

Abstract
Background
Subtle functional deficiencies in highly conserved DNA repair or growth regulatory processes resulting from polymorphic variation may increase genetic susceptibility to breast cancer. Polymorphisms in DNA repair genes can impact protein function leading to genomic instability facilitated by growth stimulation and increased cancer risk. Thus, 19 single nucleotide polymorphisms (SNPs) in eight genes involved in base excision repair (XRCC1, APEX, POLD1), BRCA1 protein interaction (BRIP1, ZNF350, BRCA2), and growth regulation (TGFß1, IGFBP3) were evaluated.
Methods
Genomic DNA samples were used in Taqman 5'-nuclease assays for most SNPs. Breast cancer risk to ages 50 and 70 were estimated using the kin-cohort method in which genotypes of relatives are inferred based on the known genotype of the index subject and Mendelian inheritance patterns. Family cancer history data was collected from a series of genotyped breast cancer cases (N = 748) identified within a cohort of female US radiologic technologists. Among 2,430 female first-degree relatives of cases, 190 breast cancers were reported.
Results
Genotypes associated with increased risk were: XRCC1 R194W (WW and RW vs. RR, cumulative risk up to age 70, risk ratio (RR) = 2.3; 95% CI 1.3–3.8); XRCC1 R399Q (QQ vs. RR, cumulative risk up to age 70, RR = 1.9; 1.1–3.9); and BRIP1 (or BACH1) P919S (SS vs. PP, cumulative risk up to age 50, RR = 6.9; 1.6–29.3). The risk for those heterozygous for BRCA2 N372H and APEX D148E were significantly lower than risks for homozygotes of either allele, and these were the only two results that remained significant after adjusting for multiple comparisons. No associations with breast cancer were observed for: APEX Q51H; XRCC1 R280H; IGFPB3 -202A>C; TGFß1 L10P, P25R, and T263I; BRCA2 N289H and T1915M; BRIP1 -64A>C; and ZNF350 (or ZBRK1) 1845C>T, L66P, R501S, and S472P.
Conclusion
Some variants in genes within the base-excision repair pathway (XRCC1) and BRCA1 interacting proteins (BRIP1) may play a role as low penetrance breast cancer risk alleles. Previous association studies of breast cancer and BRCA2 N372H and functional observations for APEX D148E ran counter to our findings of decreased risks. Due to the many comparisons, cautious interpretation and replication of these relationships are warranted.
Keywords: Breast cancer, kin-cohort, genetic variation, epidemiology, methods, risk factors
Background
Subtle functional deficiencies in highly conserved DNA repair or growth regulatory processes resulting from germ-line genetic variation have been proposed as possible mechanisms for increased genetic susceptibility to breast cancer [1-3], especially since it is estimated that the two known susceptibility genes BRCA1 and BRCA2 may account for less than 4% of all breast cancers [4,5]. Genetic epidemiologic studies suggest that BRCA1/2 account for no more than 20% of the familial risk of breast cancer, and the residual component is likely to be due polygenic inheritance of multiple low-penetrance susceptibility alleles [4-12].
We are conducting genetic studies of breast cancer among a cohort of U. S. radiologic technologists (USRT), in which the primary carcinogen under study is occupational exposure to ionizing radiation. Because the direct and indirect damaging effects of external radiation include oxidized bases and DNA single and double strand breaks, we are investigating 19 candidate variants in eight genes that are either involved in base excision repair, interact with the BRCA1 gene, or regulate cell growth. These genes are also attractive candidates as general breast cancer susceptibility factors because the repair process is not limited solely to exogenous radiation damage and includes carcinogenic chemicals, dietary constituents, and estrogens. In addition, growth deregulation is common to many developing tumors.
In the USRT case-control study, blood samples were collected initially from breast cancer cases during 1999–2001 and are currently being collected from a comparable control series. The available genotype data of the breast cancer cases provided an opportunity to perform kin-cohort analyses [13-15] using the case's family history data. In kin-cohort analyses, a cohort of relatives of index subjects is followed for disease occurrence. The genotypes of the relatives are unknown and are inferred based on the known genotype of the corresponding index subject (or proband) and Mendelian inheritance patterns. The kin-cohort method estimates risks in homozygous or heterozygous carriers and non-carriers of a variant and statistically accounts for the uncertainty in the indirect genotype information.
The kin-cohort analysis offers several unique aspects that distinguish it from the standard case-control analyses we plan to conduct in the future. First, in a kin-cohort analysis, risk estimates are obtained from the cancer history of the index cases' family members and thus provide data independent from the typical case-control analysis [16]. Second, kin-cohort analyses afford an opportunity to evaluate potential survival bias when analyzing the breast cancer case-control data that may be present given the prevalence sample in our study. The kin-cohort method is robust against such survival bias as cancer history among all family members is analyzed irrespective of the relative's vital status. Third, the underlying population for the case-control study is the cohort of radiologic technologists, who have been exposed to low levels of radiation from their occupation. The relatives of the radiologic technologists, however, are unlikely to have experienced occupational radiation exposure. Thus, separate risk estimates from the kin-cohort analysis and the breast cancer case-control study will provide information on whether risk from the genetic variants differs within two populations that have different background risks from radiation exposure.
Using the kin-cohort analytic method, we evaluated 19 single nucleotide polymorphism (SNP) variants in the following eight genes: XRCC1, APEX, and POLD1 in the base excision repair pathway; BRCA2, BRIP1 (or BACH1), and ZNF350 (or ZBRK1) as BRCA1 interacting proteins; and the growth factor genes TGFß1 and IGFBP3, and their relation to breast cancer risk.
Methods
Study population
In 1982, the U. S. National Cancer Institute, in collaboration with the University of Minnesota and the American Registry of Radiologic Technologists, initiated a study of cancer incidence and mortality among 146,022 U.S. radiologic technologists who were certified for at least two years between 1926 and 1982. The primary objectives of the study are to describe the carcinogenic risks of long-term, low- to moderate-dose, fractionated occupational radiation exposures and to determine factors associated with radiation sensitivity or cancer susceptibility. The current median age for cohort members is 53.4 years, and 73% are female. During 1984–1989 and during 1993–1998, two postal surveys were administered and included detailed questions related to work history as a radiologic technologist, lifestyle characteristics, other cancer risk factors, and health outcomes, including breast cancer. Approximately 90,000 technologists responded to each survey. All female technologists reporting a primary breast cancer that was subsequently confirmed based on medical records (pathology report, physician's notes, hospital discharge summary or physician correspondence) were eligible for inclusion if still living. In December 1999, when biospecimen collection began, there were 1345 living breast cancer cases with diagnosis years ranging from 1955 to 1998. By the end of December 2001, 748 breast cancer cases had provided informed consent, a blood sample, and responded to a telephone interview that collected updated cancer risk factor and family cancer history information and selected work history data. Another 143 cases could not be located or had an unlisted telephone number and did not respond to repeated correspondence inviting participation, 29 were too ill, 238 refused, 21 could not arrange a blood draw or the draw was unsuccessful, and 166 were still in process. This study has been approved annually by the human subjects review boards of the National Cancer Institute and the University of Minnesota.
Birth and death (if applicable) dates and breast cancer diagnosis dates were obtained for all first-degree female relatives. Data were evaluated for inconsistencies in age, reported generational intervals (all mothers had to be at least 11 years old before the birth of a child), and all breast cancers must have occurred at an age younger than current age or age at death. Initially, there were 2497 relatives in the data set and 194 of these relatives were reported to have breast cancer. For a total of 16 of the breast cancers reported in female relatives, the age at diagnosis was unknown and was imputed using the median age of breast cancer onset in all the relatives for which age was known. There were 60 half-sisters and seven relatives with unknown or un-imputable ages at last observation who were subsequently excluded. After these exclusions, 2430 relatives were retained in the analyses consisting of 190 with breast cancer and 2240 without.
Genotyping
Approximately 10 nanograms of genomic DNA extracted from peripheral lymphocytes were used as template in Taqman 5'-nuclease assays for all SNPs except for TGFß1 P25R, for which a PCR-RFLP assay was used. Taqman assays were performed using 450 nanomolar primer concentrations and 100 nanomolar probe concentrations and Universal Master Mix (Applied Biosystems). Probes specific for each SNP were designed with Primer Express software (Applied Biosystems) and labeled with either 6-FAM, TET, or VIC as reporter dyes and either Black Hole Quencher-1 (IDT, Inc.) or MGB-NFQ (Applied Biosystems) as quenchers. The primer and probe sequences and PCR conditions for each SNP are available (on request from JPS at http://lpg.nci.nih.gov/LPG/struewing/pubs). Most assays were performed in 20 microliter reactions in 96-well trays using a 7700 instrument (Applied Biosystems), but some were performed in 5 microliter reactions in 384-well trays using a 7900HT instrument (Applied Biosystems).
Subjects with each of the three possible genotypes (unless no homozygous variant subjects have ever been identified) for each SNP were confirmed by sequencing and included on each genotyping tray. In addition, approximately 5% of samples (41 samples, distributed as 2 to 7 aliquots of DNA from 8 different anonymous subjects) were included to monitor quality control (QC), with laboratory personnel blinded as to which were the QC samples. The genotypes for each of the duplicate QC samples from a subject matched exactly for all SNPs except one, and in this case, uncovered a systematic error in coding the results. This assay was repeated entirely and the QC samples then matched exactly.
Statistical methods
The analysis was based on a cohort of first degree female relatives of case probands, i.e., breast cancer patients, who were followed retrospectively over time for breast cancer incidence. Although the relatives' genotypes were not observed, the probability distribution could be inferred from the observed genotype of the corresponding case probands and Mendelian inheritance patterns. A marginal likelihood approach [15] was used to estimate age-specific cumulative risks associated with different genotypes while accounting for the uncertainty introduced by using indirect information about the relatives' genotype. Separate analyses were performed for each locus. For loci with rare variant frequencies and therefore low power to discern risk differences, we grouped heterozygote and homozygote variant genotypes together when the prevalence was less than 10%. For loci with common variants, we first estimated cumulative risks associated with the three genotypes separately. For two such loci (IGFBP3 -202A>C and TGFß1 L10P), visual inspection of the age-specific cumulative risk graphs revealed no difference in risk between homozygotes and heterozygous variant carriers and thus variant genotypes were combined.
As a summary measure for risk associated with variant genotypes, we obtained cumulative risk ratios (RR) at ages 50 and 70 with the homozygous wild type genotype considered the referent category. (Relative risks for breast cancer up to any age can be calculated using the kin-cohort method. We chose to graphically display risks up to age 80 to represent "lifetime" risk. In the tables we chose to provide risks with confidence intervals (CI) up to ages 50 and 70 to depict any differences that could be associated with earlier vs. later age-at-onset breast cancer [17].) The variance of the estimated RR was assessed by bootstrap sampling of families. The 95% confidence intervals (CI) for the estimated RR were based on the 2.5th and 97.5th percentiles of the distribution of 1,000 bootstrap replicates of the RR; the p-values for the estimated RR were two-sided and also based on 1,000 bootstrap replications. Adjustment for multiple comparisons was performed by either controlling the probability of at least one falsely rejected null hypothesis (the so called family-wise error rate [18]) or by controlling the expected proportion of falsely rejected null hypotheses among all rejected null hypotheses (the so called false discovery rate [19]).
Results
Descriptive characteristics of the radiologic technologists with breast cancer (index probands) and their first degree female relatives (with and without breast cancer) are shown in Table 1. The calendar year of birth and year of breast cancer diagnosis ranges more widely for relatives than for probands because relatives spanned three generations (mother, sister, daughter). Age at first breast cancer diagnosis was higher among relatives as compared to probands and reflects the younger ages represented in the cohort. Nearly equal numbers of breast cancers occurred in mothers (98) and sisters (85) with seven breast cancers in daughters.
Table 1.
Descriptive characteristics of breast cancer cases (probands) from the US Radiologic Technologist study and their first degree relatives with and without breast cancer.
| Characteristic at time of interview (1999–2001) | Radiologic technologists with breast cancer* (n = 748) | First degree female relatives with breast cancer (n = 190) | First degree female relatives without breast cancer (N = 2240) | |||
| N | % | N | % | N | % | |
| Year of birth | ||||||
| <1900 | 0 | 0.0 | 19 | 10.0 | 165 | 7.4 |
| 1900–29 | 155 | 20.7 | 110 | 57.9 | 702 | 31.3 |
| 1930–39 | 202 | 27.0 | 28 | 14.7 | 234 | 10.5 |
| 1940–49 | 276 | 36.9 | 20 | 10.5 | 271 | 12.1 |
| 1950–59 | 115 | 15.4 | 10 | 5.3 | 304 | 13.6 |
| ≥1960+ | 0 | 0.0 | 3 | 1.6 | 564 | 25.2 |
| Calendar year of breast cancer diagnosis | ||||||
| <1975 | 43 | 5.8 | 56 | 29.5 | NA** | --- |
| 1975–79 | 42 | 5.6 | 16 | 8.4 | NA | --- |
| 1980–84 | 98 | 13.1 | 20 | 10.5 | NA | --- |
| 1985–89 | 172 | 23.0 | 35 | 18.4 | NA | --- |
| 1990–94 | 313 | 41.8 | 31 | 16.3 | NA | --- |
| ≥1995 | 80 | 10.7 | 32 | 16.8 | NA | --- |
| Age at diagnosis | ||||||
| <40 | 114 | 15.2 | 22 | 11.6 | NA | --- |
| 40–49 | 299 | 40.0 | 28 | 14.7 | NA | --- |
| 50–59 | 203 | 27.1 | 42 | 22.1 | NA | --- |
| 60–69 | 90 | 12.0 | 49 | 25.8 | NA | --- |
| ≥70 | 42 | 5.6 | 49 | 25.8 | NA | --- |
| Relationship to the radiologic technologist (index proband) | ||||||
| Mother | NA | --- | 98 | 51.6 | 648 | 28.9 |
| Sister | NA | --- | 85 | 44.7 | 882 | 39.4 |
| Daughter | NA | --- | 7 | 3.7 | 710 | 31.7 |
* Includes invasive breast cancer and ductal carcinoma in situ **NA is Not applicable
From among 748 radiologic technologists with breast cancer, 99.6% or more of the samples were successfully genotyped (Table 2). Two samples failed repeatedly, leaving 746 consistently genotyped at least 75% of the time. The genotype frequencies for cases are also shown in Table 2. Except for BRCA2 N372H and ZBRK1 1845C>T, all distributions were consistent with Hardy Weinberg Equilibrium.
Table 2.
Polymorphic variant frequencies among breast cancer cases (probands) from the US Radiologic Technologist Health Study
| Gene | Polymorphism* | Total** number | Common homozygote frequency | Heterozygote frequency | Rare homozygote frequency | |||
| n | % | n | % | n | % | |||
| Base Excision Repair Genes | ||||||||
| XRCC1 | R194W(rs1799782) | 748 | 664 | 88.8 | 82 | 11.0 | 2 | 0.3 |
| XRCC1 | R280H(rs25489) | 748 | 676 | 90.4 | 71 | 9.5 | 1 | 0.1 |
| XRCC1 | R399Q(rs25478) | 748 | 321 | 42.9 | 335 | 44.8 | 92 | 12.3 |
| APEX | Q51H(rs1048945) | 746 | 687 | 92.1 | 58 | 7.8 | 1 | 0.1 |
| APEX | D148E(rs1130409) | 745 | 219 | 29.4 | 387 | 51.9 | 139 | 18.7 |
| POLD1 | R119H(rs1726801) | 748 | 650 | 86.9 | 92 | 12.3 | 6 | 0.8 |
| BRCA1 interacting proteins | ||||||||
| BRCA2 | N289H(rs2421655) | 748 | 698 | 93.3 | 50 | 6.7 | 0 | 0.0 |
| BRCA2 | N372H(rs144848)*** | 747 | 405 | 54.2 | 274 | 36.7 | 68 | 9.1 |
| BRCA2 | T1915I(rs4987717) | 748 | 713 | 95.3 | 35 | 4.7 | 0 | 0.0 |
| BRIP1 | -64 G>A(rs2048718) | 748 | 211 | 28.2 | 370 | 49.5 | 167 | 22.3 |
| BRIP1 | P919S(rs4986764) | 745 | 268 | 36.0 | 355 | 47.7 | 122 | 16.4 |
| ZBRK1 | L66P(rs2278420) | 744 | 535 | 71.9 | 190 | 25.5 | 19 | 2.6 |
| ZBRK1 | S472P(rs4986771) | 748 | 694 | 92.8 | 52 | 7.0 | 2 | 0.3 |
| ZBRK1 | R501S(rs2278415) | 746 | 576 | 77.2 | 154 | 20.6 | 16 | 2.1 |
| ZBRK1 | 1845 C>T (rs4986770)*** | 747 | 640 | 85.7 | 107 | 14.3 | 0 | 0.0 |
| Growth factor genes | ||||||||
| TGFß1 | L10P(rs1982073) | 745 | 263 | 35.3 | 357 | 47.9 | 125 | 16.8 |
| TGFß1 | P25R(rs1800471) | 745 | 629 | 84.4 | 109 | 14.6 | 7 | 0.9 |
| TGFß1 | T263I(rs1800472) | 748 | 709 | 94.8 | 39 | 5.2 | 0 | 0 |
| IGFΒP3 | -202A>C(rs2854744) | 746 | 194 | 26.0 | 360 | 48.3 | 192 | 25.7 |
* Amino acids and their symbols: R: Arginine, W: Tryptophan, H: Histidine, Q: Glutamine, D: Aspartic Acid, E: Glutamic Acid, N: Asparagine, P: Proline, S: Serine, L: Leucine, I: Isoleucine, T: Threonine. dbSNP reference sequence number in parentheses. ** Numbers vary because not all the cases could be genotyped due to technical issues with the sample. Also some genotyped cases were dropped from kin-cohort analyses because telephone interviews had not been completed at the time of blood collection. *** Test for significant deviation from Hardy Weinberg Equilibrium, p = 0.05
Many of the variants analyzed showed no appreciable relationship with breast cancer occurrence (Table 3). However, several variants were associated with increased risk of breast cancer, including XRCC1 R194W (WW or RW vs. RR to age 70; RR = 2.3, 95% CI: 1.3–3.8), XRCC1 R399Q (QQ vs RR to age 50, RR = 3.4; 1.3–9.3), BRIP1 P919S (SS vs PP to age 50; RR = 6.9; 1.6–29.3), and POLD1 R119H (HH or RH vs. RR to age 70, RR = 1.8; 1.0–2.9). Risks were significantly reduced for APEX D148E heterozygotes (to ages 50 and 70; RR = 0.2; 0.1–0.8 and RR = 0.2; 0.1–0.5, respectively) and BRCA2 N372H heterozygotes (to age 70; HR = 0.2; 0.1–0.5) compared to common homozygotes. When we adjusted the p-values for multiple comparisons, only the heterozygous associations of decreased risk up to age 70 in APEX D148E and BRCA2 N372H remained significant at p < 0.001.
Table 3.
Cumulative breast cancer risk ratios to age 50 and to age 70 by genotype among 2430 first degree female relatives of breast cancer cases (probands), 190 of whom were reported to have breast cancer
| Rare allele frequency | To age 50 | To age 70 | |||||||
| Polymorphism* | N** | RR | 95% CI | p-value | N** | RR | 95% CI | p-value | |
| Base excision repair genes | |||||||||
| XRCC1 R194W | 0.06 | ||||||||
| RR | 51 | 1.0 | --- | 125 | 1.0 | --- | |||
| RW or WW | 6 | 1.4 | 0.4–4.2 | 0.408 | 22 | 2.3 | 1.3–3.8 | 0.004 | |
| XRCC1 R280H | 0.03 | ||||||||
| RR | 52 | 1.0 | --- | 134 | 1.0 | --- | |||
| RH or HH | 5 | 0.7 | 0.0–2.4 | 0.736 | 13 | 1.2 | 0.5–2.5 | 0.522 | |
| XRCC1 R399Q | 0.37 | ||||||||
| RR | 21 | 1.0 | --- | 59 | 1.0 | --- | |||
| RQ | 22 | 0.7 | 0.1–2.0 | 0.444 | 64 | 1.1 | 0.4–1.6 | 0.636 | |
| 14 | 3.4 | 1.3–9.3 | 0.014 | 24 | 1.9 | 1.1–3.9 | 0.010 | ||
| APEX Q51H | 0.04 | ||||||||
| 49 | 1.0 | --- | 134 | 1.0 | --- | ||||
| QH or HH | 7 | 3.0 | 0.5–6.7 | 0.216 | 12 | 1.7 | 0.7–3.1 | 0.236 | |
| APEX D148E | 0.49 | ||||||||
| DD | 20 | 1.0 | --- | 49 | 1.0 | --- | |||
| DE | 27 | 0.2 | 0.1–0.8 | 0.028 | 65 | 0.2 | 0.1–0.5 | <0.001 | |
| EE | 9 | 0.8 | 0.2–1.9 | 0.546 | 32 | 1.0 | 0.6–1.7 | 0.860 | |
| POLD1 R119H | 0.06 | ||||||||
| RR | 44 | 1.0 | --- | 123 | 1.0 | --- | |||
| RH or HH | 13 | 2.5 | 0.8–5.0 | 0.106 | 24 | 1.8 | 1.0–2.9 | 0.058 | |
| BRCA1 interacting proteins | |||||||||
| BRCA2 N289H | 0.03 | ||||||||
| MM | 57 | 1.0 | --- | 141 | 1.0 | --- | |||
| NH or HH | 0 | 0 | --- | ---- | 6 | 1.6 | 0.5–2.9 | 0.370 | |
| BRCA2 N372H | 0.32 | ||||||||
| NN | 30 | 1.0 | --- | 89 | 1.0 | --- | |||
| NH | 23 | 0.4 | 0.1–1.3 | 0.140 | 42 | 0.2 | 0.1–0.5 | <0.001 | |
| HH | 4 | 1.5 | 0.4–4.4 | 0.322 | 16 | 1.3 | 0.8–2.4 | 0.232 | |
| BRCA2 T1915M | 0.05 | ||||||||
| TT | 53 | 1.0 | --- | 139 | 1.0 | --- | |||
| TM or MM | 4 | 1.7 | 0.0–4.0 | 0.664 | 8 | 2.1 | 0.6–3.6 | 0.215 | |
| BRIP1 -64 G>A | 0.46 | ||||||||
| GG | 17 | 1.0 | --- | 42 | 1.0 | --- | |||
| GA | 24 | 0.4 | 0.1–1.2 | 0.126 | 70 | 0.7 | 0.2–1.2 | 0.112 | |
| AA | 16 | 1.5 | 0.5–4.3 | 0.424 | 35 | 1.6 | 0.8–2.8 | 0.218 | |
| BRIP1 P919S | 0.43 | ||||||||
| PP | 10 | 1.0 | --- | 52 | 1.0 | --- | |||
| PS | 34 | 4.5 | 0.8–12.2 | 0.096 | 69 | 0.6 | 0.3–1.2 | 0.134 | |
| SS | 12 | 6.9 | 1.6–29.3 | 0.018 | 25 | 1.3 | 0.8–2.8 | 0.220 | |
| ZBRK1 L66P | 0.15 | ||||||||
| LL | 43 | 1.0 | --- | 108 | 1.0 | --- | |||
| LP or PP | 14 | 0.9 | 0.3–2.0 | 0.712 | 38 | 1.2 | 0.7–1.9 | 0.304 | |
| ZBRK1 S472P | 0.12 | ||||||||
| SS | 55 | 1.0 | --- | 137 | 1.0 | --- | |||
| SP or PP | 2 | 0.8 | 0.0–2.2 | 0.614 | 10 | 1.6 | 0.6–2.9 | 0.312 | |
| ZBRK1 R501S | 0.12 | ||||||||
| RR | 46 | 1.0 | --- | 116 | 1.0 | --- | |||
| RS or SS | 11 | 0.8 | 0.2–1.8 | 0.534 | 31 | 1.2 | 0.7–2.1 | 0.434 | |
| ZBRK1 1845 C>T | 0.06 | ||||||||
| CC | 45 | 1.0 | --- | 121 | 1.0 | --- | |||
| CT or TT | 12 | 2.2 | 0.5–4.3 | 0.218 | 25 | 1.6 | 0.8–2.8 | 0.162 | |
| Growth factor genes | |||||||||
| TGFß1 L10P | 0.38 | ||||||||
| LL | 25 | 1.0 | --- | 54 | 1.0 | --- | |||
| LP or PP | 31 | 0.5 | 0.2–1.2 | 0.148 | 92 | 0.8 | 0.5–1.5 | 0.536 | |
| TGFß1 P25R | 0.10 | ||||||||
| PP | 48 | 1.0 | --- | 126 | 1.0 | --- | |||
| PR or RR | 8 | 1.0 | 0.1–3.1 | 0.952 | 20 | 1.2 | 0.6–2.1 | 0.554 | |
| TGFß1 T263I | 0.04 | ||||||||
| TT | 54 | 1.0 | --- | 138 | 1.0 | --- | |||
| TI or II | 3 | 2.1 | 0.0–4.9 | 0.492 | 9 | 1.7 | 0.6–3.1 | 0.312 | |
| IGFBP3 -202 A>C | 0.53*** | ||||||||
| AA | 11 | 1.0 | --- | 36 | 1.0 | --- | |||
| AC or CC | 45 | 1.0 | 0.3–3.5 | 0.848 | 110 | 0.7 | 0.4–1.3 | 0.228 | |
* Amino acids and their symbols: R: Arginine, W: Tryptophan, H: Histidine, Q: Glutamine, D: Aspartic Acid, E: Glutamic Acid, N: Asparagine, P: Proline, S: Serine, L: Leucine, I: Isoleucine, T: Threonine. ** Numbers represent those first degree female relatives who had breast cancer up to age 50 or 70 by the genotype of the case (index proband). The numbers are not the probabilistic assignment of the kin-cohort calculation to a specific genotype. *** Represents the frequency of the C allele, although not technically "rare".
Estimates of the cumulative risk by age are graphically displayed for each SNP (arranged in alphabetical order) in Figure 1. The SNPs in Figure 1 are either rare (prevalence of the homozygous variant was roughly less than 10%) or little curve separation was seen in the non-parametric approach, and so heterozygous and homozygous variant carriers were combined. For SNPs with a higher prevalence, estimated cumulative risks are shown in Figure 2 for each genotype separately. The BRCA2 N372H and BRIP1 -64 G>A plots show individuals homozygous for the common allele with risks intermediate between heterozygotes (lowest breast cancer cumulative risk) and the homozygous carriers of the rare variant (highest breast cancer cumulative risk). For APEX D148E, the risk in homozygous common or variant carriers is nearly identical. The XRCC1 R399Q plot shows nearly completely overlapping risk for the homozygous common allele and the heterozygotes, but the homozygous variant risk is elevated across the age range. The curves for the BRCA2 N372H and APEX D148E polymorphisms illustrate the extremely low estimated risk for heterozygous carriers.
Figure 1.
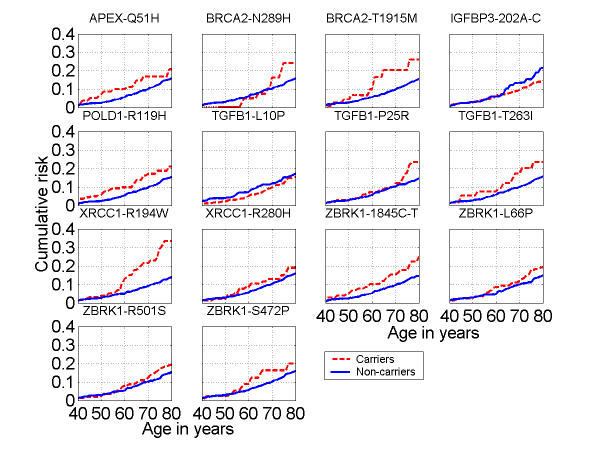
Kin-cohort breast cancer cumulative risk estimates up to age 80 by genotype for less frequent single nucleotide polymorphic variants among female relatives of probands. (Amino acids and their symbols: R: Arginine, W: Tryptophan, H: Histidine, Q: Glutamine, D: Aspartic Acid, E: Glutamic Acid, N: Asparagine, P: Proline, S: Serine, L: Leucine, I: Isoleucine, T: Threonine.)
Figure 2.
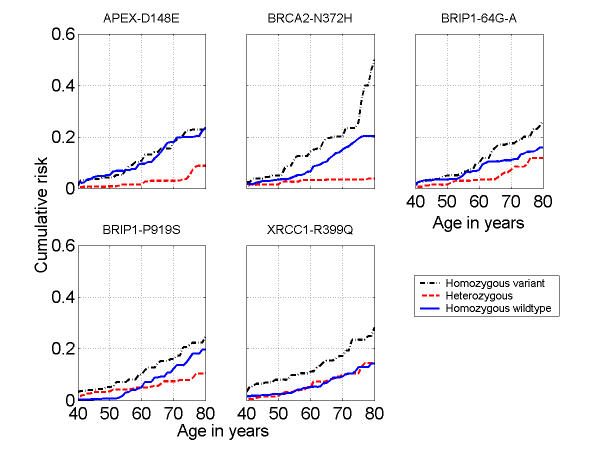
Kin-cohort breast cancer cumulative risk estimates up to age 80 for more frequent single nucleotide polymorphic variants among female relatives of probands. (Amino acids and their symbols: D: Aspartic Acid, E: Glutamic acid, R: Arginine, H: Histidine, Q: Glutamine, N: Asparagine, P: Proline, S: Serine.)
Discussion
We found evidence of a differential breast cancer risk associated with the variants XRCC1 R194W, XRCC1 R399Q, BRIP1 P919S, APEX D148E, and BRCA2 N372H using the kin-cohort analytic method. Multiple forms of DNA repair are found in mammalian species, of which the base excision repair pathway is one type involving complex protein interactions with non-bulky lesions in DNA. Since XRCC1 is a scaffolding protein integral to base excision repair [reviewed in [20]], the polymorphic loci in XRCC1 (R194W, R280H and R399Q) have been evaluated for risk at various cancer sites because their location within the gene or their conserved status make them ideal candidates with functional significance more likely [21]. Although most studies of cancer have found a decreased risk for the XRCC1 194W allele [reviewed in [22]] including a large study of breast cancer [23], we found the opposite relationship. Our results are in agreement with two recent breast cancer case-control studies that found non-significantly elevated 1.6- and 2-fold risks for at least one variant allele of XRCC1 194 [24,25]. Several studies of female breast cancer have reported elevated risk associated with the XRCC1 399Q allele among African Americans [23] and Koreans [26], but not among Caucasian [23-25,27] or Chinese women [28]. The mixed findings for the relationship of XRCC1 polymorphisms with breast cancer are difficult to reconcile, but may simply represent variability around the null for a non-susceptibility allele. Depending on the model system chosen, functional testing results indicate either a reduced DNA repair capacity associated with the XRCC1 399Gln allele [29], an increase in mitotic delay among healthy women with a family history of breast cancer after a γ-ray challenge [30], increased DNA adduct levels [31], or no difference related to the polymorphism in single strand break repair ability or cell survival in an isogenic background [32]. We evaluated two polymorphisms in the AP endonuclease APEX (also called APE1, HAP1, REF1) Q51H and D148E since this multifunctional endonuclease recognizes and begins the process of removing abasic sites in DNA [reviewed in [33]]. Further, the variant form of APEX 148 was functionally characterized as exhibiting post-irradiation challenge prolonged mitotic delay [30] and we expected the 148E allele would be associated with increased risk, but instead we found heterozygote carriers had significantly decreased breast cancer risk. Additional information, both functional and genotypic, is worth pursuing for the APEX 148 variant. POLD1 (polymerase δ) participates in a possibly redundant sub-pathway within the base excision repair "long patch" process [reviewed in [33]]. This particular variant has not been as well characterized in regard to cancer risk, compared to other SNPs in base excision repair genes, but observed RRs in our study were around two-fold up to ages 50 and 70 such that this SNP deserves further study to confirm or refute its role in breast cancer risk.
We evaluated six SNPs recently characterized in the BRCA1-interacting genes ZNF350 (ZBRK1) and BRIP1 (BACH1) [34]. Among those, the nonconservative BRIP1 P919S substitution showed a strong association with 4.5-fold and 6.9-fold (for PS and PP vs. SS, respectively) increased risks of breast cancer up to age 50. However, the association was markedly attenuated when observation was extended to age 70. This could be a chance finding, or, the variant may be associated predominantly with risk of pre-menopausal breast cancer. Further evaluation of this SNP in other study populations will be required.
Two growth factor genes, IGFBP3 and TGFß1, were evaluated because of their roles in controlling cellular growth and changes associated with malignant progression. Previous reports suggested an association between the -202A>C IGFBP3 SNP and risk of pre-menopausal breast cancer, primarily through its effect on circulating IGFBP3 levels [35] and/or IGF-1 levels [36]. Although we did not directly assess the relationship between the IGFBP3-202A>C variant and circulating IGFBP3 and IGF-1 levels, we did not observe an association between this SNP (as an indicator of IGF-1 levels) and breast cancer risk in our study. Three SNPs in the TGFß1 gene (L10P, P25R, and T263I) have previously been described [37], with the L10P variant showing some association with increased risk of breast cancer [38,39]. Our data do not support an association between these TGFß1 SNPs and breast cancer risk in this cohort.
Of all the SNPs evaluated, BRCA2 N372H had shown the most consistent relationship to breast cancer in previous studies: in both a European study [40] and an Australian study [41], HH homozygotes had significantly elevated odds ratios for breast cancer of 1.3 to 1.5 compared to NN homozygotes, and in a study of ovarian cancer from the UK and Australia [42], risk was similarly elevated (OR = 1.4) for HH homozygotes. In all three of these studies, there was no evidence of increased (or decreased) risk among heterozygotes, with ORs of 1.01 to 1.03, compared to NN homozygotes. In our study, HH homozygotes had a slightly increased RR of 1.5 (95% CI, 0.4–4.4) and 1.3 (95% CI, 0.8 – 2.4) over NN homozygotes up to ages 50 and 70, respectively. Unlike the previous studies, heterozygotes had a significantly reduced breast cancer risk compared to NN homozygotes. The minor allele frequency was somewhat higher in our study (0.32) of cases, versus approximately 0.28 in the UK\Australian study [42]. In the initial study [40], it was noted that the SNP in female controls was not in Hardy-Weinberg equilibrium (HWE), with an excess of heterozygotes and a deficit of both homozygotes. In our case series, the SNP was not in HWE, with 23 fewer heterozygotes than expected and more than 20% over the expected numbers of HH homozygotes under HWE; this is the trend one might have expected if the H allele increases risk. In addition, the 9.1% HH genotype frequency in our breast cancer cases was very similar to that observed in the Australian breast cancer study (9.2%) [41]. Whether the breast cancer risk is lower in heterozygotes, as is clearly evident in the risk curves from our analyses, or whether the inconsistent findings are still due to chance (even after multiple comparisons adjustment) are not known and will require further study in case-control analyses in this cohort or other large epidemiologic studies.
Because the striking heterozygous advantage in BRCA2 N372H and APEX D148E were unexpected and difficult to interpret biologically, we performed analyses stratified by type of relative (mother, sister). We discovered that, in general, the kin-cohort model for three genotype categories is not identifiable when restricted to mothers of the index cases because the matrix of Mendelian genotype probabilities for relatives conditional on the genotype and type of relative of the index case is rank deficient. This meant that calculations restricted to mothers could not be performed, but we could determine the relationship between individual SNPs and breast cancer among sisters. Therefore, we relied on the analysis restricted to sisters to corroborate patterns observed for all relatives combined. For sisters only, the analyses revealed the same patterns as shown in Figure 2, which were based on all female relatives, except for APEX D148E, where the results for sisters only showed similar risks for homozygous common and heterozygous genotypes and an increased risk for homozygous variant genotypes (data not shown). Due to the biological inconsistency of the results for APEX D148E and because of the differences between analyses based on sisters only and all relatives, we suggest caution in interpreting this result, despite the statistical significance. For BRCA2 N372H, results for sisters only were very similar to results for all relatives combined, lending credence to our observations, despite the difficult interpretation.
There are several study limitations. Fifty-six per cent of the women eligible donated a blood sample before the arbitrary genotyping cut-off date (December 31, 2001). Reasons for eligible women not providing a blood sample were that they could not be located, refused, or were too ill. The distribution of demographic and known breast cancer risk factors such as education, age at menarche, age at first live birth, age at breast cancer diagnosis, and year of birth were similar for participants and non-participants. Unsurprisingly, women over age 80 in 1999 were less likely to provide a blood sample (63% did not provide a blood sample compared to 48% for those under age 80). Regarding employment characteristics, more women who began to work in the 1950s tended to donate a sample (57%) compared to women who began work after 1970 (43%). Interestingly, women who reported a first degree relative with breast cancer were less inclined to donate a sample (44% vs. 53% of those with no family breast cancer history). However, for selection bias to have caused spurious associations, the differential participation would also need to be related to genotype, a generally improbable scenario. Another limitation was that the kin-cohort method is less powerful for common genotypes, such that discrimination in risk is reduced because the "at risk" allele assignment among relatives becomes less precise with increasing prevalence. Statistical power, in the presence of null results, is important to report, but these calculations for the kin-cohort method are computationally difficult. Since the statistical power to detect a two-fold increased or decreased effect in a case-control study with 190 breast cancer cases and 2240 controls for the homozygous variant genotype or the combined homozygous variant and heterozygous genotypes (for polymorphisms with rare homozygous variant genotype) ranges from 0.30 to 0.99 (assuming a = 0.05, two-sided test, and the SNP frequency varies between 0.01 to 0.25), we conclude the statistical power of our kin-cohort study was even less because the genotypes of the relatives were not known with certainty and multiple genotypes were evaluated. Breast cancers among first degree family members were not independently confirmed by medical records, however we considered that breast cancer was likely to be accurately reported in family members of breast cancer cases [43] and possibly even more so because the cases are trained to work in the medical field as radiologic technologists. It is not possible to adjust for breast cancer risk factors among family members, although all of the relatives, by definition, have a first-degree relative with breast cancer. Relative risk estimates from the kin-cohort analyses should not be affected, however absolute risk estimates could be inflated above the true values.
There were a large number of comparisons evaluated (n = 46), such that one or more relationships reported here could be due solely to chance. Of all the hypotheses tested in Table 3, only those corresponding to the two p-values < 0.001 (BRCA2 N372H heterozygous vs. homozygous common and APEX D148E heterozygous vs. homozygous common) can be rejected while controlling the family-wise error rate, i.e. the probability of a least one false positive at 0.05 [18] or while controlling the false discovery rate, i.e. the expected proportion of false positives among the positives at 0.05 [19]. It has to be noted that the hypotheses tested may not be independent. For example, SNPs in the same gene may be in linkage disequilibrium with each other and the risk to age 50 and risk to age 70 are presumably not independent. In this situation, our correction for multiple comparisons is likely overly conservative. However, the associations that we found significant at 0.05, but not 0.001, are certainly suggestive and serve as a means of hypothesis generation. Even though the kin-cohort design allows for the evaluation of gene-gene interactions [17], we did not perform such an analysis because of its very low statistical power and because of a lack of strong prior biologic hypotheses about the joint effect of SNPs in the genes studied.
The study and the kin-cohort method have several strengths and advantages. The strengths are that risks are determined by the breast cancer experience in relatives, who are included whether living or deceased, reducing concerns of selection bias from recruiting prevalent cases. The study uses information on risk from a group outside the parent study, essentially providing a rationale for later testing using other designs and increases confidence if similar associations are observed in the upcoming case-control study. In addition, the breast cancer risks among female relatives are independent of specific occupational exposures (in this study, medical radiation exposure from work as a radiologic technologist) that were the reason for the cohort assembly and follow-up. A very important feature is that a control series is not required, such that this method could easily be implemented among hospital-based cases. Once the genotypes are known for the index case series, risks for other common cancer outcomes (such as prostate, lung, or colon cancer) can readily be computed using family cancer history information collected at the time of blood sampling. Thus, the kin-cohort study provides additional independent supplemental data to an existing (or in progress) case-control study [16].
In summary, differences in breast cancer risk were associated with XRCC1 R194W, XRCC1 R399Q, BRIP1 P919S, APEX D148E and BRCA2 N372H, and were suggestive for several others. Although HH homozygotes for the BRCA2 N372H SNP had approximately 30% greater odds of breast cancer compared to NN homozygotes, as had been consistently observed in two previous studies of breast cancer [40,41], this association was weak, and we observed a significantly decreased risk among heterozygotes, a relationship that had not been suggested previously. We express caution in the interpretation of the decreased breast cancer risk observed for APEX D148E heterozygotes. It is possible that one or more of the XRCC1 R194W, XRCC1 R399Q, BRIP1 P919S variants could eventually be regarded as low-penetrance risk alleles for breast cancer, but after adjustment for multiple comparisons, none remained statistically significant. Ultimately, results from this kin-cohort analysis can be combined with findings from the standard case-control study for a more consistent interpretation of the risk associated with common genetic variants.
Conclusions
Some variants in genes within the base-excision repair pathway (XRCC1) and BRCA1 interacting proteins (BRIP1) may play a role as low penetrance breast cancer risk alleles. Previous association studies of increased breast cancer risk for BRCA2 N372H and decreased function for APEX D148E variants were not in agreement with our findings of decreased risks. Due to the many comparisons, cautious interpretation and replication of these relationships are warranted.
Competing interests
None declared.
Authors' contributions
All authors collaborated extensively on the manuscript, including research design (AJS, BHA, MMD, JPS), data collection (AJS, BHA, MMD, MH), laboratory assays (JLR, JPS), data preparation (AJS, BHA, MMD, NC, MH, JLR, JPS), and data analysis (MH, NC). All authors contributed to interpretation of the results and each wrote sections of the manuscript. All the authors read and have approved the final version.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Contributor Information
Alice J Sigurdson, Email: sigurdsa@mail.nih.gov.
Michael Hauptmann, Email: Hauptmam@mail.nih.gov.
Nilanjan Chatterjee, Email: Chattern@mail.nih.gov.
Bruce H Alexander, Email: balex@umn.edu.
Michele Morin Doody, Email: doodym@mail.nih.gov.
Joni L Rutter, Email: rutterj@mail.nih.gov.
Jeffery P Struewing, Email: struewij@mail.nih.gov.
References
- Mohrenweiser HW, Jones IM. Variation in DNA repair is a factor in cancer susceptibility: a paradigm for the promises and perils of individual and population risk estimation? Mutat Res. 1998;400:15–24. doi: 10.1016/S0027-5107(98)00059-1. [DOI] [PubMed] [Google Scholar]
- Roberts SA, Spreadborough AR, Bulman B, Barber JBP, Evans DGR, Scott D. Heritability of cellular radiosensitivity: a marker of low-penetrance predisposition genes in breast cancer? Am J Hum Genet. 1999;65:784–794. doi: 10.1086/302544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teare MD, Wallace SA, Harris M, Howell A, Birch JM. Cancer experience in the relatives of an unselected series of breast cancer patients. Br J Cancer. 1994;70:102–111. doi: 10.1038/bjc.1994.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anglian Breast Cancer Study Group Prevalence and penetrance of BRCA1 and BRCA2 in a population based series of breast cancer cases. Br J Cancer. 2000;83:1301–1308. doi: 10.1054/bjoc.2000.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peto J, Collins N, Barfoot R, Seal S, Warren W, Rahman N, Easton DF, Evans C, Deacon J, Stratton MR. Prevalence of BRCA1 and BRCA2 gene mutations in patients with early-onset breast cancer. J Natl Cancer Inst. 1999;91:943–949. doi: 10.1093/jnci/91.11.943. [DOI] [PubMed] [Google Scholar]
- Antoniou AC, Pharoah PDP, McMullen G, Day NE, Ponder BAJ, Easton DF. Evidence for further breast cancer susceptibility genes in addition to BRCA1 and BRCA2 in a population based study. Genet Epidemiol. 2002;21:1–18. doi: 10.1002/gepi.1014. [DOI] [PubMed] [Google Scholar]
- de Jong MM, Nolte IM, te Meerman GJ, van der Graaf WTA, Oosterwijk JC, Kleibeuker JH, Schaapveld M, de Vries EGE. Genes other than BRCA1 and BRCA2 involved in breast cancer susceptibility. J Med Genet. 2002;39:225–242. doi: 10.1136/jmg.39.4.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathanson KL, Wooster R, Weber BL. Breast cancer genetics: what we know and what we need. Nature Medicine. 2001;7:552–556. doi: 10.1038/87876. [DOI] [PubMed] [Google Scholar]
- Pharoah PDP, Antoniou A, Bobrow M, Zimmern RL, Easton DF, Ponder BAJ. Polygenic susceptibility to breast cancer and implications for prevention. Nature Genet. 2002;31:33–36. doi: 10.1038/ng853. [DOI] [PubMed] [Google Scholar]
- Mohrenweiser HW, Xi T, Vazquez-Matias J, Jones IM. Identification of 127 Amino Acid substitution variants in screening 37 DNA repair genes in humans. Cancer Epidemiol Biomarkers Prev. 2002;11:1054–1064. [PubMed] [Google Scholar]
- Mohrenweiser HW, Wilson DM, III, Jones IM. Challenges and complexities in estimating both the functional impact and the disease risk associated with the extensive genetic variation in human DNA repair genes. Mutat Res. 2003;526:93–125. doi: 10.1016/S0027-5107(03)00049-6. [DOI] [PubMed] [Google Scholar]
- Easton DF. How many more breast cancer predisposition genes are there? Breast Cancer Res. 1999;1:14–17. doi: 10.1186/bcr6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacholder S, Hartge P, Struewing JP, Pee D, McAdams M, Brody L, Tucker M. The kin-cohort study for estimating penetrance. Am J Epidemiol. 1998;148:623–630. doi: 10.1093/aje/148.7.623. [DOI] [PubMed] [Google Scholar]
- Gail MH, Pee D, Carroll R. Kin-cohort designs for gene characterization. J Natl Cancer Inst Monogr. 1999;26:55–60. doi: 10.1093/oxfordjournals.jncimonographs.a024227. [DOI] [PubMed] [Google Scholar]
- Chatterjee N, Wacholder S. A marginal likelihood approach for estimating penetrance from kin-cohort designs. Biometrics. 2001;57:245–252. doi: 10.1111/j.0006-341x.2001.00245.x. [DOI] [PubMed] [Google Scholar]
- Saunders CL, Begg CB. Kin-cohort evaluation of relative risks of genetic variants. Genet Epidemiol. 2003;24:220–229. doi: 10.1002/gepi.10235. [DOI] [PubMed] [Google Scholar]
- Rutter JL, Chatterjee N, Wacholder S, Struewing JP. The HER2 I655V polymorphism and breast cancer risk in Ashkenazim. Epidemiology. 2003;14:694–700. doi: 10.1097/01.ede.0000083227.74669.7b. [DOI] [PubMed] [Google Scholar]
- Hochberg Y. A sharper Bonferroni procedure for multiple tests of significance. Biometrika. 1988;75:800–80. [Google Scholar]
- Benjamini Y, Liu W. A distribution-free multiple test procedure that controls the false discovery rate. Department of Statistics and O.R., Tel Aviv University. Technical report RP-SOR-99-3 Tel Aviv. 1999.
- Thompson LH, West MG. XRCC1 keeps DNA from getting stranded (review) Mutation Res. 2000;459:1–18. doi: 10.1016/S0921-8777(99)00058-0. [DOI] [PubMed] [Google Scholar]
- Shen MR, Jones IM, Mohrenweiser H. Nonconservative amino acid substitution variants exist at polymorphic frequency in DNA genes in healthy humans. Cancer Res. 1998;58:604–608. [PubMed] [Google Scholar]
- Goode EL, Ulrich CM, Potter JD. Polymorphisms in DNA repair genes and associations with cancer risk. Cancer Epidemiol Biomarkers Prev. 2002;11:1513–1530. [PubMed] [Google Scholar]
- Duell EJ, Millikan , Pittman GS, Winkel S, Lunn RM, Tse C-KJ, Eaton A, Mohrenweiser HW, Newman B, Bell DA. Polymorphisms in the DNA repair gene XRCC1 and breast cancer. Cancer Epidemiol Biomarkers Prev. 2001;10:217–222. [PubMed] [Google Scholar]
- Smith TR, Miller MS, Lohman K, Lange EM, Case LD, Mohrenweiser HW, Hu JJ. Polymorphisms of XRCC1 and XRCC3 genes and susceptibility to breast cancer. Cancer Lett. 2003;190:183–190. doi: 10.1016/S0304-3835(02)00595-5. [DOI] [PubMed] [Google Scholar]
- Smith TR, Levine EA, Perrier ND, Miller MS, Freimanis RI, Lohman K, Case LD, Xu J, Mohrenweiser HW, Hu JJ. DNA-repair genetic polymorphisms and breast cancer risk. Cancer Epidemiol Biomarkers Prev. 2003;12:1200–1204. [PubMed] [Google Scholar]
- Kim SU, Park SK, Yoo KY, Yoon KS, Choi JY, Seo JS, Park WY, Kim JH, Noh DY, Ahn SH, Choe KJ, Strickland PT, Hirvonen A, Kang D. XRCC1 genetic polymorphism and breast cancer risk. Pharmacogenetics. 2002;12:335–338. doi: 10.1097/00008571-200206000-00010. [DOI] [PubMed] [Google Scholar]
- Moullan N, Cox DG, Angele S, Romestaing P, Gerard J-P, Hall J. Polymorphisms in the DNA repair genes XRCC1, breast cancer risk, and response to radiotherapy. Cancer Epidemiol Biomarkers Prev. 2003;12:1200–1204. [PubMed] [Google Scholar]
- Shu X-O, Cai Q, Gao Y-T, Wen W, Jin F, Zheng W. A population-based case-control study of the Arg399Gln polymorphism in DNA repair gene XRCC1 and risk of breast cancer. Cancer Epidemiol Biomarkers Prev. 2003;12:1462–1467. [PubMed] [Google Scholar]
- Lunn RM, Langlois RG, Hsieh LL, Thompson CL, Bell DA. XRCC1 polymorphisms: Effects on Aflatoxin B1-DNA adducts and glycophorin A variant frequency. Cancer Res. 1999;59:2557–2561. [PubMed] [Google Scholar]
- Hu JJ, Smith TR, Miller MS, Mohrenweiser HW, Golden A, Case LD. Amino acid substitution variants of APE1 and XRCC1 genes associated with ionizing radiation sensitivity. Carcinogenesis. 2001;22:917–922. doi: 10.1093/carcin/22.6.917. [DOI] [PubMed] [Google Scholar]
- Matullo G, Peluso M, Polidoro S, Guarrera S, Munnia A, Krogh V, Masala G, Berrino F, Panico S, Tumino R, Vineis P, Palli D. Combination of DNA repair gene single nucleotide polymorphisms and increased levels of DNA adducts in a population-based study. Cancer Epidemiol Biomarkers Prev. 2003;12:674–647. [PubMed] [Google Scholar]
- Taylor RM, Thistlethwaite A, Caldecott KW. Central role for the XRCC1 BRCT I domain in mammalian DNA single-strand break repair. Mol Cell Biol. 2002;22:2556–2563. doi: 10.1128/MCB.22.8.2556-2563.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson DM, III, Thompson LH. Life without DNA repair. Proc Natl Acad Sci. 1997;94:12754–12757. doi: 10.1073/pnas.94.24.12754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutter JL, Smith A, Davila M, Sigurdson AJ, Doody MM, Tucker MA, Greene M, Struewing JP. Mutational analysis of BRCA1-interacting genes ZNF350/ZBRK1 and BRIP1/BACH1 among BRCA1 and BRCA2-negative probands from breast-ovarian cancer families and among early-onset breast cancer cases and reference individuals. Hum Mutat. 2003;22:121–128. doi: 10.1002/humu.10238. [DOI] [PubMed] [Google Scholar]
- Schernhammer ES, Hankinson SE, Hunter DJ, Blouin MJ, Pollak MN. Polymorphic variation at the -202 locus in IGFBP3: Influence on serum levels of insulin-like growth factors, interaction with plasma retinol and vitamin D and breast cancer risk. Int J Cancer. 2003;107:60–64. doi: 10.1002/ijc.11358. [DOI] [PubMed] [Google Scholar]
- Bohlke K, Cramer DW, Trichopoulos D, Mantzoros CS. Insulin-like growth factor-I in relation to premenopausal ductal carcinoma in situ of the breast. Epidemiology. 1998;9:570–573. [PubMed] [Google Scholar]
- Cambien F, Ricard S, Troesch A, Mallet C, Generenaz L, Evans A, Arveiler D, Luc G, Ruidavets JB, Poirer O. Polymorphisms of the transforming growth factor b1 gene in relation to myocardial infarction and blood pressure. Hypertension. 1996;28:881–887. doi: 10.1161/01.hyp.28.5.881. [DOI] [PubMed] [Google Scholar]
- Ziv E, Cauley J, Morin PA, Saiz R, Browner WS. Association between the T29-->C polymorphism in the transforming growth factor beta 1 gene and breast cancer among elderly white women: the study of osteoporotic fractures. JAMA. 2001;285:2859–2863. doi: 10.1001/jama.285.22.2859. [DOI] [PubMed] [Google Scholar]
- Dunning AM, Ellis Pd, McBride S, Kirschenlohr HL, Healey CS, Kemp PR, Luben RN, Chang-Claude J, Mannermaa A, Kataja V, Pharoah PD, Easton DF, Ponder BA, Metcalf JC. A transforming growth factor ß1 signal peptide variant increases secretion in vitro and is associated with increased incidence of invasive breast cancer. Cancer Res. 2003;63:2610–2615. [PubMed] [Google Scholar]
- Healey CS, Dunning AM, Teare MD, Chase D, Parker L, Burn J, Chang-Claude J, Mannermaa A, Kataja V, Huntsman DG, Pharoah PD, Luben RN, Easton DF, Ponder BA. A common variant in BRCA2 is associated with both breast cancer risk and prenatal viability. Nat Genet. 2000;26:362–364. doi: 10.1038/81691. [DOI] [PubMed] [Google Scholar]
- Spurdle AB, Hopper JL, Chen X, Dite GS, Cui J, McCredie MR, Giles GG, Ellis-Steinborner S, Venter DJ, Newman B, Southey MC, Chenevix-Trench G. The BRCA2 372 HH genotype is associated with risk of breast cancer in Australian women under age 60 years. Cancer Epidemiol Biomarkers Prev. 2002;11:413–416. [PubMed] [Google Scholar]
- Auranen A, Spurdle AB, Chen X, Lipscombe J, Purdie DM, Hopper JL, Green A, Healey CS, Redman K, Dunning AM, Pharoah PD, Easton DF, Ponder BA, Chenevix-Trench G, Novik KL. BRCA2 Arg372His polymorphism and epithelial ovarian cancer risk. Int J Cancer. 2003;103:427–430. doi: 10.1002/ijc.10814. [DOI] [PubMed] [Google Scholar]
- Airewele G, Adatto P, Cunningham J, Mastromarino C, Spencer C, Sharp M, Sigurdson A, Bondy M. Family history of cancer in patients with glioma: a validation study of accuracy. J Natl Cancer Inst. 1998;90:543–544. doi: 10.1093/jnci/90.7.543. [DOI] [PubMed] [Google Scholar]