with the ER22/23EK polymorphism have a lower tendency to develop impaired glucose tolerance, type-2 diabetes, and cardiovascular disease suggesting the relative resistance of ER22/23EK carriers to endogenous glucocorticoids may result in a more favorable metabolic profile. The N363S polymorphism occurs in ~4% of the population and results in an asparagine to serine substitution in the NTD of GR (50). In contrast to ER22/23EK, the presence of the N363S polymorphism enhances the transcriptional activity of GR and is associated with glucocorticoid hypersensitivity. In addition, a genome-wide microarray analysis has revealed a unique pattern of gene regulation for the N363S polymorphism (52). N363S carriers have been reported to have an increased body mass index and a tendency towards decreased bone mineral density (51). These GR polymorphisms may account at least in part for the variability in the glucocorticoid response observed among individuals treated with these steroids.
GR SPLICE VARIANTS
The human GR gene is composed of 9 exons. The GR NTD is encoded primarily by exon 2, the DBD is encoded by exons 3 and 4, and the hinge region and LBD are encoded by exons 5-9 (Figure 4). Alternative splicing in exon 9 near the end of the GR primary transcript generates two receptor isoforms, termed GRα and GRβ (Figure 4) (53, 54). GRα is derived from the end of exon 8 being joined to the beginning of exon 9, whereas GRβ results from the end of exon 8 being joined to downstream sequences in exon 9. These two proteins are identical through amino acid 727 but then diverge. GRα, the classic GR protein that mediates the actions of glucocorticoids, contains an additional 50 amino acids. The splice variant GRβ contains an additional, nonhomologous 15 amino acids. The unique GRβ carboxyl-terminal sequence confers several distinct properties to this receptor isoform. GRβ does not bind glucocorticoid agonists, resides constitutively in the nucleus of cells, and by itself is inactive on glucocorticoid-responsive reporter genes (55, 56). However, when co-expressed with GRα, GRβ functions as a dominant negative inhibitor and antagonizes the activity of GRα on many glucocorticoid-responsive target genes. Competition for GRE binding, competition for transcriptional coregulators, and formation of inactive GRα/GRβ heterodimers have each been proposed to underlie the antagonism mediated by GRβ.
Figure 4.
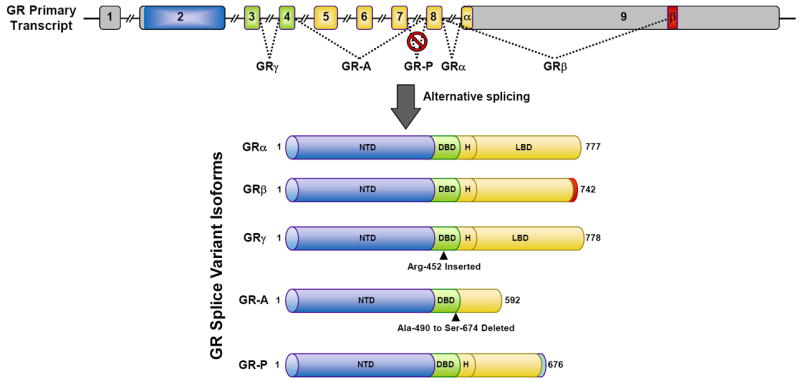
GR splice variants. The GR primary transcript is composed of 9 exons. Exon 2 encodes the NTD, exons 3-4 encode the DBD, and exons 5-9 encode the hinge region (H) and LBD. GRα results from splicing exon 8 to the beginning of exon 9. GRβ, GRγ, GR-A, and GR-P are generated by the depicted alternative splicing events.
The ability of GRβ to inhibit the activity of GRα suggests that high levels of GRβ may lead to glucocorticoid resistance. GRβ is prevalent in many cells and tissues but generally is found at lower levels than GRα. However, GRβ is abundant in certain cell-types such as neutrophils and epithelial cells. Moreover, the expression of GRβ can be selectively increased by proinflammatory cytokines and other immune activators and lead to reduced glucocorticoid sensitivity (57-59). Elevated levels of GRβ have been associated with glucocorticoid resistance in a variety of inflammatory diseases including asthma, rheumatoid arthritis, ulcerative colitis, nasal polyposis, systemic lupus erythematous, sepsis, acute lymphoblastic leukemia, and chronic lymphocytic leukemia (56). Upregulation of GRβ and diminished GRα signaling is also observed in erythroid cells expanded from polycythemia vera patients (60). The molecular factors that control GRβ expression are poorly understood, but several studies have implicated the involvement of the splicing factor SRp30c (61-63). Treatment of cells with agents that induce SRp30c expression leads to a selective increase in GRβ and consequent glucocorticoid insensitivity. A naturally occurring polymorphism (A3669G) in the 3’ untranslated region of the GRβ mRNA also leads to increased expression of GRβ (64, 65). The polymorphism disrupts an mRNA destabilization motif (AUUUA) resulting in a prolonged half-life of the GRβ mRNA. A3669G carriers have an elevated risk for pathologies with known inflammatory underpinnings such as autoimmune disease, myocardial infarction, coronary artery disease, and heart failure (66-68). These findings suggest that the rise in GRβ in persons with the A3669G polymorphism may attenuate the beneficial immunosuppressive and anti-inflammatory actions of GRα.
Recent discoveries suggest an even broader role for GRβ in cell signaling and physiology. Employing genome wide microarrays on cells engineered to selectively express GRβ, multiple laboratories have shown that GRβ directly induces and represses the expression of a large number of genes independent of its dominant negative activity on GRα (69, 70). The ability of GRβ to recruit histone deacetylases and close local chromatin structure appears to be involved in its repression of certain genes such as interleukin 5 and interleukin 13 (71, 72). GRβ can also bind the glucocorticoid antagonist mifepristone (RU486) (70). In the presence of RU486, many of the GRβ-mediated changes in gene expression are abolished. These findings indicate that GRβ can function as a bonafide transcription factor, and they raise the possibility that GRβ modulates glucocorticoid responses by genomic actions distinct from its antagonism of GRα. Indeed, GRβ has recently been shown to directly reduce the expression of histone deacetylase 2 and promote glucocorticoid insensitivity in bronchoalveolar lavage cells from patients with steroid resistant asthma (73). Another interesting finding has been the discovery of GRβ in zebrafish, mice, and rats (74-76). These isoforms are similar in structure and function to human GRβ but originate from a mechanism of alternative splicing that differs from that in humans. Knockout of GRβ in these species promises to shed new light into the physiological and pathological importance of this splice variant.
In addition to GRβ, alternative splicing of the GR gene gives rise to other receptor isoforms with distinct signaling properties (Figure 4). The use of an alternative splice donor site in the intron separating exons 3 and 4 results in a receptor isoform that contains an insertion of a single arginine residue between the 2 zinc fingers of the GR DBD (77). This receptor variant, termed GRγ, is widely expressed and binds glucocorticoids and DNA in a manner similar to GRα. However, GRγ is impaired in its ability to regulate glucocorticoid-responsive reporter genes and exhibits a transcriptional profile distinct from GRα on a subset of endogenous genes regulated by glucocorticoids. GRγ expression is associated with glucocorticoid resistance in small cell lung carcinoma, corticotroph adenomas, and childhood acute lymphoblastic leukemia (77-79). Two GR splice variants missing large regions of the LBD were initially discovered in glucocorticoid-resistant multiple myeloma cells (80). GR-A is missing middle exons 5-7 which encode the amino-terminal half of the LBD, and GR-P is missing the terminal exons 8-9 which encode the carboxyl-terminal half of the LBD. As expected from these changes in the LBD, GR-A and GR-P do not bind glucocorticoids. Little is currently known about GR-A; however, GR-P has been shown to modulate the transcriptional activity of GRα in a cell-type specific manner (81-83). GR-P is found in many tissues and appears to be the predominant receptor variant expressed in glucocorticoid resistant cancer cells.
GR TRANSLATIONAL ISOFORMS
Alternative translation initiation from the single GRα mRNA transcript produces an additional cohort of diverse GR proteins (Figure 5). Eight AUG start codons derived from exon 2 of the GR gene give rise to 8 GRα subtypes with progressively shorter NTDs (GRα-A, GRα-B, GRα-C1, GRα-C2, GRα-C3, GRα-D1, GRα-D2, and GRα-D3) (15, 84). All 8 sites are highly conserved across species. GRα-A is the full-length receptor that is generated from the first initiator codon. Production of the truncated isoforms from internal start codons involves both ribosomal leaky scanning and ribosomal shunting mechanisms. The mRNA species encoding the splice variants discussed above (GRβ, GRγ, GR-A, and GR-P) also contain the identical set of start codons and would therefore be expected to give rise to a similar complement of translational isoforms.
Figure 5.
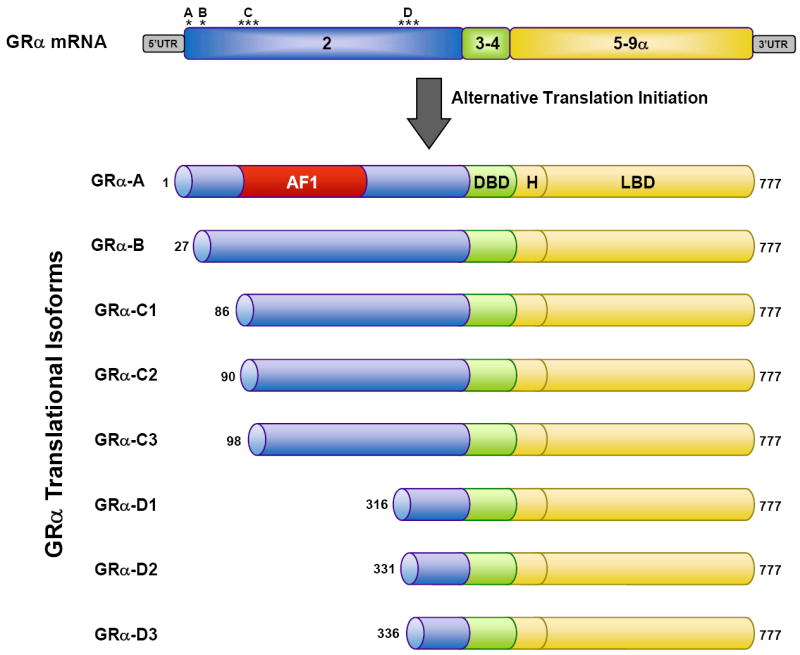
GR translational isoforms. Translation initiation from 8 different AUG start codons in the single GRα mRNA (location of the AUG start codons indicated by an asterisk) produces 8 receptor isoforms with progressively shorter amino NTDs. The numbers for the initiator methionines and AF1 region (amino acids 77-262) are for the human GRα protein.
The GRα translational isoforms display a similar affinity for glucocorticoids and a similar capacity to interact with GREs following ligand activation; however, they show marked differences in other properties (8, 15). The GRα-A, GRα-B, and GRα-C isoforms are localized in the cytoplasm of cells in the absence of hormone and translocate to the nucleus upon glucocorticoid binding. In contrast, the GRα-D isoforms reside constitutively in the nucleus of cells. In addition, nuclear localized GRα-D associates with GRE-containing promoters of specific target genes independent of glucocorticoid treatment. Most striking, however, is that each GRα subtype possesses a distinct gene regulatory profile. When the individual isoforms are expressed at similar levels in U2OS osteosarcoma or Jurkat T lymphoblastic leukemia cells, they each regulate a unique set of genes (8, 85). Less than 10% of the genes were commonly regulated by all the subtypes, indicating that the vast majority of genes were selectively regulated by different GRα isoforms. These isoform-unique gene regulatory profiles lead to functional differences in glucocorticoid-induced apoptosis (8, 85, 86). Cells expressing GRα-C3 were the most sensitive to the cell-killing effects of glucocorticoids whereas cells expressing GRα-D3 were the most resistant. The heightened activity of the GRα-C3 isoform has also been observed on glucocorticoid responsive reporters and has been linked to an N-terminal motif (residues 98-115) that is sterically hindered in the larger receptor isoforms (15, 87). When unobstructed in GRα-C3, this motif increases the activity of the AF1 domain presumably through enhanced recruitment of various coregulators. The complete absence of the AF-1 domain likely underlies the reduced transcriptional activity of the GRα-D isoforms. GRα-D3 does not efficiently repress the expression of multiple anti-apoptotic genes due to deficits in its ability to interact with the p65 subunit of NF-κB and to be recruited to the promoters of these glucocorticoid-responsive genes (86).
The discovery that each GRα translational isoform regulates a unique transcriptome suggests that the cellular response to glucocorticoids will be governed by the expressed complement of receptor subtypes. The GRα-A and GRα-B isoforms are the most abundant GR proteins in many cell-types. However, trabecular meshwork cells from the human eye preferentially express the GRα-C and GRα-D isoforms (88). In addition, immature dendritic cells predominantly express the GRα-D isoforms whereas mature dendritic cells predominantly express the GRα-A subtype (89). In rodents, GRα-C isoforms are highest in pancreas and colon and the GRα-D subtypes are highest in spleen and lung (15). Recent studies have also demonstrated that the composition of GRα subtypes in a given cell or tissue can change in response to different conditions. Treatment of differentiated murine skeletal muscle cells with a selective estrogen-related receptor β/γ agonist induced a preferential increase in the GRα-D isoforms (90), and the GRα-C isoforms are selectively upregulated following mitogen activation of human primary T cells (85). In the human brain, alterations in the expression of the GR subtypes were observed during development and during the ageing process (91). Moreover, a selective increase in GRα-D was measured in specific regions of the brain in patients with schizophrenia and bipolar disorder (92, 93). Since preferential increases in the expression of the GRα-C or GRα-D isoforms may lead to glucocorticoid hypersensitivity or hyposensitivity, respectively, an important goal of future studies will be to define the operative cellular mechanisms that control the expressed complement of translational isoforms. Currently, the efficiency with which a particular start codon is used has been reported to be influenced by the ER22/23EK polymorphism in the GR gene and heterogeneity in the 5’untranslated region of the GRα mRNA (94, 95). Post translational modifications that differentially affect the half-life of the various receptor subtypes may also contribute to alterations in the expression profile of the GRα isoforms.
POST-TRANSLATIONAL MODIFICATION OF GR ISOFORMS
Each individual GR isoform originating from alternative processing of the GR gene is subject to a variety of post-translational modifications that further modulate its function and expand the repertoire of receptor subtypes available for glucocorticoid signaling. The first identified and most extensively studied covalent modification of GR is phosphorylation (96-98). Human GRα is phosphorylated on at least 7 serine residues (Ser-113, Ser-134, Ser-141, Ser-203, Ser-211, Ser-226, and Ser-404), all of which are located in the NTD of the receptor (Figure 2). The major kinases that phosphorylate GRα include MAPKs, cyclin dependent kinases, casein kinase II, and GSK-3β. Many of the sites exhibit a low level of basal phosphorylation and become hyperphosphorylated following the binding of glucocorticoids (99). The structure of the bound glucocorticoid can influence both the pattern and extent of GRα phosphorylation (100). Ser-134 is unique in that it is phosphorylated in a glucocorticoid-independent manner by stress-activating stimuli (101).
One of the major effects of GRα phosphorylation is that it changes the transcriptional activity of the receptor. Early studies demonstrated that phosphorylation-deficient GRα mutants were impaired in their ability to activate some glucocorticoid-responsive promoters but not others (102). Subsequent reports showed that phosphorylation at Ser-211 correlated with increased transcriptional activity of GRα whereas phosphorylation at Ser-226 decreased the signaling capacity of the receptor (99, 103, 104). A deficiency in Ser-211 phosphorylation may contribute to the resistance to glucocorticoid-induced apoptosis that develops in malignant lymphoid cells (105, 106). Conversely, increased phosphorylation at Ser-226 may account for the impaired glucocorticoid signaling in the pathophysiology of depression (107). Phosphorylation of Ser-404 also has major consequences on glucocorticoid responses, as the ability of GRα to both activate and repress target genes is diminished by phosphorylation at this site (108). In cells expressing a GRα mutant incapable of Ser-404 phosphorylation, the global transcriptional response to glucocorticoids is redirected to favor the activation of distinct signaling pathways. Consistent with many of the phosphorylation sites residing within the AF1 domain, differences in cofactor recruitment appear to be responsible for the transcriptional alterations that accompany GRα phosphorylation. GRα recruitment of the coactivator MED14 is enhanced by glucocorticoid-dependent phosphorylation at Ser-211 (104), whereas the interaction of GRα with the coactivator p300/CBP and the p65 subunit of NF-κB are both diminished by phosphorylation at Ser-404 (108).
Phosphorylation alters other properties of GR that impact the profile of glucocorticoid signaling. Degradation of the GRα protein is enhanced by glucocorticoid-dependent phosphorylation of the receptor, as phosphorylation deficient mutants are stabilized in the presence of glucocorticoids (102). In addition, the ligand-free GRα is protected from degradation by its association with the tumor suppressor gene TSG101 which preferentially interacts with the non-phosphorylated receptor (109). The cellular distribution of GRα is also altered by receptor phosphorylation. GRα phosphorylated on Ser-203, Ser-226, or Ser-404 spends less time in the nuclear compartment due to greater cytoplasmic retention and/or enhanced nucleocytoplasmic transport. As a consequence, GRα phosphorylated on either of these residues exhibits reduced transcriptional activity on glucocorticoid-responsive target genes (99, 103, 108, 110).
Additional post-translational modifications of GR have been described that regulate the function of the receptor. Ubiquitin is a 76 amino acid protein that when attached to specific lysine residues marks proteins for proteasomal degradation. Ubiquitination of GRα at a conserved lysine residue (Lys-419) has been shown to target the receptor for turnover by the proteasome (Figure 2) (111, 112). Mutant receptors that cannot be ubiquitinated at this residue are resistant to ligand-dependent downregulation and exhibit potentiated transcriptional activity on glucocorticoid-responsive reporter genes. Moreover, changes in the expression of an E3 ubiquitin ligase for GRα leads to alterations in both receptor levels and cellular sensitivity to glucocorticoids (113). GRα is also post-translationally modified by sumoylation, a process in which SUMO (small ubiquitin-related modifier) peptides are covalently attached to specific lysine residues (Lys-277, Lys-293, Lys-703) within the receptor (Figure 2). The addition of SUMO peptides to the receptor occurs in the absence of ligand and is increased by the binding of glucocorticoids. Depending on the site of sumoylation, the transcriptional activity of GRα can be enhanced or repressed via alterations in the recruitment and/or activity of specific coregulators (114-119). Finally, it has been suggested that GRα becomes acetylated on lysine residues (Lys-494 and Lys-495) located within the hinge region in response to glucocorticoid binding (Figure 2) (120). Deacetylation of GR by histone deacetylase 2 was reported to be required for the receptor to efficiently repress the transcriptional activity of NF-κB, suggesting that acetylation of GR limits the inhibitory actions of glucocorticoids on NF-κB signaling. More recently, studies have shown that the clock transcription factor acetylates GRα and attenuates its ability to both activate and repress glucocorticoid responsive genes, conferring glucocorticoid insensitivity to target tissues (121). Clearly, post translational modifications can regulate multiple aspects of GRα function, providing cells with additional receptor heterogeneity for controlling glucocorticoid responses. An important goal of future research will be to determine to what extent the various splicing and translational isoforms of GR are subject to and regulated by these post translational modifications.
SUMMARY AND FUTURE PERSPECTIVES
Glucocorticoids act through GR to regulate numerous physiological processes, and synthetic derivatives of these hormones are widely prescribed for treating inflammatory diseases, autoimmune disorders, and various cancers. A challenge to clinicians and patients alike is that cell, tissue, and individual responses to glucocorticoids are markedly different and can change over time and in response to various extracellular cues. The discovery that multiple GR isoforms arise from a single gene has advanced our understanding of the molecular basis for the diversity in glucocorticoid signaling. GR isoforms with unique expression, gene regulatory, and functional profiles are generated by alternative splicing of the primary transcript, alternative translation initiation of the mature mRNA, and post-translational modifications of the encoded protein. The potential for these GR subtypes to undergo various post-translational modifications and to function as monomers, homodimers, and heterodimers provides cells with a wealth of possibilities for generating diverse glucocorticoid responses with fine-tuned precision. Alterations in the relative levels of the GR subtypes may underlie pathologies characterized by hyposensitivity or hypersensitivity to glucocorticoids.
The cellular response to glucocorticoids will depend not only on the GR isoform composition but also on the glucocorticoid that binds and activates the receptor as well as the concentration of the administered steroid. Not all glucocorticoids are created equal, as structurally different but similarly potent steroids used in the clinic regulate both common and unique sets of genes (88, 122). The distinct transcriptional signature of these glucocorticoids suggests that their binding confers unique conformations on GR that lead to differences in DNA binding, chromatin remodeling, and/or coregulator recruitment. Recent discoveries also suggest that the concentration of glucocorticoids needed to achieve the desired transcriptional response can vary in a context-specific manner. For example, GR repression of pro-inflammatory genes via tethering to NF-κB occurs at much lower glucocorticoid concentrations than the induction of gene expression (123). Global GR recruitment assays have revealed that different GREs and their associated target genes also exhibit profound differences in their sensitivity to glucocorticoids (29). In addition, dose-response studies in the livers of male and female rats have discovered several genes in females that are 10- to 100-fold less sensitive to glucocorticoid regulation than they are in males (43). Remarkably, even the polarity of the transcriptional response of certain GR-regulated genes can vary depending on the dose of glucocorticoid (124). Collectively, these findings suggest that as the specific genes underlying various pathologies become identified, careful consideration of the structure and dose of the employed glucocorticoid may help fine-tune the glucocorticoid response and lead to improved benefit/risk ratios for patients on steroid therapy.
Glucocorticoid resistance remains a major barrier to the effective treatment of a variety of immune and inflammatory diseases (125, 126). Advances in our understanding of glucocorticoid signaling have uncovered a variety of mechanisms that contribute to reduced glucocorticoid responsiveness, including increased expression of the GRβ and GRα-D isoforms, changes in GR phosphorylation, and homologous down-regulation of the receptor. Understanding the molecular mechanisms of resistance permits not only the prediction of patient responsiveness to glucocorticoids but also the design of novel therapeutic strategies for combatting the insensitivity. For instance, altered patterns of GR phosphorylation thought to contribute to glucocorticoid resistance in patients with severe asthma can be reversed by co-administration of long acting β2AR agonists or p38 MAPK inhibitors (125). Development of glucocorticoid analogues deficient in their ability to down regulate the expression of GR may also prove to be an effective strategy for overcoming steroid resistance.
The harmful side effects of medicinal glucocorticoids also remain a frustration for clinicians and their patients. Therefore, intense efforts have been made over the last decade to develop novel GR ligands, termed dissociated or selective GR agonists (SEGRA), that retain the negative regulation of gene expression thought to be important for many of the anti-inflammatory actions of glucocorticoids but lose the positive regulation of gene expression thought to underlie many of their adverse effects. A number of SEGRAs have been developed but few have made it to clinical trials, and several recent studies have challenged both the premise and utility of these analogues as therapeutic agents (127, 128). One potential short-coming of SEGRAs is that mounting evidence indicates that glucocorticoid-dependent activation of many genes also plays a critical role in the anti-inflammatory mechanisms of glucocorticoids. Clearly, more research is needed to unravel the molecular details by which glucocorticoids repress the inflammatory response and induce undesirable side effects. A greater understanding of the heterogeneity in GR signaling responses in both healthy and diseased tissues will aid in the development of safer and more effective glucocorticoid therapies.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, NIEHS.
References
- 1.Barnes PJ. Anti-inflammatory actions of glucocorticoids: molecular mechanisms. Clin Sci (Lond) 1998;94(6):557–72. doi: 10.1042/cs0940557. [DOI] [PubMed] [Google Scholar]
- 2.Sapolsky RM, Romero LM, Munck AU. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev. 2000;21(1):55–89. doi: 10.1210/edrv.21.1.0389. [DOI] [PubMed] [Google Scholar]
- 3.Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids--new mechanisms for old drugs. N Engl J Med. 2005;353(16):1711–23. doi: 10.1056/NEJMra050541. [DOI] [PubMed] [Google Scholar]
- 4.Busillo JM, Cidlowski JA. The five Rs of glucocorticoid action during inflammation: ready, reinforce, repress, resolve, and restore. Trends Endocrinol Metab. 2013 doi: 10.1016/j.tem.2012.11.005. Epub 2013/01/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miner JN, Hong MH, Negro-Vilar A. New and improved glucocorticoid receptor ligands. Expert Opin Investig Drugs. 2005;14(12):1527–45. doi: 10.1517/13543784.14.12.1527. [DOI] [PubMed] [Google Scholar]
- 6.Evans RM. The steroid and thyroid hormone receptor superfamily. Science. 1988;240(4854):889–95. doi: 10.1126/science.3283939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galon J, Franchimont D, Hiroi N, Frey G, Boettner A, Ehrhart-Bornstein M, et al. Gene profiling reveals unknown enhancing and suppressive actions of glucocorticoids on immune cells. Faseb J. 2002;16(1):61–71. doi: 10.1096/fj.01-0245com. [DOI] [PubMed] [Google Scholar]
- 8.Lu NZ, Collins JB, Grissom SF, Cidlowski JA. Selective regulation of bone cell apoptosis by translational isoforms of the glucocorticoid receptor. Mol Cell Biol. 2007;27(20):7143–60. doi: 10.1128/MCB.00253-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ren R, Oakley RH, Cruz-Topete D, Cidlowski JA. Dual Role for Glucocorticoids in Cardiomyocyte Hypertrophy and Apoptosis. Endocrinology. 2012 doi: 10.1210/en.2012-1563. Epub 2012/09/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cole TJ, Blendy JA, Monaghan AP, Krieglstein K, Schmid W, Aguzzi A, et al. Targeted disruption of the glucocorticoid receptor gene blocks adrenergic chromaffin cell development and severely retards lung maturation. Genes Dev. 1995;9(13):1608–21. doi: 10.1101/gad.9.13.1608. [DOI] [PubMed] [Google Scholar]
- 11.Kino T, De Martino MU, Charmandari E, Mirani M, Chrousos GP. Tissue glucocorticoid resistance/hypersensitivity syndromes. J Steroid Biochem Mol Biol. 2003;85(2-5):457–67. doi: 10.1016/s0960-0760(03)00218-8. [DOI] [PubMed] [Google Scholar]
- 12.Lamberts SW, Huizenga AT, de Lange P, de Jong FH, Koper JW. Clinical aspects of glucocorticoid sensitivity. Steroids. 1996;61(4):157–60. doi: 10.1016/0039-128x(96)00005-0. [DOI] [PubMed] [Google Scholar]
- 13.Gorovits R, Ben-Dror I, Fox LE, Westphal HM, Vardimon L. Developmental changes in the expression and compartmentalization of the glucocorticoid receptor in embryonic retina. Proc Natl Acad Sci U S A. 1994;91(11):4786–90. doi: 10.1073/pnas.91.11.4786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hsu SC, DeFranco DB. Selectivity of cell cycle regulation of glucocorticoid receptor function. J Biol Chem. 1995;270(7):3359–64. doi: 10.1074/jbc.270.7.3359. [DOI] [PubMed] [Google Scholar]
- 15.Lu NZ, Cidlowski JA. Translational regulatory mechanisms generate N-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Mol Cell. 2005;18(3):331–42. doi: 10.1016/j.molcel.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 16.Oakley RH, Cidlowski JA. Cellular processing of the glucocorticoid receptor gene and protein: new mechanisms for generating tissue-specific actions of glucocorticoids. J Biol Chem. 2011;286(5):3177–84. doi: 10.1074/jbc.R110.179325. Epub 2010/12/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar R, Thompson EB. Gene regulation by the glucocorticoid receptor: structure:function relationship. J Steroid Biochem Mol Biol. 2005;94(5):383–94. doi: 10.1016/j.jsbmb.2004.12.046. [DOI] [PubMed] [Google Scholar]
- 18.Bledsoe RK, Montana VG, Stanley TB, Delves CJ, Apolito CJ, McKee DD, et al. Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell. 2002;110(1):93–105. doi: 10.1016/s0092-8674(02)00817-6. [DOI] [PubMed] [Google Scholar]
- 19.Grad I, Picard D. The glucocorticoid responses are shaped by molecular chaperones. Mol Cell Endocrinol. 2007;275(1-2):2–12. doi: 10.1016/j.mce.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 20.Pratt WB, Toft DO. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr Rev. 1997;18(3):306–60. doi: 10.1210/edrv.18.3.0303. [DOI] [PubMed] [Google Scholar]
- 21.Seckl JR. 11beta-hydroxysteroid dehydrogenases: changing glucocorticoid action. Curr Opin Pharmacol. 2004;4(6):597–602. doi: 10.1016/j.coph.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 22.Yang H, Dou W, Lou J, Leng Y, Shen J. Discovery of novel inhibitors of 11beta-hydroxysteroid dehydrogenase type 1 by docking and pharmacophore modeling. Bioorganic & medicinal chemistry letters. 2008;18(4):1340–5. doi: 10.1016/j.bmcl.2008.01.020. Epub 2008/02/05. [DOI] [PubMed] [Google Scholar]
- 23.Beato M. Gene regulation by steroid hormones. Cell. 1989;56(3):335–44. doi: 10.1016/0092-8674(89)90237-7. [DOI] [PubMed] [Google Scholar]
- 24.Freedman LP. Anatomy of the steroid receptor zinc finger region. Endocr Rev. 1992;13(2):129–45. doi: 10.1210/edrv-13-2-129. [DOI] [PubMed] [Google Scholar]
- 25.Uhlenhaut NH, Barish GD, Yu RT, Downes M, Karunasiri M, Liddle C, et al. Insights into negative regulation by the glucocorticoid receptor from genome-wide profiling of inflammatory cistromes. Mol Cell. 2013;49(1):158–71. doi: 10.1016/j.molcel.2012.10.013. Epub 2012/11/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Surjit M, Ganti KP, Mukherji A, Ye T, Hua G, Metzger D, et al. Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell. 2011;145(2):224–41. doi: 10.1016/j.cell.2011.03.027. Epub 2011/04/19. [DOI] [PubMed] [Google Scholar]
- 27.Hudson WH, Youn C, Ortlund EA. The structural basis of direct glucocorticoid-mediated transrepression. Nature structural & molecular biology. 2013;20(1):53–8. doi: 10.1038/nsmb.2456. Epub 2012/12/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.John S, Sabo PJ, Thurman RE, Sung MH, Biddie SC, Johnson TA, et al. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nature genetics. 2011;43(3):264–8. doi: 10.1038/ng.759. Epub 2011/01/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reddy TE, Gertz J, Crawford GE, Garabedian MJ, Myers RM. The hypersensitive glucocorticoid response specifically regulates period 1 and expression of circadian genes. Mol Cell Biol. 2012;32(18):3756–67. doi: 10.1128/MCB.00062-12. Epub 2012/07/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burd CJ, Archer TK. Chromatin architecture defines the glucocorticoid response. Mol Cell Endocrinol. 2013 doi: 10.1016/j.mce.2013.03.020. Epub 2013/04/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oakley RH, Revollo J, Cidlowski JA. Glucocorticoids regulate arrestin gene expression and redirect the signaling profile of G protein-coupled receptors. Proc Natl Acad Sci U S A. 2012;109(43):17591–6. doi: 10.1073/pnas.1209411109. Epub 2012/10/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramamoorthy S, Cidlowski JA. Ligand-induced repression of the glucocorticoid receptor gene is mediated by an NCoR1 repression complex formed by long-range chromatin interactions with intragenic glucocorticoid response elements. Mol Cell Biol. 2013;33(9):1711–22. doi: 10.1128/MCB.01151-12. Epub 2013/02/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McNally JG, Muller WG, Walker D, Wolford R, Hager GL. The glucocorticoid receptor: rapid exchange with regulatory sites in living cells. Science. 2000;287(5456):1262–5. doi: 10.1126/science.287.5456.1262. [DOI] [PubMed] [Google Scholar]
- 34.Jenkins BD, Pullen CB, Darimont BD. Novel glucocorticoid receptor coactivator effector mechanisms. Trends Endocrinol Metab. 2001;12(3):122–6. doi: 10.1016/s1043-2760(00)00357-x. [DOI] [PubMed] [Google Scholar]
- 35.Lonard DM, O’Malley BW. Expanding functional diversity of the coactivators. Trends Biochem Sci. 2005;30(3):126–32. doi: 10.1016/j.tibs.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 36.Rosenfeld MG, Glass CK. Coregulator codes of transcriptional regulation by nuclear receptors. J Biol Chem. 2001;276(40):36865–8. doi: 10.1074/jbc.R100041200. [DOI] [PubMed] [Google Scholar]
- 37.Meijsing SH, Pufall MA, So AY, Bates DL, Chen L, Yamamoto KR. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science. 2009;324(5925):407–10. doi: 10.1126/science.1164265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ronacher K, Hadley K, Avenant C, Stubsrud E, Simons SS, Jr, Louw A, et al. Ligand-selective transactivation and transrepression via the glucocorticoid receptor: role of cofactor interaction. Mol Cell Endocrinol. 2009;299(2):219–31. doi: 10.1016/j.mce.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 39.Rogatsky I, Ivashkiv LB. Glucocorticoid modulation of cytokine signaling. Tissue Antigens. 2006;68(1):1–12. doi: 10.1111/j.1399-0039.2006.00599.x. [DOI] [PubMed] [Google Scholar]
- 40.Nissen RM, Yamamoto KR. The glucocorticoid receptor inhibits NFkappaB by interfering with serine-2 phosphorylation of the RNA polymerase II carboxy-terminal domain. Genes Dev. 2000;14(18):2314–29. doi: 10.1101/gad.827900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang-Yen HF, Chambard JC, Sun YL, Smeal T, Schmidt TJ, Drouin J, et al. Transcriptional interference between c-Jun and the glucocorticoid receptor: mutual inhibition of DNA binding due to direct protein-protein interaction. Cell. 1990;62(6):1205–15. doi: 10.1016/0092-8674(90)90396-v. Epub 1990/09/21. [DOI] [PubMed] [Google Scholar]
- 42.Chinenov Y, Gupte R, Dobrovolna J, Flammer JR, Liu B, Michelassi FE, et al. Role of transcriptional coregulator GRIP1 in the anti-inflammatory actions of glucocorticoids. Proc Natl Acad Sci U S A. 2012;109(29):11776–81. doi: 10.1073/pnas.1206059109. Epub 2012/07/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Duma D, Collins JB, Chou JW, Cidlowski JA. Sexually dimorphic actions of glucocorticoids provide a link to inflammatory diseases with gender differences in prevalence. Sci Signal. 2010;3(143):ra74. doi: 10.1126/scisignal.2001077. Epub 2010/10/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Groeneweg FL, Karst H, de Kloet ER, Joels M. Mineralocorticoid and glucocorticoid receptors at the neuronal membrane, regulators of nongenomic corticosteroid signalling. Mol Cell Endocrinol. 2012;350(2):299–309. doi: 10.1016/j.mce.2011.06.020. Epub 2011/07/09. [DOI] [PubMed] [Google Scholar]
- 45.Samarasinghe RA, Witchell SF, DeFranco DB. Cooperativity and complementarity: synergies in non-classical and classical glucocorticoid signaling. Cell Cycle. 2012;11(15):2819–27. doi: 10.4161/cc.21018. Epub 2012/07/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Croxtall JD, Choudhury Q, Flower RJ. Glucocorticoids act within minutes to inhibit recruitment of signalling factors to activated EGF receptors through a receptor-dependent, transcription-independent mechanism. Br J Pharmacol. 2000;130(2):289–98. doi: 10.1038/sj.bjp.0703272. Epub 2000/05/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Solito E, Mulla A, Morris JF, Christian HC, Flower RJ, Buckingham JC. Dexamethasone induces rapid serine-phosphorylation and membrane translocation of annexin 1 in a human folliculostellate cell line via a novel nongenomic mechanism involving the glucocorticoid receptor, protein kinase C, phosphatidylinositol 3-kinase, and mitogen-activated protein kinase. Endocrinology. 2003;144(4):1164–74. doi: 10.1210/en.2002-220592. Epub 2003/03/18. [DOI] [PubMed] [Google Scholar]
- 48.Matthews L, Berry A, Ohanian V, Ohanian J, Garside H, Ray D. Caveolin mediates rapid glucocorticoid effects and couples glucocorticoid action to the antiproliferative program. Mol Endocrinol. 2008;22(6):1320–30. doi: 10.1210/me.2007-0154. Epub 2008/03/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Samarasinghe RA, Di Maio R, Volonte D, Galbiati F, Lewis M, Romero G, et al. Nongenomic glucocorticoid receptor action regulates gap junction intercellular communication and neural progenitor cell proliferation. Proc Natl Acad Sci U S A. 2011;108(40):16657–62. doi: 10.1073/pnas.1102821108. Epub 2011/09/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gross KL, Cidlowski JA. Tissue-specific glucocorticoid action: a family affair. Trends Endocrinol Metab. 2008;19(9):331–9. doi: 10.1016/j.tem.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van Rossum EF, Lamberts SW. Polymorphisms in the glucocorticoid receptor gene and their associations with metabolic parameters and body composition. Recent Prog Horm Res. 2004;59:333–57. doi: 10.1210/rp.59.1.333. Epub 2004/01/30. [DOI] [PubMed] [Google Scholar]
- 52.Jewell CM, Cidlowski JA. Molecular evidence for a link between the N363S glucocorticoid receptor polymorphism and altered gene expression. J Clin Endocrinol Metab. 2007;92(8):3268–77. doi: 10.1210/jc.2007-0642. Epub 2007/05/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bamberger CM, Bamberger AM, de Castro M, Chrousos GP. Glucocorticoid receptor beta, a potential endogenous inhibitor of glucocorticoid action in humans. J Clin Invest. 1995;95(6):2435–41. doi: 10.1172/JCI117943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oakley RH, Sar M, Cidlowski JA. The human glucocorticoid receptor beta isoform. Expression, biochemical properties, and putative function. J Biol Chem. 1996;271(16):9550–9. doi: 10.1074/jbc.271.16.9550. [DOI] [PubMed] [Google Scholar]
- 55.Kino T, Su YA, Chrousos GP. Human glucocorticoid receptor isoform beta: recent understanding of its potential implications in physiology and pathophysiology. Cell Mol Life Sci. 2009;66(21):3435–48. doi: 10.1007/s00018-009-0098-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lewis-Tuffin LJ, Cidlowski JA. The physiology of human glucocorticoid receptor beta (hGRbeta) and glucocorticoid resistance. Ann N Y Acad Sci. 2006;1069:1–9. doi: 10.1196/annals.1351.001. [DOI] [PubMed] [Google Scholar]
- 57.Hauk PJ, Hamid QA, Chrousos GP, Leung DY. Induction of corticosteroid insensitivity in human PBMCs by microbial superantigens. J Allergy Clin Immunol. 2000;105(4):782–7. doi: 10.1067/mai.2000.105807. [DOI] [PubMed] [Google Scholar]
- 58.Tliba O, Cidlowski JA, Amrani Y. CD38 expression is insensitive to steroid action in cells treated with tumor necrosis factor-alpha and interferon-gamma by a mechanism involving the up-regulation of the glucocorticoid receptor beta isoform. Mol Pharmacol. 2006;69(2):588–96. doi: 10.1124/mol.105.019679. [DOI] [PubMed] [Google Scholar]
- 59.Webster JC, Oakley RH, Jewell CM, Cidlowski JA. Proinflammatory cytokines regulate human glucocorticoid receptor gene expression and lead to the accumulation of the dominant negative beta isoform: a mechanism for the generation of glucocorticoid resistance. Proc Natl Acad Sci U S A. 2001;98(12):6865–70. doi: 10.1073/pnas.121455098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Varricchio L, Masselli E, Alfani E, Battistini A, Migliaccio G, Vannucchi AM, et al. The dominant negative beta isoform of the glucocorticoid receptor is uniquely expressed in erythroid cells expanded from polycythemia vera patients. Blood. 2011;118(2):425–36. doi: 10.1182/blood-2010-07-296921. Epub 2011/03/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jain A, Wordinger RJ, Yorio T, Clark AF. Spliceosome protein (SRp) regulation of glucocorticoid receptor isoforms and glucocorticoid response in human trabecular meshwork cells. Invest Ophthalmol Vis Sci. 2012;53(2):857–66. doi: 10.1167/iovs.11-8497. Epub 2011/12/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xu Q, Leung DY, Kisich KO. Serine-arginine-rich protein p30 directs alternative splicing of glucocorticoid receptor pre-mRNA to glucocorticoid receptor beta in neutrophils. J Biol Chem. 2003;278(29):27112–8. doi: 10.1074/jbc.M300824200. [DOI] [PubMed] [Google Scholar]
- 63.Zhu J, Gong JY, Goodman OB, Jr, Cartegni L, Nanus DM, Shen R. Bombesin attenuates pre-mRNA splicing of glucocorticoid receptor by regulating the expression of serine-arginine protein p30c (SRp30c) in prostate cancer cells. Biochim Biophys Acta. 2007;1773(7):1087–94. doi: 10.1016/j.bbamcr.2007.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Derijk RH, Schaaf MJ, Turner G, Datson NA, Vreugdenhil E, Cidlowski J, et al. A human glucocorticoid receptor gene variant that increases the stability of the glucocorticoid receptor beta-isoform mRNA is associated with rheumatoid arthritis. J Rheumatol. 2001;28(11):2383–8. [PubMed] [Google Scholar]
- 65.Schaaf MJ, Cidlowski JA. AUUUA motifs in the 3’UTR of human glucocorticoid receptor alpha and beta mRNA destabilize mRNA and decrease receptor protein expression. Steroids. 2002;67(7):627–36. doi: 10.1016/s0039-128x(02)00015-6. [DOI] [PubMed] [Google Scholar]
- 66.Geelhoed JJ, van Duijn C, van Osch-Gevers L, Steegers EA, Hofman A, Helbing WA, et al. Glucocorticoid receptor-9beta polymorphism is associated with systolic blood pressure and heart growth during early childhood. The Generation R Study. Early human development. 2011;87(2):97–102. doi: 10.1016/j.earlhumdev.2010.11.006. Epub 2010/12/15. [DOI] [PubMed] [Google Scholar]
- 67.Otte C, Wust S, Zhao S, Pawlikowska L, Kwok PY, Whooley MA. Glucocorticoid receptor gene, low-grade inflammation, and heart failure: the Heart and Soul study. J Clin Endocrinol Metab. 2010;95(6):2885–91. doi: 10.1210/jc.2009-2251. Epub 2010/04/08. [DOI] [PubMed] [Google Scholar]
- 68.van den Akker EL, Koper JW, van Rossum EF, Dekker MJ, Russcher H, de Jong FH, et al. Glucocorticoid receptor gene and risk of cardiovascular disease. Arch Intern Med. 2008;168(1):33–9. doi: 10.1001/archinternmed.2007.41. [DOI] [PubMed] [Google Scholar]
- 69.Kino T, Manoli I, Kelkar S, Wang Y, Su YA, Chrousos GP. Glucocorticoid receptor (GR) beta has intrinsic, GRalpha-independent transcriptional activity. Biochem Biophys Res Commun. 2009;381(4):671–5. doi: 10.1016/j.bbrc.2009.02.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lewis-Tuffin LJ, Jewell CM, Bienstock RJ, Collins JB, Cidlowski JA. The Human Glucocorticoid Receptor {beta} (hGR{beta}) Binds RU-486 and is Transcriptionally Active. Mol Cell Biol. 2007 doi: 10.1128/MCB.01439-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kelly A, Bowen H, Jee YK, Mahfiche N, Soh C, Lee T, et al. The glucocorticoid receptor beta isoform can mediate transcriptional repression by recruiting histone deacetylases. J Allergy Clin Immunol. 2008;121(1):203–8 e1. doi: 10.1016/j.jaci.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 72.Kim SH, Kim DH, Lavender P, Seo JH, Kim YS, Park JS, et al. Repression of TNF-alpha-induced IL-8 expression by the glucocorticoid receptor-beta involves inhibition of histone H4 acetylation. Exp Mol Med. 2009;41(5):297–306. doi: 10.3858/emm.2009.41.5.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li LB, Leung DY, Martin RJ, Goleva E. Inhibition of histone deacetylase 2 expression by elevated glucocorticoid receptor beta in steroid-resistant asthma. Am J Respir Crit Care Med. 2010;182(7):877–83. doi: 10.1164/rccm.201001-0015OC. Epub 2010/06/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.DuBois DC, Sukumaran S, Jusko WJ, Almon RR. Evidence for a glucocorticoid receptor beta splice variant in the rat and its physiological regulation in liver. Steroids. 2013;78(2):312–20. doi: 10.1016/j.steroids.2012.11.014. Epub 2012/12/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hinds TD, Jr, Ramakrishnan S, Cash HA, Stechschulte LA, Heinrich G, Najjar SM, et al. Discovery of glucocorticoid receptor-beta in mice with a role in metabolism. Mol Endocrinol. 2010;24(9):1715–27. doi: 10.1210/me.2009-0411. Epub 2010/07/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schaaf MJ, Champagne D, van Laanen IH, van Wijk DC, Meijer AH, Meijer OC, et al. Discovery of a functional glucocorticoid receptor beta-isoform in zebrafish. Endocrinology. 2008;149(4):1591–9. doi: 10.1210/en.2007-1364. [DOI] [PubMed] [Google Scholar]
- 77.Ray DW, Davis JR, White A, Clark AJ. Glucocorticoid receptor structure and function in glucocorticoid-resistant small cell lung carcinoma cells. Cancer Res. 1996;56(14):3276–80. [PubMed] [Google Scholar]
- 78.Beger C, Gerdes K, Lauten M, Tissing WJ, Fernandez-Munoz I, Schrappe M, et al. Expression and structural analysis of glucocorticoid receptor isoform gamma in human leukaemia cells using an isoform-specific real-time polymerase chain reaction approach. Br J Haematol. 2003;122(2):245–52. doi: 10.1046/j.1365-2141.2003.04426.x. [DOI] [PubMed] [Google Scholar]
- 79.Rivers C, Levy A, Hancock J, Lightman S, Norman M. Insertion of an amino acid in the DNA-binding domain of the glucocorticoid receptor as a result of alternative splicing. J Clin Endocrinol Metab. 1999;84(11):4283–6. doi: 10.1210/jcem.84.11.6235. [DOI] [PubMed] [Google Scholar]
- 80.Moalli PA, Pillay S, Krett NL, Rosen ST. Alternatively spliced glucocorticoid receptor messenger RNAs in glucocorticoid-resistant human multiple myeloma cells. Cancer Res. 1993;53(17):3877–9. [PubMed] [Google Scholar]
- 81.de Lange P, Segeren CM, Koper JW, Wiemer E, Sonneveld P, Brinkmann AO, et al. Expression in hematological malignancies of a glucocorticoid receptor splice variant that augments glucocorticoid receptor-mediated effects in transfected cells. Cancer Res. 2001;61(10):3937–41. [PubMed] [Google Scholar]
- 82.Gaitan D, DeBold CR, Turney MK, Zhou P, Orth DN, Kovacs WJ. Glucocorticoid receptor structure and function in an adrenocorticotropin-secreting small cell lung cancer. Mol Endocrinol. 1995;9(9):1193–201. doi: 10.1210/mend.9.9.7491111. [DOI] [PubMed] [Google Scholar]
- 83.Krett NL, Pillay S, Moalli PA, Greipp PR, Rosen ST. A variant glucocorticoid receptor messenger RNA is expressed in multiple myeloma patients. Cancer Res. 1995;55(13):2727–9. [PubMed] [Google Scholar]
- 84.Yudt MR, Jewell CM, Bienstock RJ, Cidlowski JA. Molecular origins for the dominant negative function of human glucocorticoid receptor beta. Mol Cell Biol. 2003;23(12):4319–30. doi: 10.1128/MCB.23.12.4319-4330.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wu I, Shin SC, Cao Y, Bender IK, Jafari N, Feng G, et al. Selective glucocorticoid receptor translational isoforms reveal glucocorticoid-induced apoptotic transcriptomes. Cell death & disease. 2013;4:e453. doi: 10.1038/cddis.2012.193. Epub 2013/01/11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gross KL, Oakley RH, Scoltock AB, Jewell CM, Cidlowski JA. Glucocorticoid Receptor {alpha} Isoform-Selective Regulation of Antiapoptotic Genes in Osteosarcoma Cells: A New Mechanism for Glucocorticoid Resistance. Mol Endocrinol. 2011;25(7):1087–99. doi: 10.1210/me.2010-0051. Epub 2011/04/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bender IK, Cao Y, Lu NZ. Determinants of the Heightened Activity of Glucocorticoid Receptor Translational Isoforms. Mol Endocrinol. 2013 doi: 10.1210/me.2013-1009. Epub 2013/07/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nehme A, Lobenhofer EK, Stamer WD, Edelman JL. Glucocorticoids with different chemical structures but similar glucocorticoid receptor potency regulate subsets of common and unique genes in human trabecular meshwork cells. BMC Med Genomics. 2009;2:58. doi: 10.1186/1755-8794-2-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cao Y, Bender IK, Konstantinidis AK, Shin SC, Jewell CM, Cidlowski JA, et al. Glucocorticoid receptor translational isoforms underlie maturational stage-specific glucocorticoid sensitivities of dendritic cells in mice and humans. Blood. 2013;121(9):1553–62. doi: 10.1182/blood-2012-05-432336. Epub 2013/01/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang SC, Myers S, Dooms C, Capon R, Muscat GE. An ERRbeta/gamma agonist modulates GRalpha expression, and glucocorticoid responsive gene expression in skeletal muscle cells. Mol Cell Endocrinol. 2009 doi: 10.1016/j.mce.2009.07.012. [DOI] [PubMed] [Google Scholar]
- 91.Sinclair D, Webster MJ, Wong J, Weickert CS. Dynamic molecular and anatomical changes in the glucocorticoid receptor in human cortical development. Mol Psychiatry. 2010:1–12. doi: 10.1038/mp.2010.28. [DOI] [PubMed] [Google Scholar]
- 92.Sinclair D, Webster MJ, Fullerton JM, Weickert CS. Glucocorticoid receptor mRNA and protein isoform alterations in the orbitofrontal cortex in schizophrenia and bipolar disorder. BMC psychiatry. 2012;12:84. doi: 10.1186/1471-244X-12-84. Epub 2012/07/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sinclair D, Tsai SY, Woon HG, Weickert CS. Abnormal glucocorticoid receptor mRNA and protein isoform expression in the prefrontal cortex in psychiatric illness. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2011;36(13):2698–709. doi: 10.1038/npp.2011.160. Epub 2011/09/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pedersen KB, Geng CD, Vedeckis WV. Three mechanisms are involved in glucocorticoid receptor autoregulation in a human T-lymphoblast cell line. Biochemistry. 2004;43(34):10851–8. doi: 10.1021/bi049458u. [DOI] [PubMed] [Google Scholar]
- 95.Russcher H, van Rossum EF, de Jong FH, Brinkmann AO, Lamberts SW, Koper JW. Increased expression of the glucocorticoid receptor-A translational isoform as a result of the ER22/23EK polymorphism. Mol Endocrinol. 2005;19(7):1687–96. doi: 10.1210/me.2004-0467. [DOI] [PubMed] [Google Scholar]
- 96.Beck IM, Vanden Berghe W, Vermeulen L, Yamamoto KR, Haegeman G, De Bosscher K. Crosstalk in inflammation: the interplay of glucocorticoid receptor-based mechanisms and kinases and phosphatases. Endocr Rev. 2009;30(7):830–82. doi: 10.1210/er.2009-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Galliher-Beckley AJ, Cidlowski JA. Emerging roles of glucocorticoid receptor phosphorylation in modulating glucocorticoid hormone action in health and disease. IUBMB Life. 2009;61(10):979–86. doi: 10.1002/iub.245. Epub 2009/09/30. [DOI] [PubMed] [Google Scholar]
- 98.Kumar R, Calhoun WJ. Differential regulation of the transcriptional activity of the glucocorticoid receptor through site-specific phosphorylation. Biologics. 2008;2(4):845–54. doi: 10.2147/btt.s3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang Z, Frederick J, Garabedian MJ. Deciphering the phosphorylation “code” of the glucocorticoid receptor in vivo. J Biol Chem. 2002;277(29):26573–80. doi: 10.1074/jbc.M110530200. [DOI] [PubMed] [Google Scholar]
- 100.Avenant C, Ronacher K, Stubsrud E, Louw A, Hapgood JP. Role of ligand-dependent GR phosphorylation and half-life in determination of ligand-specific transcriptional activity. Mol Cell Endocrinol. 2010;327(1-2):72–88. doi: 10.1016/j.mce.2010.06.007. Epub 2010/06/22. [DOI] [PubMed] [Google Scholar]
- 101.Galliher-Beckley AJ, Williams JG, Cidlowski JA. Ligand-independent phosphorylation of the glucocorticoid receptor integrates cellular stress pathways with nuclear receptor signaling. Mol Cell Biol. 2011;31(23):4663–75. doi: 10.1128/MCB.05866-11. Epub 2011/09/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Webster JC, Jewell CM, Bodwell JE, Munck A, Sar M, Cidlowski JA. Mouse glucocorticoid receptor phosphorylation status influences multiple functions of the receptor protein. J Biol Chem. 1997;272(14):9287–93. doi: 10.1074/jbc.272.14.9287. [DOI] [PubMed] [Google Scholar]
- 103.Blind RD, Garabedian MJ. Differential recruitment of glucocorticoid receptor phospho-isoforms to glucocorticoid-induced genes. J Steroid Biochem Mol Biol. 2008;109(1-2):150–7. doi: 10.1016/j.jsbmb.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chen W, Dang T, Blind RD, Wang Z, Cavasotto CN, Hittelman AB, et al. Glucocorticoid receptor phosphorylation differentially affects target gene expression. Mol Endocrinol. 2008 doi: 10.1210/me.2007-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Miller AL, Garza AS, Johnson BH, Thompson EB. Pathway interactions between MAPKs, mTOR, PKA, and the glucocorticoid receptor in lymphoid cells. Cancer Cell Int. 2007;7:3. doi: 10.1186/1475-2867-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Miller AL, Webb MS, Copik AJ, Wang J, Johnson BH, Kumar R, et al. p38 Mitogen-activated protein kinase (MAPK) is a key mediator in glucocorticoid-induced apoptosis of lymphoid cells: correlation between p38 MAPK activation and site-specific phosphorylation of the human glucocorticoid receptor at serine 211. Mol Endocrinol. 2005;19(6):1569–83. doi: 10.1210/me.2004-0528. [DOI] [PubMed] [Google Scholar]
- 107.Simic I, Maric NP, Mitic M, Soldatovic I, Pavlovic Z, Mihaljevic M, et al. Phosphorylation of leukocyte glucocorticoid receptor in patients with current episode of major depressive disorder. Progress in neuro-psychopharmacology & biological psychiatry. 2013;40:281–5. doi: 10.1016/j.pnpbp.2012.10.021. Epub 2012/11/06. [DOI] [PubMed] [Google Scholar]
- 108.Galliher-Beckley AJ, Williams JG, Collins JB, Cidlowski JA. Glycogen synthase kinase 3beta-mediated serine phosphorylation of the human glucocorticoid receptor redirects gene expression profiles. Mol Cell Biol. 2008;28(24):7309–22. doi: 10.1128/MCB.00808-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ismaili N, Blind R, Garabedian MJ. Stabilization of the unliganded glucocorticoid receptor by TSG101. J Biol Chem. 2005;280(12):11120–6. doi: 10.1074/jbc.M500059200. [DOI] [PubMed] [Google Scholar]
- 110.Itoh M, Adachi M, Yasui H, Takekawa M, Tanaka H, Imai K. Nuclear export of glucocorticoid receptor is enhanced by c-Jun N-terminal kinase-mediated phosphorylation. Mol Endocrinol. 2002;16(10):2382–92. doi: 10.1210/me.2002-0144. [DOI] [PubMed] [Google Scholar]
- 111.Deroo BJ, Rentsch C, Sampath S, Young J, DeFranco DB, Archer TK. Proteasomal inhibition enhances glucocorticoid receptor transactivation and alters its subnuclear trafficking. Mol Cell Biol. 2002;22(12):4113–23. doi: 10.1128/MCB.22.12.4113-4123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wallace AD, Cidlowski JA. Proteasome-mediated glucocorticoid receptor degradation restricts transcriptional signaling by glucocorticoids. J Biol Chem. 2001;276(46):42714–21. doi: 10.1074/jbc.M106033200. [DOI] [PubMed] [Google Scholar]
- 113.Wang X, DeFranco DB. Alternative effects of the ubiquitin-proteasome pathway on glucocorticoid receptor down-regulation and transactivation are mediated by CHIP, an E3 ligase. Mol Endocrinol. 2005;19(6):1474–82. doi: 10.1210/me.2004-0383. [DOI] [PubMed] [Google Scholar]
- 114.Druker J, Liberman AC, Antunica-Noguerol M, Gerez J, Paez-Pereda M, Rein T, et al. RSUME enhances glucocorticoid receptor SUMOylation and transcriptional activity. Mol Cell Biol. 2013;33(11):2116–27. doi: 10.1128/MCB.01470-12. Epub 2013/03/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Davies L, Karthikeyan N, Lynch JT, Sial EA, Gkourtsa A, Demonacos C, et al. Cross talk of signaling pathways in the regulation of the glucocorticoid receptor function. Mol Endocrinol. 2008;22(6):1331–44. doi: 10.1210/me.2007-0360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Holmstrom S, Van Antwerp ME, Iniguez-Lluhi JA. Direct and distinguishable inhibitory roles for SUMO isoforms in the control of transcriptional synergy. Proc Natl Acad Sci U S A. 2003;100(26):15758–63. doi: 10.1073/pnas.2136933100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Holmstrom SR, Chupreta S, So AY, Iniguez-Lluhi JA. Sumo-Mediated Inhibition of Glucocorticoid Receptor Synergistic Activity Depends on Stable Assembly at the Promoter but Not on Daxx. Mol Endocrinol. 2008 doi: 10.1210/me.2007-0581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Le Drean Y, Mincheneau N, Le Goff P, Michel D. Potentiation of glucocorticoid receptor transcriptional activity by sumoylation. Endocrinology. 2002;143(9):3482–9. doi: 10.1210/en.2002-220135. [DOI] [PubMed] [Google Scholar]
- 119.Tian S, Poukka H, Palvimo JJ, Janne OA. Small ubiquitin-related modifier-1 (SUMO-1) modification of the glucocorticoid receptor. Biochem J. 2002;367(Pt 3):907–11. doi: 10.1042/BJ20021085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ito K, Yamamura S, Essilfie-Quaye S, Cosio B, Ito M, Barnes PJ, et al. Histone deacetylase 2-mediated deacetylation of the glucocorticoid receptor enables NF-kappaB suppression. J Exp Med. 2006;203(1):7–13. doi: 10.1084/jem.20050466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Charmandari E, Chrousos GP, Lambrou GI, Pavlaki A, Koide H, Ng SS, et al. Peripheral CLOCK regulates target-tissue glucocorticoid receptor transcriptional activity in a circadian fashion in man. PLoS One. 2011;6(9):e25612. doi: 10.1371/journal.pone.0025612. Epub 2011/10/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Fan BJ, Wang DY, Tham CC, Lam DS, Pang CP. Gene expression profiles of human trabecular meshwork cells induced by triamcinolone and dexamethasone. Invest Ophthalmol Vis Sci. 2008;49(5):1886–97. doi: 10.1167/iovs.07-0414. [DOI] [PubMed] [Google Scholar]
- 123.Adcock IM, Nasuhara Y, Stevens DA, Barnes PJ. Ligand-induced differentiation of glucocorticoid receptor (GR) trans-repression and transactivation: preferential targetting of NF-kappaB and lack of I-kappaB involvement. Br J Pharmacol. 1999;127(4):1003–11. doi: 10.1038/sj.bjp.0702613. Epub 1999/08/05. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 124.Chen SH, Masuno K, Cooper SB, Yamamoto KR. Incoherent feed-forward regulatory logic underpinning glucocorticoid receptor action. Proc Natl Acad Sci U S A. 2013;110(5):1964–9. doi: 10.1073/pnas.1216108110. Epub 2013/01/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Barnes PJ. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2013;131(3):636–45. doi: 10.1016/j.jaci.2012.12.1564. Epub 2013/01/31. [DOI] [PubMed] [Google Scholar]
- 126.Yang N, Ray DW, Matthews LC. Current concepts in glucocorticoid resistance. Steroids. 2012;77(11):1041–9. doi: 10.1016/j.steroids.2012.05.007. Epub 2012/06/26. [DOI] [PubMed] [Google Scholar]
- 127.Clark AR, Belvisi MG. Maps and legends: the quest for dissociated ligands of the glucocorticoid receptor. Pharmacol Ther. 2012;134(1):54–67. doi: 10.1016/j.pharmthera.2011.12.004. Epub 2012/01/04. [DOI] [PubMed] [Google Scholar]
- 128.Vandevyver S, Dejager L, Tuckermann J, Libert C. New insights into the anti-inflammatory mechanisms of glucocorticoids: an emerging role for glucocorticoid-receptor-mediated transactivation. Endocrinology. 2013;154(3):993–1007. doi: 10.1210/en.2012-2045. Epub 2013/02/07. [DOI] [PubMed] [Google Scholar]