

Abstract
Background
Persistent anxiety-like symptoms may have an inflammatory-related pathophysiology. Our previous work using repeated social defeat (RSD) in mice showed that recruitment of peripheral myeloid cells to the brain is required for the development of anxiety. Here, we aimed to determine if 1) RSD promotes prolonged anxiety through redistribution of myeloid cells and 2) prior exposure to RSD sensitizes the neuroimmune axis to secondary subthreshold stress.
Methods
Mice were subjected to RSD and several immune and behavioral parameters were determined 0.5, 8, or 24 days later. In follow-up studies, control and RSD mice were subjected to subthreshold stress at 24 days.
Results
Repeated social defeat-induced macrophage recruitment to the brain corresponded with development and maintenance of anxiety-like behavior 8 days after RSD, but neither remained at 24 days. Nonetheless, social avoidance and an elevated neuroinflammatory profile were maintained at 24 days. Subthreshold social defeat in RSD-sensitized mice increased peripheral macrophage trafficking to the brain that promoted re-establishment of anxiety. Moreover, subthreshold social defeat increased social avoidance in RSD-sensitized mice compared with naïve mice. Stress-induced monocyte trafficking was linked to redistribution of myeloid progenitor cells in the spleen. Splenectomy before subthreshold stress attenuated macrophage recruitment to the brain and prevented anxiety-like behavior in RSD-sensitized mice.
Conclusions
These data indicate that monocyte trafficking from the spleen to the brain contributes re-establishment of anxiety in stress-sensitized mice. These findings show that neuroinflammatory mechanisms promote mood disturbances following stress-sensitization and outline novel neuroimmune interactions that underlie recurring anxiety disorders such as posttraumatic stress disorder.
Keywords: Anxiety, microglia, monocytes, neuroinflammation, PTSD, stress
Pychological stress contributes to the development of depression and anxiety disorders (1,2). The etiology of mood disorders is diverse (3,4), but stress-induced immune dysregulation promotes neuroinflammation underlying anxiety- and depressive-like behaviors (5–8). Indeed, increased neuroinflammatory signaling following stress augments neuroplasticity and leads to neuronal adaptations underlying anxiety and depressive symptoms (9–12). Thus, stress-induced neuroinflammation can contribute to the development of chronic mood disorders, such as posttraumatic stress disorder. While many have postulated that anxiety disorders have an inflammatory-related pathophysiology (13,14), no studies have examined the role of neuroinflammation in prolonged and recurrent anxiety.
Clinically relevant models of stress, including repeated social defeat (RSD), cause immune dysregulation, neuroinflammation, and anxiety-like behavior (15–18). For example, RSD significantly increased myeloid (CD11b+) cells in the blood and spleen that maintain an inflammatory (i.e., primed) and glucocorticoid-insensitive phenotype (19–22). These findings are mirrored in clinical studies of stress where isolated mononuclear cells from stressed individuals have an enhanced inflammatory profile and have impaired glucocorticoid regulation. Redistribution of primed myeloid cells generates peripheral and central inflammation that has deleterious effects on behavior. For instance, RSD-induced anxiety-like behavior was associated with microglia activation, macrophage recruitment to the brain, and elevated proinflammatory cytokine production (16,18). Moreover, our recent findings indicate that peripheral myeloid cell trafficking to the brain was instrumental in development of RSD-induced anxiety. Indeed, repeated exposure to social defeat increased circulating monocytes and brain macrophages, which corresponded with development of anxiety. In addition, mice lacking the chemokine receptors, CCR2 and CX3CR1, had limited macrophage recruitment to the brain and did not develop anxiety-like behavior after RSD (15). Thus, we aimed to 1) determine how long neuroinflammatory responses and anxiety-like behavior persisted following RSD; and 2) determine if subthreshold stress re-established myeloid cell trafficking and anxiety in RSD-sensitized mice.
Here, we show that RSD activated the neuroimmune axis to promote anxiety and key peripheral and central components of the neuroimmune axis remain sensitized. For instance, increased microglia activation and increased myeloid progenitors in the spleen persisted after RSD. These components were sensitized because subthreshold stress (i.e., acute social defeat) caused robust inflammation in RSD-sensitized mice (24 days) with increased peripheral monocyte/macrophage trafficking in the brain and elevated proinflammatory cytokine expression by microglia/macrophages. In addition, subthreshold stress in RSD-sensitized mice reestablished anxiety and reinforced social avoidance. Splenectomy in RSD-sensitized mice before subthreshold stress disrupted monocyte release/trafficking to the brain and blocked the re-establishment of anxiety.
Methods and Materials
Animals
Male C57BL/6 (6–8 weeks) and CD-1 (12 months) mice were obtained from Charles River Laboratories (Wilmington, MA). C57BL/6-Tg(CAG-EGFP) 131Osb/LeySopJ mice were purchased from Jackson Laboratories (Bar Harbor, ME). All procedures were in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals and were approved by the Ohio State University Institutional Laboratory Animal Care and Use Committee.
Repeated social defeat was performed as described in Supplement 1 and in previous reports (15,16,23). In brief, an intruder male CD-1 mouse was introduced into home cages of male C57BL/6 mice (three per cage) for 2 hours on 6 consecutive nights. Acute social defeat consisted of a single 2-hour exposure to the intruder mouse. Control mice (CON) were left undisturbed until sacrificed.
Anxiety-like behavior in the open field was determined as previously described (15). Mice were placed individually into the Plexiglas test apparatus (40 × 40 × 25 cm) and activity was recorded for 5 minutes. Time spent in the center and latency to enter the center of the open field were determined using an automated system (AccuScan Instruments Columbus, OH).
Social avoidance was determined as previously described (24,25). In the empty trial, an experimental mouse was placed into the arena with an empty wire mesh cage and activity was recorded for 2.5 minutes. In the social trial, an unfamiliar CD-1 mouse was placed in the wire mesh cage and the experimental mouse was placed in the arena and activity was recorded for 2.5 minutes. Activity was video-recorded and analyzed using Noldus EthoVision Software (Leesburg, VA).
Green fluorescent protein (GFP)+ bone marrow (BM)-Chimera mice were generated as described in Supplement 1 and in previous reports (15). In brief, mice were injected twice with low-dose busulfan, and 48 hours later GFP+ bone marrow was transferred into recipients by tail vein injection. Recipient mice were undisturbed for 4 weeks to allow engraftment. This protocol resulted in approximately 45% GFP+ cells in the bone marrow and 50% GFP+ cells in the circulation 4 weeks after reconstitution (Figure S1 in Supplement 1).
Brain microglia enrichment and isolation of blood, bone marrow, and spleen cells were completed as described in Supplement 1. As previously described (16), microglia were enriched from brain homogenates by Percoll centrifugation in differential Percoll gradients.
Flow cytometric analysis of antigen expression was completed as described in Supplement 1 and in previous reports (15,16,23).
Interleukin-6 (IL-6) was determined from plasma using the BD OptEIA Mouse IL-6 enzyme-linked immunosorbent assay according to the manufacturer’s instructions (BD Biosciences San Jose, CA). Absorbance was read at 450 nm using a Spectramax Plus384 plate reader (Molecular Devices Sunnyvale, CA). The assay was sensitive to 10 ng/mL IL-6 and intra-assay coefficients of variation were less than 10%.
Immunohistochemistry was completed as described in Supplement 1 and in previous reports (15). In brief, fixed brain tissue was fluorescently labeled with rabbit anti-mouse ionized calcium binding adaptor molecule-1 (Iba-1) (Wako Chemicals Richmond, VA) and rat anti-mouse Ly6C (Abcam Cambridge, MA). Sections were incubated with appropriate conjugated secondary antibody. Immunofluorescence was visualized and images were captured using an epifluorescent Leica DM5000B microscope and Leica DFC300 FX camera (Buffalo Grove, IL).
Histological quantification of microglia and GFP+ macrophages was determined as described in Supplement 1 and as previously reported (15,26,27). Iba-1 proportional area was reported as the average percentage area in the positive threshold for all representative images. GFP+ cells in brain sections were categorized as perivascular or parenchymal based on morphology, Iba-1 co-localization, and spatial relationship to Ly6C+ blood vessels.
RNA isolation and real-time polymerase chain reaction were completed as described in Supplement 1. In brief, messenger RNA (mRNA) was transcribed to complementary DNA and was amplified by real-time polymerase chain reaction and normalized based on reference complementary DNA (glyceraldehyde 3-phosphate dehydrogenase). Data were analyzed with comparative threshold cycle method (28).
Splenectomy surgery was performed 8 days after RSD as described in Supplement 1. Two weeks after splenectomy, mice were exposed to acute social defeat.
Statistical Analysis
Data were subjected to the Shapiro-Wilk test using Statistical Analysis Systems (SAS) software (Cary, NC). Observations greater than three interquartile ranges from the first and third quartile were excluded from analyses. Significant main effects and interactions were determined using one- (stress, day) or two- (stress × treatment) way analysis of variance using the general linear model procedures of SAS. Differences between group means were evaluated with t test using the least significant difference procedure of SAS.
Results
RSD-Induced Anxiety-like Behavior Was Resolved by 24 Days but Social Avoidance Was Maintained
To examine how long RSD-induced behavioral and immune alterations persisted, mice were subjected to repeated social defeat and anxiety-like behavior in the open-field and social avoidance were determined 0.5, 8, or 24 days later. In control mice, baseline behavior was not significantly different at the 0.5-, 8-, or 24-day time points, so these data were combined and presented as a single CON group. In addition, this experimental design was cross-sectional, so an individual mouse was tested once for each behavioral paradigm.
In the first experiment, RSD increased latency to enter the center of the open-field at 0.5 and 8 days compared with control mice (p < .04 for each; Figure 1A) and increased time spent in the center at 0.5 days (p < .04; Figure 1B). Nonetheless, indications of RSD-induced anxiety in the open-field were resolved by 24 days. In the second experiment, social avoidance was determined using a two-trial social interaction paradigm with an empty trial followed by a social trial (24,25). Representative activity traces of social interaction are shown from CON and RSD mice (24 days) for empty and social trials (Figure 1C). There were no differences between groups in time spent in the interaction zone during the empty trial (Figure 1D). During the social trial, however, time spent in the interaction zone was significantly decreased 0.5 (p < .02), 8 (p = .08), and 24 (p < .02) days after RSD (Figure 1D). In addition, mice subjected to RSD spent more time in the corners during the social trial at 0.5 and 24 days after RSD (p < .02 for each; Figure 1D). Overall, reductions in social interaction persisted 24 days after RSD (p < .02 for each; Figure 1F). These data indicate that anxiety in the open-field was diminished by 24 days after RSD, but social avoidance was maintained.
Figure 1.
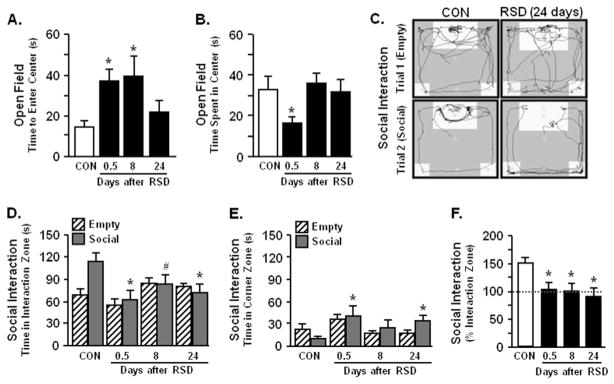
Repeated social defeat (RSD)-induced anxiety-like behavior was resolved by 24 days, but social avoidance was maintained. Male C57BL/6 mice were subjected to repeated social defeat or left undisturbed as control mice (CON). Anxiety-like behavior and social avoidance were determined 0.5, 8, or 24 days after the final cycle of RSD. Repeated social defeat significantly increased anxiety-like behavior with (A) increased time to enter the center of the open-field (F3,47 = 3.34, p < .03) and (B) decreased total time spent in the open-field (F3,47 = 2.20, p = .10). (C) Representative activity traces of CON or RSD (24 days) mice are shown for empty and social trials. Repeated social defeat caused social avoidance with (D) decreased time spent in the interaction zone (social: F3,33 = 3.92, p < .02) and (E) increased time spent in corner zones (empty: F3,34 = 2.87, p < .05, | social: F3,34 = 2.17, p = .10) (F) Repeated social defeat-induced social avoidance was maintained over time shown by decreased percent time spent in the interaction zone compared between trials [100 × (social/empty)] (F3,34 = 3.64, p < .02). Bars represent the mean ± SEM. Means with asterisk (*) are significantly different from CON (p < .05) and means with (#) tended to be different from CON (p < .06–.10).
RSD Transiently Increased Monocytes in Circulation and Macrophages in the Brain
Following behavioral testing, markers of immune alterations associated with RSD were determined, including spleen weight, plasma IL-6, and circulating CD11b+ cells (21,29). Increased spleen weight (p < .02; Figure 2A), plasma IL-6 (p < .02; Figure 2B), and circulating CD11b+ cells were observed 0.5 days after RSD (p < .002; Figure 2C), but these immune-related alterations returned to control levels by 24 days after RSD. The percentage of circulating monocytes (CD11b+/Ly6Chi) was increased 0.5 days after RSD (p < .02; Figure 2D,E) but returned to control levels by 24 days after RSD. The brain macrophage population was also increased 0.5 and 8 days after RSD (p < .01; Figure 2F,G) but returned to control levels by 24 days after RSD.
Figure 2.

Repeated social defeat (RSD) caused transient accumulation of monocytes in circulation and macrophages in the brain. Male C57BL/6 mice were subjected to six cycles of social defeat (RSD) or left undisturbed as control mice (CON). Following behavioral testing at each time point (0.5, 8, or 24 days after RSD), blood and enriched brain CD11b+ cells were collected. Repeated social defeat increased (A) spleen weight (F3,53 = 3.99, p < .01), (B) plasma interleukin-6 (IL-6) (F3,48 = 6.52, p < .0009), and (C) percentage of CD11b+ cells in the blood (F3,51 = 8.33, p < .001), but these markers were decreased by 24 days after RSD. (D) Representative flow bivariate dot plots of Ly6C/CD11b labeling in the blood show that RSD transiently elevated (E) percentage of blood monocytes (CD11b+/Ly6Chi) (F3,46 = 5.57, p < .003). (F) Representative flow bivariate dot plots of CD11b/CD45 labeling in Percoll isolated microglia/macrophages demonstrate that RSD increased (G) percentage of brain macrophages (CD11b+/CD45hi) (F3,35 = 4.61, p < .009). Bars represent the mean ± SEM. Means with asterisk (*) are significantly different from CON (p < .05) and means with (#) tended to be different from CON (p < .06–.10).
Acute Social Defeat Re-established Anxiety-like Behavior and Enhanced Social Avoidance in Stress-Sensitized Mice
The next objective was to determine if anxiety-like behavior was re-established by exposure to acute social defeat 24 days after RSD. These studies included three experimental groups of mice: 1) CON (nonstressed) mice that were not exposed to any social defeat; 2) naïve (no initial stress) mice subjected to acute social defeat 24 days later; and 3) stress-sensitized (SS) mice that were exposed to RSD and subsequently subjected to acute social defeat 24 days later (Figure 3A). Acute social defeat increased anxiety in stress-sensitized mice with increased time to enter the center and decreased total time spent in the center of the open-field but did not affect behavior in naïve mice (p < .05 for each; Figure 3B,C). Representative traces of naïve and stress-sensitized mice are shown for the social interaction paradigm (Figure 3D). As observed in Figure 1D, CON mice spent more time in the interaction zone when the mouse CON-specific was present (Figure 3E). Compared with CON, naïve mice tended to spend less time in the interaction zone during the social trial (p = .06; Figure 3E). In stress-sensitized mice, acute defeat reduced time in the interaction zone (p < .0003; Figure 3E) and increased time spent in the corner zone (p < .05; Figure 3F) during social conditions, which resulted in pronounced social avoidance (p < .008; Figure 3G). Taken together, acute social defeat re-established anxiety in the open-field and social avoidance in stress-sensitized mice.
Figure 3.

Acute social defeat re-established anxiety-like behavior and exaggerated social avoidance in SS mice. These studies included three experimental groups of mice: 1) CON (nonstressed) mice that were not exposed to any social defeat; 2) naïve (no initial stress) mice subjected to acute social defeat 24 days later; and 3) SS mice that were exposed to repeated social defeat (RSD) and subjected to acute social defeat 24 days later. Anxiety-like behavior and social avoidance were determined 0.5 days after acute social defeat. (A) Experimental schematic is shown. Acute social defeat caused anxiety-like behavior in SS mice with (B) increased time to enter the center of the open-field (F2,32 = 3.41, p < .05) and (C) decreased total time spent in the open-field (F2,32 = 2.77, p = .07). (D) Representative activity traces of naïve and stress-sensitized mice are shown for empty and social trials. Acute social defeat in naïve mice promoted social avoidance but exacerbated social withdrawal in SS mice with (E) decreased time spent in the interaction zone (F2,24 = 11.34, p < .003) and (F) increased time spent in corner zones of social avoidance test following acute social defeat (F2,24 = 4.56, p < .05). (G) Percent time spent in the interaction zone [100 × (social/empty)] was markedly reduced in stress-sensitized mice compared with control mice (F2,24 = 6.51, p < .02). Bars represent the mean ± SEM. Means with asterisk (*) are significantly different from CON (p < .05) and means with (#) tended to be different from CON (p < .06–.10).
Acute Social Defeat Enhanced the Neuroinflammatory Profile and Reinitiated Macrophage Recruitment to the Brain of Stress-Sensitized Mice
Our previous results indicate that trafficking of peripheral myeloid cells to the brain contributes to development of anxiety-like behavior after RSD (15). Therefore, myeloid cell populations were determined in the blood and brain after acute social defeat in naïve and stress-sensitized mice. Acute social defeat increased spleen weight (p < .006; Figure 4A) along with circulating CD11b+ cells (p < .006; Figure 4B) and monocytes (p < .003; Figure 4C) in stress-sensitized mice. Consistent with increased monocytes in circulation, acute defeat enhanced the proportion of macrophages in the brain of stress-sensitized mice (p < .002; Figure 4D,E).
Figure 4.

Acute social defeat enhanced the neuroinflammatory profile and reinitiated macrophage recruitment to the brain of stress-sensitized mice. These studies included three experimental groups of mice: 1) control (CON) nonstressed mice that were not exposed to any social defeat; 2) naïve (no initial stress) mice subjected to acute social defeat 24 days later; and 3) stress-sensitized (SS) mice that were exposed to repeated social defeat (RSD) and subjected to acute social defeat 24 days later. Following behavioral testing (Figure 3), spleen, blood, and brain CD11b+ cells were collected. Acute social defeat markedly increased (A) spleen weight (F2,36 = 12.51, p < .0001), (B) blood myeloid (CD11b+) cells (F2,35 = 6.01, p < .006), and (C) percentage of blood monocytes (CD11b+/Ly6Chi) in stress-sensitized mice (F2,29 = 7.23, p < .003). (D) Representative flow bivariate dot plots of CD11b/CD45 labeling in Percoll isolated microglia/macrophages show that acute social defeat also increased (E) the percentage of brain macrophages (CD11b+/CD45hi) in SS mice (F2,26 = 10.72, p < .0005). Bars represent the mean ± SEM. (F) Corresponding with increased macrophage recruitment in the brain, RSD increased relative gene expression of interleukin-1 beta (IL-1β), tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), and CD14, (p < .03 for each) and decreased CX3CR1 (p < .05) in enriched brain CD11b+ cells. In addition, acute social defeat enhanced expression of all genes analyzed in enriched brain CD11b+ cells from stress-sensitized mice (p < .05 for each). Values represent average fold change ± SEM. Means with asterisk (*) are significantly different from CON (p < .05) and means with (‡) are significantly different from naïve (p < .05). mRNA, messenger RNA.
Next, mRNA levels of interleukin-1 beta (IL-1β), tumor necrosis factor-alpha (TNF-α), IL-6, CD14, and CX3CR1 were determined 0.5, 8, and 24 days after RSD in enriched brain CD11b+ cells (microglia/macrophages). As expected, mRNA levels of IL-1β, TNF-α, and CD14 were increased 0.5 days after RSD (p < .03 for each; Figure 4F). CX3CR1 expression was significantly decreased in mice subjected to RSD (p < .007; Figure 4F). Interleukin-1 beta and TNF-α mRNA levels remained elevated 8 days after RSD (p < .0008 for each) but returned to baseline expression by 24 days (Figure 4F). Moreover, IL-6, CD14, and CX3CR1 mRNA levels were elevated at 8 days after RSD and remained elevated at 24 days (p < .06 for each). Acute social defeat 24 days later had no effect on these inflammatory markers in naïve mice but profoundly increased IL-1β, TNF-α, and CD14 mRNA levels in stress-sensitized mice comparable with levels detected 0.5 days after RSD (p < .008 for each; Figure 4F). Taken together, acute social defeat in stress-sensitized mice increased circulating monocytes, trafficking brain macrophages, and neuroinflammation.
Microglia in the Prefrontal Cortex, Amygdala, and Hippocampus Exhibited Deramified Morphology After RSD
Inflammatory cytokine production in the brain is associated with an increased presence of deramified/activated microglia after RSD (16). Figure 5A–E shows representative Iba-1 labeling of microglia and increased Iba-1 proportional area was detected in the prefrontal cortex (PFC) at 0.5, 8, and 24 days after RSD (p < .02 for each; Figure 5B,F). Increased Iba-1 immunoreactivity was also detected in the amygdala (AMYG) (Figure 5F), hippocampus (HPC)-cornu ammonis 3 (CA3) (Figure 5H), and HPC-dentate gyrus (DG) (Figure 5I) at 0.5 and 8 days after RSD (p < .0008 for each) but no longer detected by 24 days. Acute stress also altered microglia morphology in the stress-sensitized mice compared with naïve mice in all regions examined (p < .05; Figure 5F–I). There was no difference in Iba-1 reactivity in the PFC and AMYG compared with RSD (24 days) (Figure 5F,G). These findings indicate that microglia return to a surveying state in a time-dependent manner after RSD but can be provoked toward a deramified state in stress-sensitized mice.
Figure 5.

Microglia in the PFC, amygdala (AMYG), and hippocampus (HPC) exhibited deramified morphology after repeated social defeat (RSD). Brains were collected from control (CON), RSD (0.5, 8, or 24 days later), naïve, and SS mice. Representative images of ionized calcium binding adaptor molecule 1 (Iba-1) labeling in the PFC of (A) CON, (B) RSD (.5 days), (C) RSD (24 days), (D) naïve, and (E) SS mice are shown. (F) Representative section of the area in the PFC used for microglia morphology analyses (64). Repeated social defeat caused microglia activation with increased proportional area of Iba-1 immunofluorescence in the (G) PFC (F5,25 = 22.67, p < .0001), (H) AMYG (F5,25 = 18.26, p < .0001), (I) HPC-cornu ammonis 3 (CA3) (F5,25 = 13.88, p < .0001), and (J) HPC-dentate gyrus (DG) (F5,25 = 18.84, p < .0001) that was reduced over time (except PFC; p < .01). Acute social defeat modestly increased microglia activation in the PFC (p < .01), AMYG (p < .03), HPC-CA3 (p < .02), and HPC-DG (p < .02) of SS mice. Bars represent the mean ± SEM. Means with asterisk (*) are significantly different from CON (p < .05) and means with (‡) are significantly different from naïve (p < .05).
Acute Social Defeat Promoted Infiltration of BM-Derived Macrophages into the Brain of Stress-Sensitized Mice
Our recent report using GFP+ BM-chimera indicated that RSD promoted the recruitment of peripheral macrophages in specific brain regions including the PFC, AMYG, and HPC (15). Using the same BM-chimera experimental design, RSD caused significant infiltration of GFP+ macrophages into the AMYG of BM-chimera mice compared with CON (Figure 6A,B). Ramified GFP+ macrophages were present in the brain parenchyma of GFP+ BM-chimera mice at 8 days after RSD but were diminished by 24 days (Figure 6C). Acute social defeat did not cause significant macrophage trafficking in naïve mice (Figure 6D) but profoundly increased macrophage infiltration into the brain of stress-sensitized mice (Figure 6E).
Figure 6.

Acute social defeat promoted infiltration of bone marrow (BM)-derived macrophages into the brain of stress-sensitized (SS) mice. Brains were collected from control (CON), repeated social defeat (RSD) (.5, 8, or 24 days later), naïve, and stress-sensitized green fluorescent protein (GFP)+ BM-chimera mice. Representative images of peripheral GFP+ (green) cells in the amygdala (AMYG) of (A) CON, (B) RSD (.5 days), (C) RSD (24 days), (D) naïve, and (E) SS mice are shown. The inset image illustrates the area of the AMYG in which the representative images were taken (64). Sections were co-labeled with antibodies to identify resident microglia (ionized calcium binding adaptor molecule 1 [Iba-1]+, red), the vasculature (Ly6C+, blue), and parenchymal BM-macrophages (GFP+/Iba-1+, yellow). The subsequent panels in each row show merged GFP/Iba-1 or GFP/Iba-1/Ly6C images. White scale bar represents 100 μm (20×).
Repeated social defeat increased the number of perivascular and parenchymal BM-derived (GFP+) macrophages in the PFC (p < .0001; Figure 7A), AMYG (p < .01; Figure 7B), HPC-CA3 (p < .001; Figure 7C); and HPC-DG (p < .01; Figure 7D) at 0.5 and 8 days. In these regions, the number of BM-derived macrophages in the brain returned to control levels 24 days after RSD. Acute social defeat promoted recruitment of BM-derived macrophages in the PFC, AMYG, and HPC (CA3 and DG) of stress-sensitized mice but did not affect brain macrophage recruitment in naïve mice.
Figure 7.

Acute social defeat in stress-sensitized mice promoted infiltration of bone marrow (BM)-derived macrophages that coincided with microglia activation. Green fluorescent protein (GFP)+ BM-chimera mice were subjected to six cycles of social defeat (repeated social defeat [RSD]) or left undisturbed as control mice (CON). Brains were collected from CON, RSD (0.5, 8, or 24 days later), naïve, and stress-sensitized (SS) mice. The neuroanatomical distribution of GFP+ macrophages was determined and cells were classified as perivascular or parenchymal. Repeated social defeat significantly increased GFP+ perivascular and parenchymal macrophages in (A) prefrontal cortex (PFC) (F5,39 = 8.35, p < .001), (B) amygdala (AMYG) (F5,39 = 7.38, p < .0002), (C) hippocampus (HPC)-cornu ammonis 3 (CA3) (F5,39 = 6.99, p < .0001), and (D) HPC-dentate gyrus (DG) (F5,39 = 4.60, p < .003). At 24 days after RSD, GFP+ macrophages were reduced, but acute social defeat caused robust macrophage infiltration in stress-sensitized mice (p < .05 for each). Repeated social defeat also caused microglia activation with increased ionized calcium binding adaptor molecule 1 (Iba-1) proportional area (GFP+ and GFP−) in the (E) PFC (F5,31 = 9.52, p < .0001), (F) AMYG (F5,26 = 21.10, p < .0001), (G) HPC-CA3 (F5,33 = 16.38, p < .0001), and (H) HPC-DG (F5,33 = 11.35, p < .0001) of BM-chimera mice. Coinciding with macrophage infiltration, RSD-induced microglia activation was diminished 24 days later (except PFC, p < .05). Acute social defeat in stress-sensitized mice also enhanced Iba-1 proportional area in the AMYG, HPC-CA3, and HPC-DG (p < .05 for each). Bars represent the mean + SEM. Means with asterisk (*) are significantly different from CON (p < .05) and means with (‡) are significantly different from naïve (p < .05). Bars with (+) indicate means are significantly different from CON (p < .05).
To determine if macrophage infiltration was associated with microglia activation, Iba-1 proportional area was examined in the PFC, AMYG, and HPC (CA3 and DG). Coinciding with macrophage trafficking, microglia activation was increased at 0.5 and 8 days after RSD (p < .01 for each) but decreased by 24 days (Figure 5E–H). Acute social defeat also increased the presence of deramified microglia in stress-sensitized BM-chimera mice (p < .01 for each) but had no effect in naïve mice. Despite a significant increase in GFP+/Iba-1+ proportional area (macrophages) after RSD, resident microglia (GFP−/Iba-1+) comprised the majority of Iba-1 proportional area in all brain regions examined. These data indicate that acute social defeat caused significant macrophage infiltration and microglia activation in the PFC, AMYG, and HPC of RSD-sensitized mice.
RSD Enhanced Myelopoiesis and Increased Myeloid Progenitors in the Spleen
Our previous studies indicate that increased myeloid cells in the blood and spleen after RSD was mediated by sympathetic nervous system activation (17) and related to enhanced myelopoiesis in the bone marrow (21,30). To determine the source of increased circulating monocytes and brain macrophages following acute social defeat, bone marrow and spleen were collected. Repeated social defeat increased the proportion of granulocyte (p < .02; Figure 8A) and monocyte (p < .003; Figure 8B) progenitors in the BM at 0.5 days. Moreover, acute social defeat increased granulocyte bone marrow progenitors (p < .02; Figure 8A) but did not alter monocyte bone marrow progenitors in stress-sensitized mice (Figure 8B). Spleen CD11b+ cells and monocytes were increased 0.5 and 8 days after RSD (p < .05 for each) but returned to control levels by 24 days (Figure 8C,D). In addition, the proportion of splenic myeloid cells (CD11b+/Ly6C+) expressing the progenitor cell marker CD34 was significantly increased after RSD. This CD34+ myeloid progenitor population was maintained in the spleen 24 days after RSD (p < .0006 for each; Figure 8E,F). In addition, there were increased CD34+ progenitors in the spleen of stress-sensitized mice after acute defeat compared with naïve mice (p < .03; Figure 8F). Taken together, RSD-induced sensitization was associated with an increased population of myeloid progenitors in the spleen.
Figure 8.

Repeated social defeat (RSD) enhanced myelopoiesis and increased myeloid progenitors in the spleen. Male C57BL/6 mice were subjected to six cycles of social defeat (RSD) or left undisturbed as control mice (CON). Bone marrow (BM) was collected from CON, RSD (0.5, 8, or 24 days later), naïve, and stress-sensitized (SS) mice. Repeated social defeat increased (A) granulocyte BM progenitors (F5,36 = 3.06, p < 0.02) and (B) monocyte BM progenitors (F5,36 = 5.01, p < .002). Repeated social defeat-induced myelopoiesis was transient and acute social defeat caused a relative increase in granulocyte progenitors (p = .08). In follow-up studies, spleens were collected from CON and RSD (0.5, 8, or 24 days later) mice. Repeated social defeat increased (C) spleen CD11b+ cells (F5,32 = 3.44, p < .02 and (D) monocytes that were diminished 24 days after stress cessation (F5,32 = 7.87, p < .0001). (E) Representative flow bivariate dot plots of Ly6C/CD34 labeling in splenocytes. (F) Repeated social defeat also increased spleen myeloid progenitor cells (CD11b+/Ly6C+/CD34+) that were maintained over time (F5,32 = 6.49, p < .0004). Bars represent the mean + SEM. Means with asterisk (*) are significantly different from CON (p < .05) and means with (‡) are significantly different from naïve (p < .05).
Splenectomy in Stress-Sensitized Mice Disrupted Monocyte Trafficking and Prevented Anxiety-Like Behavior Induced by Acute Social Defeat
To determine if increased circulating monocytes in stress-sensitized mice were supplied by splenic reservoirs of myeloid progenitor cells, sham surgery or splenectomy was performed on naïve and SS mice before acute social defeat (Figure 9A). In SS-sham mice, acute defeat increased spleen weight and CD11b+ cells in circulation (data not shown). Similarly, acute social defeat increased circulating monocytes in SS-sham mice (p < .0002; Figure 9B) but had no effect on monocytes in SS-splenectomy mice or naïve control mice. Enhanced monocyte trafficking in SS-sham mice coincided with significant accumulation of brain macrophages (CD11b+/CD45hi) after acute social defeat (p < .006; Figure 9C). Diminished macrophage recruitment in the brain of SS-splenectomy mice was associated with reduced expression of neuroinflammatory genes, including IL-1β, TNF-α, and IL-6 compared with SS-sham mice (p < .04 for each; Figure 9D). Similar to Figure 3B, acute social defeat caused anxiety-like behavior in the open field for SS-sham mice (p < .03 for each; Figure 9E,F) but had no effect on SS-splenectomy mice. Naïve-sham and naïve-splenectomy mice did not have any anxiety in the open field. Collectively, these data indicate that subthreshold stress caused splenic-derived monocytes to traffic to the brain and re-establish anxiety in RSD-sensitized mice.
Figure 9.

Splenectomy in SS mice disrupted monocyte trafficking and prevented anxiety-like behavior induced by acute social defeat. Male C57BL/6 mice were subjected to six cycles of social defeat (repeated social defeat [RSD]) or left as nonstressed control mice. After 8 days, mice received sham surgery or splenectomy and then 16 days later (day 24) mice were subjected to an acute social defeat. Mice with prior exposure to RSD were termed SS, while control mice were naïve. (A) Experimental schematic is shown. Blood and brain were collected 0.5 days after acute social defeat. (B) Percent blood monocytes (F1,42 = 13.13, p < .0008) were increased after acute social defeat in SS mice and splenectomy significantly reduced these cells in SS mice (stress × splenectomy interaction, F1,42 = 9.65, p < .004). (C) Acute social defeat SS mice (F1,42 = 5.71, p < .02) and splenectomy attenuated this response (stress × splenectomy interaction, F1,42 = 3.20, p = .08). (D) Gene expression analyses in enriched brain CD11b+ cells showed that acute social defeat increased interleukin-1 beta (IL-1β), tumor necrosis factor-alpha (TNF-α), and interleukin-6 (IL-6) messenger RNA (mRNA) levels in SS mice, but splenectomy attenuated these effects (stress × splenectomy interaction, p < .08 for each). Anxiety-like behavior was determined 0.5 days after acute social defeat. Acute social defeat increased anxiety-like behavior in SS mice with (E) increased time to enter center of open-field (F1,40 = 3.19, p = .08) and (F) decreased time spent in center of open-field (F1,40 = 4.32 p < .05). Splenectomy in SS mice reduced anxiety-like behavior (open field time to enter, F1,40 = 7.60, p < .009 | open field center time, F1,40 = 4.11, p < .05). Values represent average fold change ± SEM. Means with asterisk (*) are significantly different from naïve-sham (p < .05).
Discussion
Our previous work indicates that RSD-induced anxiety-like behavior involves resident microglia activation and recruitment of bone marrow-derived macrophages to the brain (15,16). Here, we show that time-dependent reductions in brain macrophages were associated with resolution of anxiety-like behavior after RSD. In addition, increased proinflammatory cytokine gene expression in microglia and redistribution of myeloid progenitor cells in the spleen were maintained 24 days after RSD. Moreover, subthreshold stress (i.e., acute social defeat) reactivated the neuroimmune axis leading to increased monocyte/macrophage trafficking in the brain and elevated proinflammatory cytokine expression in microglia/macrophages. These findings indicate that RSD sensitized key peripheral and central components of the neuroimmune axis. Coinciding with enhanced neuroinflammation, subthreshold stress re-established anxiety-like behavior and reinforced social avoidance in RSD-sensitized mice. Finally, splenic-derived monocytes were critical in the re-establishment of behavioral deficits after subthreshold stress. In support of this notion, splenectomy of RSD-sensitized mice prevented macrophage trafficking to the brain and blocked the re-establishment of anxiety-like behavior after subthreshold stress.
An important finding in this study was the temporal relationship between the recruitment of BM-derived macrophages to the brain and anxiety. Repeated social defeat caused anxiety in the open-field that was associated with robust microglia activation, neuroinflammation, and macrophage trafficking to the brain. Similar to our recent report, macrophage infiltration and enhanced microglia activation were detected in stress-responsive brain regions, including the prefrontal cortex, amygdala, and hippocampus (15). Increased anxiety and brain macrophages persisted for 8 days after RSD (in wild-type and bone marrow-chimera mice) (18) but were no longer detectable by 24 days. Thus, resolution of anxiety-like behavior was paralleled by reductions in brain macrophages. Consistent with our previous study, bone marrow-chimera mice generated with low-dose busulfan provided an effective model to study myeloid cell trafficking in the brain (15). Similar to other studies, chemical ablation with low-dose busulfan caused limited nonspecific myeloid cell trafficking in the brain of control mice (31,32) and reduced potential confounds associated with irradiation (31–33). It is also important to point out that GFP+ bone marrow-derived macrophages were diminished in mice 24 days after RSD. These data are supported by other reports in models of stress and neurological disease where infiltrating GFP+ macrophages were reduced in the brain or spinal cord after 1 month (34,35). It is unclear if these peripheral macrophages leave or undergo apoptosis, but consistent with other reports, bone marrow-derived monocytes did not contribute significantly to the resident microglia population (36,37). Overall, our findings indicate that there is a critical role of peripherally derived brain macrophages in development and persistence of RSD-induced anxiety like behavior.
Although brain macrophages and anxiety-like behavior were diminished over time, resident microglia maintained elevations in inflammatory gene expression and altered morphology 24 days after RSD. The sensitized phenotype of microglia following RSD are similar to models of aging in which primed microglia are characterized by enhanced pro-inflammatory gene expression along with increased reactivity to immune stimulation (38–41). Consistent with this idea of stress-induced neuroimmune sensitization, secondary immune challenge with lipopolysaccharide caused exaggerated microglia/macrophage activation in mice subjected to RSD leading to prolonged sickness behavior (23). Persistent inflammatory changes in microglia after stress likely caused neuronal adaptations that lead to mood disorders (42–44). Notably, we show here that social avoidance persisted despite reductions in brain macrophages, indicating that subtle microglia activation may underlie extended impairments in social interaction.
Another key finding was that re-establishment of anxiety by subthreshold stress was associated with increased macrophage trafficking and elevated neuroinflammation. Moreover, splenectomy in stress-sensitized mice blocked the acute stress-induced myeloid cell redistribution, neuroinflammation, and anxiety-like behavior. These findings indicate that splenic-derived monocytes promote recurrent anxiety responses through elevated neuro-immune activation after subthreshold stress. These data support previous findings that peripherally derived macrophages that traffic to the brain are critical determinants of RSD-induced behavioral responses (15). Related to these points, our previous work has established that RSD caused proliferation of myeloid progenitor cells in the bone marrow that, in turn, are released into circulation. This RSD-induced myelopoiesis and release of monocytes into circulation was dependent on repeated social defeat exposures, granulocyte-monocyte colony stimulating factor signaling, and sympathetic nervous system activation (17,20,21,30). Here, in RSD-sensitized mice (24 days after RSD), increased circulating monocytes and brain macrophages were detected after subthreshold stress. Elevated circulating myeloid cells in stress-sensitized mice after acute stress, however, were not linked to proliferation of monocyte bone marrow progenitor cells. Moreover, removal of the spleen in RSD-sensitized mice blocked elevations in circulating monocytes and brain macrophages following acute stress. These data are interpreted to indicate that enhanced levels of circulating monocytes and brain macrophages were derived from residual myeloid progenitors in the spleen 24 days after RSD. Other reports also implicate splenic-derived monocytes in the pathophysiology of inflammatory-related conditions including atherosclerosis (45–47). Moreover, release of splenic immune cells after acute stress may be mediated by noradrenergic signaling from direct sympathetic nervous system innervation of the spleen (17,48). It is unclear if splenic-derived monocytes maintain a primed, glucocorticoid-insensitive profile, but their presence in the brain led to neuroinflammation. These findings have translational relevance because psychosocial stress in humans also altered the distribution and promoted proinflammatory cytokine signaling inflammatory status in peripheral mononuclear cells (49–52).
The sensitization of resident microglia with RSD may also play a role in the recruitment of splenic myeloid cells to the brain by acute stress. Similar to other reports (53), it is plausible that localized microglia activation and enhanced chemokine expression mediated recruitment of primed macrophages to the brain leading to neuroimmune activation. Because re-establishment of anxiety-like behavior was dependent on brain macrophage trafficking, we surmise that primed monocytes/macrophages recruited to the brain from the spleen alter neurocircuitry in the limbic system to promote anxiety-like behavior. It is important to consider that neuroinflammatory signaling may not underlie behavioral consequences reported in other models of stress sensitization. Indeed, recent studies indicate that central noradrenergic pathways are hyperactive following reminders of stress (54). However, it is relevant to note that proinflammatory cytokines (IL-1β, TNF-α, and IL-6) influence neurotransmission and may contribute to receptor sensitivity (55–57). Furthermore, several studies demonstrate that microglia activation and brain macrophages directly influence behavior and neurophysiology via cytokines and secondary mediators (9,58,59). Thus, it is pertinent to determine the neurobiological consequences (i.e., neuroplasticity) associated with RSD-induced neuroinflammation and subsequent re-exposure to social defeat.
In summary, the current study provides significant evidence that redistribution of myeloid cells leads to long-term sensitization of neuroimmune and behavioral responses to subthreshold stressors. Thus, increased susceptibility to immune and behavioral complications observed in stressed individuals may be related to priming and redistribution of peripheral myeloid cells (60–63). These findings have implications for persistent anxiety disorders, such as posttraumatic stress disorder, and provide an innovative target to prevent recurrent anxiety symptoms.
Supplementary Material
Acknowledgments
This research was supported by National Institute of Mental Health grants (R01-MH-093473 and R01-097243) to JFS and a National Institute on Aging grant (R01-AG033028) to JPG. ESW was supported by a National Institute of Mental Health predoctoral fellowship (F31-MH09547301).
We thank Yan Huang, Jenna Patterson, and Vikram Sharma for their technical assistance.
Footnotes
The authors declare no biomedical financial interests or potential conflicts of interest.
Supplementary material cited in this article is available online at http://dx.doi.org/10.1016/j.biopsych.2013.11.029.
References
- 1.Kendler KS, Karkowski LM, Prescott CA. Causal relationship between stressful life events and the onset of major depression. Am J Psychiatry. 1999;156:837–841. doi: 10.1176/ajp.156.6.837. [DOI] [PubMed] [Google Scholar]
- 2.Gilman SE, Trinh NH, Smoller JW, Fava M, Murphy JM, Breslau J. Psychosocial stressors and the prognosis of major depression: A test of Axis IV. Psychol Med. 2013;43:303–316. doi: 10.1017/S0033291712001080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Charney DS. Psychobiological mechanisms of resilience and vulnerability: Implications for successful adaptation to extreme stress. Am J Psychiatry. 2004;161:195–216. doi: 10.1176/appi.ajp.161.2.195. [DOI] [PubMed] [Google Scholar]
- 4.Mathew SJ, Price RB, Charney DS. Recent advances in the neurobiology of anxiety disorders: Implications for novel therapeutics. Am J Med Genet C Semin Med Genet. 2008;148C:89–98. doi: 10.1002/ajmg.c.30172. [DOI] [PubMed] [Google Scholar]
- 5.Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: When the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haroon E, Raison CL, Miller AH. Psychoneuroimmunology meets neuropsychopharmacology: Translational implications of the impact of inflammation on behavior. Neuropsychopharmacology. 2012;37:137–162. doi: 10.1038/npp.2011.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beumer W, Gibney SM, Drexhage RC, Pont-Lezica L, Doorduin J, Klein HC, et al. The immune theory of psychiatric diseases: A key role for activated microglia and circulating monocytes. J Leukoc Biol. 2012;92:959–975. doi: 10.1189/jlb.0212100. [DOI] [PubMed] [Google Scholar]
- 8.Miller AH, Maletic V, Raison CL. Inflammation and its discontents: The role of cytokines in the pathophysiology of major depression. Biol Psychiatry. 2009;65:732–741. doi: 10.1016/j.biopsych.2008.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tanaka K, Furuyashiki T, Kitaoka S, Senzai Y, Imoto Y, Segi-Nishida E, et al. Prostaglandin E2-mediated attenuation of mesocortical dopaminergic pathway is critical for susceptibility to repeated social defeat stress in mice. J Neurosci. 2012;32:4319–4329. doi: 10.1523/JNEUROSCI.5952-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Christoffel DJ, Golden SA, Dumitriu D, Robison AJ, Janssen WG, Ahn HF, et al. IkappaB kinase regulates social defeat stress-induced synaptic and behavioral plasticity. J Neurosci. 2011;31:314–321. doi: 10.1523/JNEUROSCI.4763-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koo JW, Russo SJ, Ferguson D, Nestler EJ, Duman RS. Nuclear factor-kappaB is a critical mediator of stress-impaired neurogenesis and depressive behavior. Proc Natl Acad Sci U S A. 2010;107:2669–2674. doi: 10.1073/pnas.0910658107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koo JW, Duman RS. IL-1beta is an essential mediator of the antineurogenic and anhedonic effects of stress. Proc Natl Acad Sci U S A. 2008;105:751–756. doi: 10.1073/pnas.0708092105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Andrews JA, Neises KD. Cells, biomarkers, and post-traumatic stress disorder: Evidence for peripheral involvement in a central disease. J Neurochem. 2012;120:26–36. doi: 10.1111/j.1471-4159.2011.07545.x. [DOI] [PubMed] [Google Scholar]
- 14.Pace TW, Heim CM. A short review on the psychoneuroimmunology of posttraumatic stress disorder: From risk factors to medical comorbidities. Brain Behav Immun. 2012;25:6–13. doi: 10.1016/j.bbi.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 15.Wohleb ES, Powell ND, Godbout JP, Sheridan JF. Stress-induced recruitment of bone marrow-derived monocytes to the brain promotes anxiety-like behavior. J Neurosci. 2013;33:13820–13833. doi: 10.1523/JNEUROSCI.1671-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wohleb ES, Hanke ML, Corona AW, Powell ND, Stiner LM, Bailey MT, et al. β-Adrenergic receptor antagonism prevents anxiety-like behavior and microglial reactivity induced by repeated social defeat. J Neurosci. 2011;31:6277–6288. doi: 10.1523/JNEUROSCI.0450-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanke ML, Powell ND, Stiner LM, Bailey MT, Sheridan JF. β-adrenergic blockade decreases the immunomodulatory effects of social disruption stress. Brain Behav Immun. 2012;26:1150–1159. doi: 10.1016/j.bbi.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kinsey SG, Bailey MT, Sheridan JF, Padgett DA, Avitsur R. Repeated social defeat causes increased anxiety-like behavior and alters splenocyte function in C57BL/6 and CD-1 mice. Brain Behav Immun. 2007;21:458–466. doi: 10.1016/j.bbi.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bailey MT, Kinsey SG, Padgett DA, Sheridan JF, Leblebicioglu B. Social stress enhances IL-1beta and TNF-alpha production by Porphyromonas gingivalis lipopolysaccharide-stimulated CD11b+ cells. Physiol Behav. 2009;98:351–358. doi: 10.1016/j.physbeh.2009.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Engler H, Engler A, Bailey MT, Sheridan JF. Tissue-specific alterations in the glucocorticoid sensitivity of immune cells following repeated social defeat in mice. J Neuroimmunol. 2005;163:110–119. doi: 10.1016/j.jneuroim.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 21.Engler H, Bailey MT, Engler A, Sheridan JF. Effects of repeated social stress on leukocyte distribution in bone marrow, peripheral blood and spleen. J Neuroimmunol. 2004;148:106–115. doi: 10.1016/j.jneuroim.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 22.Avitsur R, Kavelaars A, Heijnen C, Sheridan JF. Social stress and the regulation of tumor necrosis factor-alpha secretion. Brain Behav Immun. 2005;19:311–317. doi: 10.1016/j.bbi.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 23.Wohleb ES, Fenn AM, Pacenta AM, Powell ND, Sheridan JF, Godbout JP. Peripheral innate immune challenge exaggerated microglia activation, increased the number of inflammatory CNS macrophages, and prolonged social withdrawal in socially defeated mice. Psychoneuroendocrinology. 2012;37:1491–1505. doi: 10.1016/j.psyneuen.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krishnan V, Han MH, Graham DH, Berton O, Renthal W, Russo SJ, et al. Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell. 2007;131:391–404. doi: 10.1016/j.cell.2007.09.018. [DOI] [PubMed] [Google Scholar]
- 25.Berton O, McClung CA, Dileone RJ, Krishnan V, Renthal W, Russo SJ, et al. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 2006;311:864–868. doi: 10.1126/science.1120972. [DOI] [PubMed] [Google Scholar]
- 26.Vallieres L, Sawchenko PE. Bone marrow-derived cells that populate the adult mouse brain preserve their hematopoietic identity. J Neurosci. 2003;23:5197–5207. doi: 10.1523/JNEUROSCI.23-12-05197.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Donnelly DJ, Gensel JC, Ankeny DP, van Rooijen N, Popovich PG. An efficient and reproducible method for quantifying macrophages in different experimental models of central nervous system pathology. J Neurosci Methods. 2009;181:36–44. doi: 10.1016/j.jneumeth.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 29.Stark JL, Avitsur R, Padgett DA, Campbell KA, Beck FM, Sheridan JF. Social stress induces glucocorticoid resistance in macrophages. Am J Physiol Regul Integr Comp Physiol. 2001;280:R1799–R1805. doi: 10.1152/ajpregu.2001.280.6.R1799. [DOI] [PubMed] [Google Scholar]
- 30.Powell ND, Sloan EK, Bailey MT, Arevalo JM, Miller GE, Chen E, et al. Social stress up-regulates inflammatory gene expression in the leukocyte transcriptome via beta-adrenergic induction of myelopoiesis. Proc Natl Acad Sci U S A. 2013;110:16574–16579. doi: 10.1073/pnas.1310655110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kierdorf K, Katzmarski N, Haas CA, Prinz M. Bone marrow cell recruitment to the brain in the absence of irradiation or parabiosis bias. PLoS One. 2013;8:e58544. doi: 10.1371/journal.pone.0058544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lampron A, Lessard M, Rivest S. Effects of myeloablation, peripheral chimerism, and whole-body irradiation on the entry of bone marrow-derived cells into the brain. Cell Transplant. 2012;21:1149–1159. doi: 10.3727/096368911X593154. [DOI] [PubMed] [Google Scholar]
- 33.Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch UK, Mack M, et al. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat Neurosci. 2007;10:1544–1553. doi: 10.1038/nn2015. [DOI] [PubMed] [Google Scholar]
- 34.Brevet M, Kojima H, Asakawa A, Atsuchi K, Ushikai M, Ataka K, et al. Chronic foot-shock stress potentiates the influx of bone marrow-derived microglia into hippocampus. J Neurosci Res. 2010;88:1890–1897. doi: 10.1002/jnr.22362. [DOI] [PubMed] [Google Scholar]
- 35.Ajami B, Bennett JL, Krieger C, McNagny KM, Rossi FM. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci. 2011;14:1142–1149. doi: 10.1038/nn.2887. [DOI] [PubMed] [Google Scholar]
- 36.Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012;336:86–90. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- 37.Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Corona AW, Huang Y, O’Connor JC, Dantzer R, Kelley KW, Popovich PG, Godbout JP. Fractalkine receptor (CX3CR1) deficiency sensitizes mice to the behavioral changes induced by lipopolysaccharide. J Neuroinflammation. 2010;7:93. doi: 10.1186/1742-2094-7-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Henry CJ, Huang Y, Wynne AM, Godbout JP. Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1beta and anti-inflammatory IL-10 cytokines. Brain Behav Immun. 2009;23:309–317. doi: 10.1016/j.bbi.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Godbout JP, Moreau M, Lestage J, Chen J, Sparkman NL, O’Connor J, et al. Aging exacerbates depressive-like behavior in mice in response to activation of the peripheral innate immune system. Neuropsychopharmacology. 2008;33:2341–2351. doi: 10.1038/sj.npp.1301649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Godbout JP, Johnson RW. Age and neuroinflammation: A lifetime of psychoneuroimmune consequences. Neurol Clin. 2006;24:521–538. doi: 10.1016/j.ncl.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 42.Iwata M, Ota KT, Duman RS. The inflammasome: Pathways linking psychological stress, depression, and systemic illnesses. Brain Behav Immun. 2012;31:105–114. doi: 10.1016/j.bbi.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koo JW, Duman RS. Evidence for IL-1 receptor blockade as a therapeutic strategy for the treatment of depression. Curr Opin Investig Drugs. 2009;10:664–671. [PMC free article] [PubMed] [Google Scholar]
- 44.Duman RS. Neuronal damage and protection in the pathophysiology and treatment of psychiatric illness: Stress and depression. Dialogues Clin Neurosci. 2009;11:239–255. doi: 10.31887/DCNS.2009.11.3/rsduman. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seifert HA, Hall AA, Chapman CB, Collier LA, Willing AE, Pennypacker KR. A transient decrease in spleen size following stroke corresponds to splenocyte release into systemic circulation. J Neuroimmune Pharmacol. 2012;7:1017–1024. doi: 10.1007/s11481-012-9406-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dutta P, Courties G, Wei Y, Leuschner F, Gorbatov R, Robbins CS, et al. Myocardial infarction accelerates atherosclerosis. Nature. 2012;487:325–329. doi: 10.1038/nature11260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–616. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sloan EK, Capitanio JP, Tarara RP, Cole SW. Social temperament and lymph node innervation. Brain Behav Immun. 2008;22:717–726. doi: 10.1016/j.bbi.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cole SW, Arevalo JM, Takahashi R, Sloan EK, Lutgendorf SK, Sood K, et al. Computational identification of gene-social environment interaction at the human IL6 locus. Proc Natl Acad Sci U S A. 2010;107:5681–5686. doi: 10.1073/pnas.0911515107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mehta D, Klengel T, Conneely KN, Smith AK, Altmann A, Pace TW, et al. Childhood maltreatment is associated with distinct genomic and epigenetic profiles in posttraumatic stress disorder. Proc Natl Acad Sci U S A. 2013;110:8302–8307. doi: 10.1073/pnas.1217750110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miller GE, Chen E, Sze J, Marin T, Arevalo JM, Doll R, et al. A functional genomic fingerprint of chronic stress in humans: Blunted glucocorticoid and increased NF-kappaB signaling. Biol Psychiatry. 2008;64:266–272. doi: 10.1016/j.biopsych.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dhabhar FS. Enhancing versus suppressive effects of stress on immune function: Implications for immunoprotection and immunopathology. Neuroimmunomodulation. 2009;16:300–317. doi: 10.1159/000216188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.D’Mello C, Le T, Swain MG. Cerebral microglia recruit monocytes into the brain in response to tumor necrosis factoralpha signaling during peripheral organ inflammation. J Neurosci. 2009;29:2089–2102. doi: 10.1523/JNEUROSCI.3567-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Olson VG, Rockett HR, Reh RK, Redila VA, Tran PM, Venkov HA, et al. The role of norepinephrine in differential response to stress in an animal model of posttraumatic stress disorder. Biol Psychiatry. 2011;70:441–448. doi: 10.1016/j.biopsych.2010.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Qian J, Zhu L, Li Q, Belevych N, Chen Q, Zhao F, et al. Interleukin-1R3 mediates interleukin-1-induced potassium current increase through fast activation of Akt kinase. Proc Natl Acad Sci U S A. 2012;109:12189–12194. doi: 10.1073/pnas.1205207109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006;440:1054–1059. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- 57.Gruol DL, Nelson TE. Purkinje neuron physiology is altered by the inflammatory factor interleukin-6. Cerebellum. 2005;4:198–205. doi: 10.1080/14734220500199987. [DOI] [PubMed] [Google Scholar]
- 58.Serrats J, Schiltz JC, Garcia-Bueno B, van Rooijen N, Reyes TM, Sawchenko PE. Dual roles for perivascular macrophages in immune-to-brain signaling. Neuron. 2010;65:94–106. doi: 10.1016/j.neuron.2009.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Derecki NC, Quinnies KM, Kipnis J. Alternatively activated myeloid (M2) cells enhance cognitive function in immune compromised mice. Brain Behav Immun. 2011;25:379–385. doi: 10.1016/j.bbi.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dantzer R, Kelley KW. Twenty years of research on cytokine-induced sickness behavior. Brain Behav Immun. 2007;21:153–160. doi: 10.1016/j.bbi.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Anisman H. Cascading effects of stressors and inflammatory immune system activation: Implications for major depressive disorder. J Psychiatry Neurosci. 2009;34:4–20. [PMC free article] [PubMed] [Google Scholar]
- 62.McLaughlin KA, Conron KJ, Koenen KC, Gilman SE. Childhood adversity, adult stressful life events, and risk of past-year psychiatric disorder: A test of the stress sensitization hypothesis in a population-based sample of adults. Psychol Med. 2010;40:1647–1658. doi: 10.1017/S0033291709992121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pace TW, Mletzko TC, Alagbe O, Musselman DL, Nemeroff CB, Miller AH, Heim CM. Increased stress-induced inflammatory responses in male patients with major depression and increased early life stress. Am J Psychiatry. 2006;163:1630–1633. doi: 10.1176/ajp.2006.163.9.1630. [DOI] [PubMed] [Google Scholar]
- 64.Sidman RL, Kosaras B, Misra BM, Senft SL. [Accessed 9-2012];High Resolution Mouse Brain Atlas. 1999 Available at: http://www.hms.harvard.edu/research/brain.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.