

Abstract
Lung cancer remains the most common cause of cancer-related death worldwide and it continues to lack effective treatment. The increasingly large and diverse public databases of lung cancer gene expression constitute a rich source of candidate oncogenic drivers and therapeutic targets. To define novel targets for lung adenocarcinoma (ADC), we conducted a large scale meta-analysis of genes specifically overexpressed in ADC. We identified an eleven-gene signature that was overexpressed consistently in ADC specimens relative to normal lung tissue. Six genes in this signature were specifically overexpressed in ADC relative to other subtypes of non-small cell lung cancer (NSCLC). Among these genes was the little studied protein tyrosine kinase PTK7. Immunohistochemical analysis confirmed that PTK7 is highly expressed in primary ADC patient samples. RNAi-mediated attenuation of PTK7 decreased cell viability and increased apoptosis in a subset of ADC cell lines. Further, loss of PTK7 activated the MKK7-JNK stress response pathway and impaired tumor growth in xenotransplantation assays. Our work defines PTK7 as a highly and specifically expressed gene in ADC and a potential therapeutic target in this subset of NSCLC.
Keywords: lung cancer, gene expression meta-analysis, PTK7
Introduction
Lung cancer is the leading cause of cancer death in the US and worldwide 1. Despite intensive basic and clinical research, the overall 5-year survival rate of the major histological subtype, Non-small cell lung cancer (NSCLC) has only improved from 14% to 18% since 1975 1. Recently, targeted treatment based on patient-specific molecular aberrations has led to significant response rates in subsets of NSCLC patients 2, 3. However, about half of all patients do not harbor known “driver” mutations and cannot be treated with targeted agents 4. Thus, new approaches for identification of novel regulators and potential targets for treatment of lung cancer are needed.
Gene expression analysis has been used to classify cancers, predict clinical outcomes and discover disease-associated biomarkers. However, gene expression experiments are usually analyzed in isolation and are limited to a small number of samples. Meta-analysis approaches make it possible to combine multiple gene expression datasets and increase the statistical power for gene discovery. Such meta-analysis approaches have been successfully employed for cancers of the breast 5, 6, prostate 7, liver 8, and lung 9, as well as broadly across cancers 10-12. Statistically, individual studies of gene expression in cancer are limited by both biological (e.g. sampling of a particular patient population) and technical (e.g. only using one expression analysis platform) biases that hinder the broader application of their findings and ultimate translation into clinical practice. Meta-analysis can control for such confounding factors by increasing the statistical power to detect consistent changes across multiple datasets. Several groups have made available large NSCLC gene expression datasets that consist of tumor-to-normal comparisons9, 13-21. These datasets represent a large and as yet not fully tapped resource for discovering novel genes relevant to the pathogenesis of lung cancer. We reasoned that a careful meta-analysis that combined multiple patient populations across many institutions, platforms, and data procurement methods would uncover genes with functional relevance to lung cancer that may have been otherwise overlooked by the isolated analysis of individual gene expression studies.
We applied a recently proposed meta-analysis approach 22 to 13 independent NSCLC gene expression datasets consisting of 2,026 lung samples, which enabled the discovery and validation of commonly over-expressed genes in lung adenocarcinoma (ADC), the predominant subtype of NSCLC. Among the most consistently over-expressed genes was PTK7, a member of the receptor tyrosine kinase family conserved across Hydra, Drosophila, Japanese puffer fish, chicken and human 23. PTK7 contains seven immunoglobin (Ig)-domains, a transmembrane domain, and a catalytically inactive kinase domain within the cytoplasmic tail 24. It was first discovered in melanocytes 24 and subsequently found to be over-expressed in colon carcinoma 25. In Xenopus, PTK7 acts at the level of Frizzled and Dishevelled to regulate the Wnt/planar cell polarity (PCP) pathway 26. Ptk7-knockout mice die perinatally and display developmental defects of the inner ear and of neural tube closure, consistent with a role in regulating PCP 27. Over-expression of PTK7 has been described in several cancers, including colon 25, gastric 28, esophageal 29, and AML 30. However, the precise role of PTK7 in regulating oncogenesis remains unclear. In colon cancer, PTK7 may play a role in the Wnt/β-catenin pathway 31, and knock-down leads to caspase-10-mediated apoptosis 32.
Consistent with the gene expression analysis, we found consistently elevated expression of PTK7 protein in primary ADC patient samples. Knock-down of PTK7 demonstrated that it is essential for the viability of a subset of NSCLC cell lines, and PTK7 disruption increased MKK7-JNK pathway activity. Xenotransplantation studies revealed the requirement of PTK7 in tumor growth. These results demonstrate the power of using publically available patient data to uncover oncogenic drivers and suggest that PTK7 may represent a novel therapeutic target in ADC.
Methods
Data collection, pre-processing, and normalization
Gene expression data for 13 human lung cancer studies were downloaded from the NCBI GEO (accession numbers GSE10072, GSE2514, GSE7670, GSE19188, GSE11969, GSE21933, GSE42127, GSE41271, GSE37745, GSE28571, GSE20853) and websites http://www.broadinstitute.org/mpr/lung/ and https://array.nci.nih.gov/caarray/project/details.action?project.id=182. The histological phenotypes were defined as in the corresponding original publications. Datasets were curated to include only normal, adenocarcinoma (ADC), squamous cell carcinoma (SCC) and large cell carcinoma (LCC) samples. All datasets were normalized individually using gcRMA 33. For PTK7 gene expression, RNA was extracted and prepared using established protocols and hybridized to Affymetrix Human Gene 1.0 ST Array. Raw data are available in GEO (GSE50138).
Meta-analysis of gene expression data
Two meta-analysis approaches were applied to the normalized data 22. The first approach combines effect sizes from each dataset into a meta-effect size to estimate the amount of change in expression across all datasets. For each gene in each dataset, an effect size was computed using Hedges' adjusted g. If multiple probes mapped to a gene, the effect size for each gene was summarized using the fixed effect inverse-variance model. Next, study-specific effect sizes were combined to obtain the pooled effect size and its standard error using the random effects inverse-variance technique. The z-statistic was computed as a ratio of the pooled effect size to its standard error for each gene, and the result was compared to a standard normal distribution to obtain a nominal p-value. P-values were corrected for multiple hypotheses testing using Benjamini-Hochberg correction 34.
A second non-parametric meta-analysis that combines p-values from individual experiments to identify genes with a large effect size in all datasets was also used. A t-statistic was calculated for each gene in each study. After computing one-tail p-values for each gene, these were corrected for multiple hypotheses using Benjamini-Hochberg correction. Next, Fisher's sum of logs method 35 was applied. Briefly, this method sums the logarithm of corrected p-values across all datasets for each gene, and compares the sum against a chi-square distribution with 2k degrees of freedom, where k is the number of datasets used in the analysis.
Leave-one-out validation and classification
To control for the influence of single large experiments on the meta-analysis results, leave-one-out meta-analysis was performed. One dataset at a time was excluded and both meta-analysis methods were applied to the remaining datasets. We hypothesized that the minimal set of genes that are significantly over-expressed, irrespective of the set of datasets analyzed, would constitute a robust gene expression signature of adenocarcinoma across multiple independent cohorts. A very stringent threshold (FDR ≤ 1 × 10-5) for selecting differentially over-expressed genes in ADC was used. Furthermore, we analyzed heterogeneity of the effect sizes across all studies. Genes with significant heterogeneity (p ≤ 0.05) were removed from the over-expressed genes identified using stringent FDR criteria. The geometric mean of the remaining significant genes was computed and used to create a univariate binomial linear model for classifying a lung sample as a normal or ADC sample, or as ADC or SCC sample.
Immunohistochemistry
Immunohistochemistry was performed as previously described 36 with the following antibodies: rabbit antibody to phospho-Histone H3 (1:500, Upstate), rabbit antibody to cleaved caspase 3 (1:400, Cell Signaling, 9664), rabbit antibody to PTK7 (1:1000, Sigma, SAB3500340). PTK7 staining was performed using pepsin antigen retrieval.
Human primary ADC samples
This study complied with federal, state, and local regulations of the Human Research Protection Program and was approved by the Stanford Institutional Research Board. Informed consent was obtained from all patients included in the study.
Establishment of patient-derived xenograft tumors from primary human ADC
Surgically removed human NSCLC tumor tissues were kept in ice-cold HBSS (Life Technologies) until use. Tumors were cut into 1mm pieces and implanted in the subrenal capsules in NOD-SCID-IL2Rg (NSG) mice (Jackson Laboratory, Bar Harbor, Maine).
Tissue microarray
Lung adenocarcinoma tissue microarray with normal lung tissue, containing 20 cases of lung adenocarcinoma and 10 normal lung tissue (BC04119b, US Biomax) was immunohistochemically stained for PTK7. Staining was scored as negative (0), weak (1) or strong (2).
Cell culture
All NSCLC cell lines were maintained in RPMI supplemented with 10% FBS and 1% Penicillin-Streptomycin. Cell lines included NCI-H1299, NCI-H2009, NCI-H23, A549, NCI-H441, NCI-H460, NCI-H1792, NCI-H1975, NCI-H2126, NCI-H358, NCI-H727, NCI-H1568, NCI-H1650, and NCI-H2087. Immortalized normal lung epithelial cell line SALE 37 was cultured in serum free media SAGM (CC-3118, Lonza).
shRNA and virus production
Human shRNA constructs against PTK7 were purchased from OpenBiosystems. The human PTK7 shRNA set was cat #: RHS4533-NM_002821. Of this set, TRCN0000006434 and TRCN0000006435 were labeled as shPTK7-1 and 2, respectively. Control hairpins against GFP and luciferase (LUC) in the pLKO.1-backbone were used (see Supplementary Methods for target sequences). Transfection-quality DNA was extracted using Qiagen DNA kits. Lentivirus was produced by transfection into 293FT cells as previously described 38, filtered, and applied directly to cells. Puromycin selection was started two days after lentiviral infection for a duration of two days at 2μg/ml.
qRT-PCR analysis
RNA was isolated 5 days after lentiviral infection and puromycin selection with Trizol reagent (Invitrogen) following the manufacturer's protocol. cDNA was synthesized with DyNAmo cDNA synthesis kit (F470; New England Biolabs), and qRT-PCR was carried out in triplicate by SYBR Green (Quanta Biosciences) using a C1000 Thermal Cycler (BioRad). See Supplementary Methods for primer sequences.
Cell proliferation assay
Cells were trypsinized and plated in triplicate into 96-well plates (day 0). Cell viability was measured by treating cells with 3-[4,5-dimethlythiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT). Absorbance measurements were recorded using a SpectraMax 340 (Molecular Devices) at 570nm on the indicated days (Cell Proliferation Kit I; Roche). All data were normalized to experimental day 0.
FACS analysis
Cells were scraped from the plate and single cell suspensions were made by passing cells through 28G1/2 insulin syringes (BD) ten times. Cells were then washed in PBS and resuspended in PF10 (10% FBS in PBS), stained at 4°C for 30 min in the dark with rabbit-anti-PTK7 (GTX104510, GeneTex) or IgG2a control (Clone 188B isotype:mouse, BD). For annexin-V staining, cell lines were plated at 5 × 105 cells per 6 cm plates and stained with annexin-V (FITC-Annexin-V, 556419, BD Biosciences) and propidium iodide (PI, 50pl/ml final concentration) following the manufacturer's protocol at indicated time points. A C6 Flow Cytometer (Accuri) was used for FACS analysis and the manufacturer's software and FlowJo v5 for analysis.
Immunoblotting
Cells were scraped and lysed in RIPA buffer with protease inhibitor cocktail (Roche), 25mM sodium fluoride, 1mM PMSF and 1mM sodium orthovanadate (Sigma-Aldrich). Protein samples were resolved by SDS-PAGE, transferred to Amersham Hybond-P membranes (GE Healthcare), and blocking buffer (5% bovine serum albumin in TBS-T) for 1 hour prior to addition of primary antibody, which was applied overnight at 4°C. The following antibodies from Cell Signaling were used (1:1000 dilution unless otherwise noted): rabbit-anti-Cleaved PARP (#5625), mouse-anti-pERK (# 9106), rabbit-anti-ERK (#4695), rabbit-anti-pAKT (T308) (#9275; 1:500), rabbit-anti-AKT (#9272), rabbit-anti-pMKK7 (S271/T275) (#4171), rabbit-anti-MKK7 (#4172), rabbit-anti-pJNK (T183/Y185) (#9251), rabbit-anti-JNK (# 9252), rabbit-anti-pJUN (S73) (# 9164), rabbit-anti-p-P38 (T180/Y182) (#9211), and rabbit-anti-P38 (#9212). Mouse-anti-β-actin (Sigma-Aldrich, clone 1A4; 1:10,000) was used as a loading control.
Xenograft
Cell lines infected with specific shRNA were resuspended in serum free RPMI and injected subcutaneously at 1 million cells per tumor into the 2 lower flanks of nude mice (The Jackson Laboratory). Between 4-8 tumors were injected for each condition. One week after injection, tumor dimensions were measured approximately every 3 days and tumor volume was calculated using the formula: π/6 × ((length+width)/2)3 39 All animal experiments were approved by the Stanford University School of Medicine Committee on Animal Care (APLAC).
Statistical analyses
Unpaired two-tailed t-tests were used for comparisons between different groups. Error bars correspond to standard error of the mean (s.e.m). Significant p values in the text correspond to <0.05 (*), <0.01 (**) or <0.001 (***).
Results
Meta-analysis of five ADC gene expression datasets identifies 11 significantly over-expressed genes in ADC
We identified and manually curated seven published and publicly available ADC datasets9, 13-15, 17-19. Only datasets with accessible unprocessed raw data and containing both normal and ADC samples were used for further analysis. We identified five datasets that met these criteria (743 ADC, 127 normal; Supplementary Table S1) 9, 14, 17-19. We applied a recently proposed meta-analysis approach 22 to these five datasets as outlined in Figure 1A. Because an experiment with a large number of samples can have significant influence on meta-analysis results, we performed a leave-one-out analysis, where we excluded one dataset at a time and applied our meta-analysis approach on the remaining datasets (see methods). We hypothesized that the minimal set of genes identified as significantly over-expressed by both methods, irrespective of the datasets used for analysis, would constitute a robust gene expression signature of ADC across multiple independent cohorts.
Figure 1.

Meta-analysis of NSCLC gene expression datasets identified 11 significantly over-expressed genes in NSCLC. A. Meta-analysis and functional validation pipeline. In silico bioinformatics data analysis workflow consisted of the curation of five publically available datasets, data pre-processing, discovery meta-analysis, independent validation, and subtype analysis followed by validation in primary human patient samples, and in vitro and in vivo functional validation. B. The 11 genes cluster ADC vs. normal across each independent dataset. Each column is a sample and each row is the expression level of a gene. The color scale represents the raw Z-score ranging from blue (low expression) to yellow (high expression). Dendrograms above each heatmap correspond to the Pearson correlation-based hierarchical clustering of samples by expression of the 11 genes.
Because of the heterogeneity of gene expression within and among cancer samples and datasets, we used a very stringent threshold (FDR ≤ 1 × 10-5) for both meta-analysis methods in order to select over-expressed genes in ADC. This approach identified 14 genes significantly and highly over-expressed in ADC across all five datasets. In addition, we estimated between-studies heterogeneity in effect sizes. Three genes (ARHGAP8, CEACAM1, and DDR1) had significant between-studies heterogeneity (p ≤ 0.05), and were removed from the further analysis because we did not have sufficient information available to further explore heterogeneity in these genes (Table 1, Supplementary Fig. S1). As expected, hierarchical clustering of the five discovery datasets using the remaining 11 genes distinguished normal lung from ADC samples (Fig. 1B). We further validated the 11 genes in three additional independent datasets16, 20, 21 (146 ADC, 91 normal; Supplementary Table S2). All genes were significantly over-expressed in the validation datasets at FDR ≤ 5% with at least one meta-analysis method, and all except three (CACNB3, SLC2A5 and SULT1C2) were significantly over-expressed as assessed by both methods (Supplementary Fig. S2 and Supplementary Table S3). Geometric mean of the 11 over-expressed genes was significantly higher for ADC compared to normal samples in each of the three validation datasets (Fig. 2A). A univariate linear regression model using the geometric mean had the area under ROC curve ranging from 0.809 to 0.993 (Fig. 2B). However, because of a very small number of normal samples in Takeuchi et al. dataset, high AUC value should be interpreted with caution. Thus, publicly available datasets can consistently identify a small set of genes as over-expressed in human ADC. However, as only ADC samples were used in this analysis it is possible that these 11 genes are upregulated in other cancers compared to normal tissue. Therefore, we sought to determine whether the 11-gene signature was specific for ADC or whether it was also upregulated in other NSCLC subtypes.
Figure 2.

Eleven genes were significantly over-expressed in three independent ADC gene expression data sets. A. The 11 genes cluster ADC vs normal in three independent ADC data sets. Each column is a sample and each row is the expression level of a gene. The color scale represents the raw Z-score ranging from blue (low expression) to yellow (high expression). Dendrograms above each heatmap correspond to the Pearson correlation-based hierarchical clustering of samples by expression of the 11 genes. B. Swarm plots summarizing the normal and ADC expression values (log2) by geometric mean of the 11-gene signature. C. Performance of a univariate classification model for predicting ADC vs normal based on gene expression of the 11-gene signature across the independent datasets measured by ROC curves. AUC is area under the curve.
Six out of the 11 genes are over-expressed in ADC compared to other NSCLC subtypes
We analyzed an additional eight gene expression datasets consisting of 1040 human NSCLC samples16, 20, 40-43 (687 ADC, 353 SCC; Supplementary Table S4). Six of the 11 genes were significantly over-expressed in ADC compared to SCC, whereas two genes were significantly under-expressed in ADC compared to SCC (Supplementary Table S5). One of the six over-expressed gene, SULT1C2, had significant between-studies heterogeneity (p ≤ 0.05) in the eight datasets. Therefore, it was removed from further analysis. Geometric mean of the remaining 5 over-expressed genes was significantly higher for ADC compared to SCC in seven out of the eight datasets (Supplementary Fig. S3A). The geometric mean of the 5-gene signature in ADC was also significantly higher than that in LCC in the two additional datasets (46 LCC; Supplementary Fig. S3A). A univariate linear regression model using the geometric mean had the area under ROC curve ranging from 0.603 to 0.766 for discriminating between ADC and other NSCLC subtypes (Supplementary Fig. S3B).
In summary, a meta-analysis of 13 independent NSCLC datasets consisting of 2,026 human lung samples (1,430 ADC, 353 SCC, 46 LCC, 197 normal) identified five genes (ARHGEF16, CACNB3, MPZL1, PTK7, and RUNX1) that are significantly and consistently over-expressed in ADC across all datasets, and are able to distinguish ADC samples from normal lung samples or other NSCLC subtypes. Consistent over-expression of these genes despite the potential presence of biological and technological confounding factors including different cohorts, treatments, and microarray technologies, strongly suggests that at least some of these genes may be functionally important in ADC and may play a role in the pathogenesis of this disease.
The receptor tyrosine kinase, PTK7, is over-expressed in ADC
PTK7, belongs to the receptor tyrosine kinase (RTK) family. It is located on chromosomal locus 6p21, which is within the top 4% of all regions of most frequent copy number gain in an independent lung cancer dataset 44 (analyzed using Oncomine 45, data not shown). In addition, 6p21 has been associated with poor survival in NSCLC 46. PTK7 was significantly over-expressed in NSCLC samples compared to normal samples irrespective of subtypes in all discovery (Supplementary Fig. S1) and validation datasets (Supplementary Fig. S2). Furthermore, PTK7 is specifically over-expressed in ADC compared to SCC (Supplementary Fig. S3C). Therefore, we focused further investigations on PTK7.
Over-expression of PTK7 in primary human and mouse lung adenocarcinoma specimens
Increased expression of PTK7 (Fig. 3A) was assessed by immunohistochemical staining. First, two patient-derived xenografts (PDX) of human lung ADC were analyzed and noted to stain positively for PTK7 to varying intensities (Fig. 3B). Staining patterns were heterogeneous, with both nuclear and cytoplasmic staining present within each specimen. Next, a tissue microarray consisting of human samples across various stages and grades of ADC was used to determine PTK7 expression. Fourteen of the 20 ADC samples stained positively for PTK7, of which five stained strongly and nine stained weakly to moderately (Fig. 3C. Supplementary Fig. S4A and Supplementary Table S6). In contrast, all normal lung specimens were negative for epithelial staining of PTK7 (Supplementary Fig. S4A). We found no correlation between PTK7 staining and stage or grade although this may be due to limited sample size. Notably, samples with weak staining tended to have more cytoplasmic rather than nuclear PTK7 (Supplementary Table S6). Thus, PTK7 appears to be overexpressed in most human patient primary ADC samples and a significant subset demonstrates high levels of staining.
Figure 3.

PTK7 is over-expressed in human ADC. A. Forest plot of PTK7 expression across all discovery and validation meta-analysis datasets. The x-axis is the standardized mean difference between ADC and normal on a log2 scale. Thus, a value of 1 signifies a 2-fold difference in gene expression between cancer and normal. B. Representative images of PTK7 expression in two patient-derived xenograft (PDX) specimens. C. Representative images of PTK7 staining of normal lung and ADC spanning grades 1-3. Scale bars represent 50μm.
Consistent with these results, a well-established mouse model of ADC 47 also exhibited increased mRNA transcript levels of Ptk7 (Supplementary Fig. S4C) and positive PTK7 protein staining by IHC (Supplementary Fig. S4D) relative to normal lung. These results suggest that PTK7 is over-expressed in both human and mouse ADC specimens.
Loss of PTK7 in lung cancer cell lines decreases cell viability and induces apoptosis
To determine whether PTK7 could play a functional role in the maintenance of ADC, two shRNAs directed against PTK7 were used to decrease expression in a panel of lung cancer cell lines (Supplementary Fig. S5A). Importantly, PTK7 knock-down had no effect on normal lung epithelial cells (Fig. 4A). However, PTK7 depletion using two independent shRNAs led to a significant decrease in viability in a subset of NSCLC cell lines (Fig. 4A, Supplemental Table S7). The dependency on PTK7 was not determined by its basal expression (Supplementary Fig. S5B) (p = 0.51) or mutations of any known cancer genes (Supplementary Fig. S5C). However, cell lines with chromosomal gains of the locus harboring PTK7 tended to be more sensitive to PTK7 knock-down (p = 0.047) (Fig. 4B).
Figure 4.
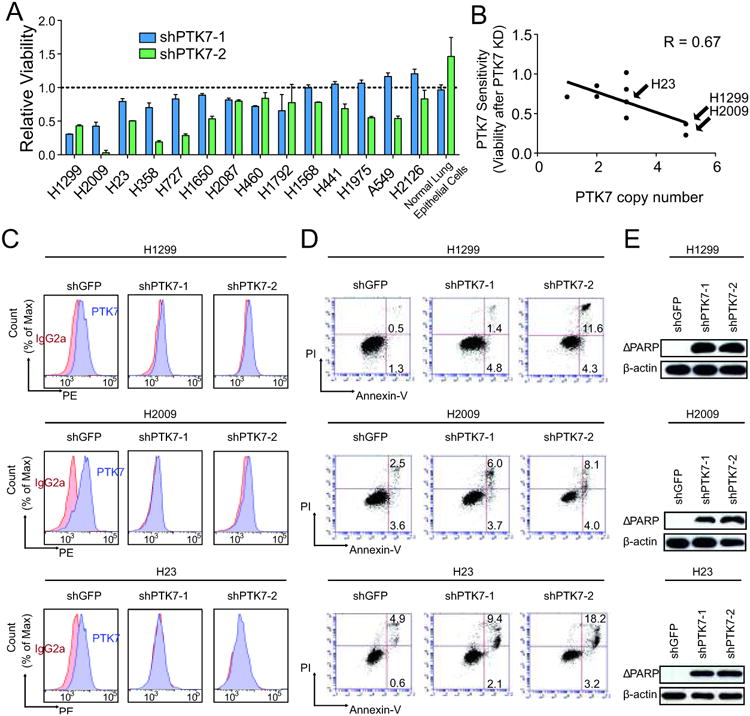
Disruption of PTK7 in three NSCLC cell lines leads to a decrease cell viability and increase in apoptosis. A. Knock-down of PTK7 with two independent hairpins across a panel of NSCLC cell lines and assessment of cell viability by MTT. B. Scatter plot of PTK7 sensitivity vs copy number in the panel of cell lines. PTK7 sensitivity was calculated by averaging the relative viability across both hairpins after PTK7 knock-down. Copy number at the 6p21 PTK7 locus was queried in the Sanger COSMIC database. R represents the linear correlation coefficient. C. PTK7 knock-down validation by FACS. D. FACS analysis of Annexin-V-positive cells. E. Cleaved poly (ADP-ribose) polymerase (ΔPARP) immunoblotting and β-actin loading control.
To further delineate how PTK7 affects cell viability, we chose the three cell lines most sensitive to PTK7 knock-down (H1299, H2009 and H23) for further analysis. Substantial PTK7 knockdown was validated at the RNA level by qRT-PCR (Supplementary Fig. S5D), coinciding with a decrease in protein expression (Fig. 4C). Knock-down of PTK7 in all three cell lines led to an increase in apoptosis as detected by annexin-V staining (Fig. 4D), as well as by cleaved PARP (Fig. 4E). Thus, knock-down of PTK7 in several ADC cell lines in vitro results in decreased cell viability and increased apoptosis whereas normal lung epithelial cells are unaffected. These results suggest that PTK7 may be involved in signaling cascades that regulate survival of a subset of ADC.
Disrupted PTK7 expression activates MKK7-JNK signaling
During normal development PTK7 is thought to play a role in mediating planar cell polarity 26, 27. However, it remains unclear why disruption of this signaling pathway is important in cancer cells as it is not evident why planar cell polarity mechanisms would be involved in oncogenesis. To understand the signaling pathways that are perturbed upon PTK7 disruption, we surveyed the ERK, AKT, JNK and p38MAPK pathways by immunoblotting (Fig. 5). We found no significant change in ERK and AKT signaling, and no consistent change in p38 phosphorylation status across the three cell lines. However, phosphorylation of Mapk kinase kinase-7 (MKK7) at Ser171/Thr275, two residues crucial for MKK7 activity, was clearly increased after PTK7 knock-down. Consistent with the activation of MKK7, phosphorylation of JNK, a downstream target of MKK7, was also increased in response to PTK7 knock-down in the three cell lines tested with two different shRNAs. Activation of MKK7 was also observed when pooled siRNAs were used to deplete PTK7 expression across all three cell lines (Supplementary Fig. S6A-F), arguing against an off-target effect.
Figure 5.
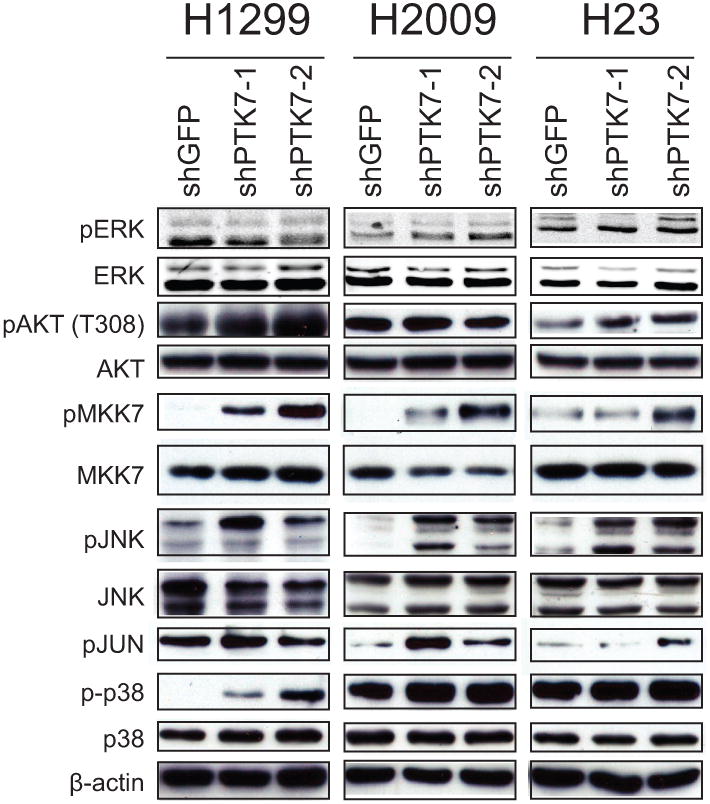
PTK7 regulates MKK7-JNK signaling. Assessment of the ERK, AKT, JNK, and p38 signaling pathways by immunoblotting in three NSCLC cell lines.
Upon phosphorylation by MKK7, JNK translocates into the nucleus, leading to the heterodimerization of c-jun with c-fos to form the AP-1 transcriptional complex. Gene expression profiling following PTK7 knock-down in the H1299 and H2009 cell lines (Supplementary Fig. S6G) revealed an over-representation of AP-1 binding sites associated with the differentially expressed genes (Supplementary Fig. S6H). Thus, in some ADC cell lines, loss of PTK7 leads to dysregulation of MKK7-JNK and possibly other signaling pathways that ultimately lead to decreased proliferation and increased apoptosis.
Xenotransplantation of lung cancer cells depleted of PTK7 reduce tumor burden
The above studies demonstrate that PTK7 is required for cell viability in a subset of ADC cell lines in vitro. To test whether PTK7 is important for tumor maintenance in vivo, we examined the effect of PTK7 depletion on xenograft ADC tumor growth. Xenotransplantation of H1299 (Fig. 6A and B), H2009 (Fig. 6C and D), and H23 (Fig. 6E and F) cell lines infected with shRNA hairpins against PTK7 into immunocompromised mice resulted in a significant decrease in tumor growth relative to control shRNA infected cells. PTK7 loss was confirmed by immunohistochemistry for the two PTK7-specific hairpins relative to control (Fig. 6G). The control experiment of infecting normal lung epithelial cell lines was not performed, as these cell lines do not readily grow in vivo. PTK7 depletion led to a significant decrease in phospho-histone H3 (pHH3), a marker of mitosis (Fig. 6G and H), and a concomitant increase in cleaved caspase 3 (CC3), a marker of apoptosis (Fig. 6G and I). Taken together these studies suggest that PTK7 expression may be required for tumor maintenance in vivo for at least a subset of ADC.
Figure 6.
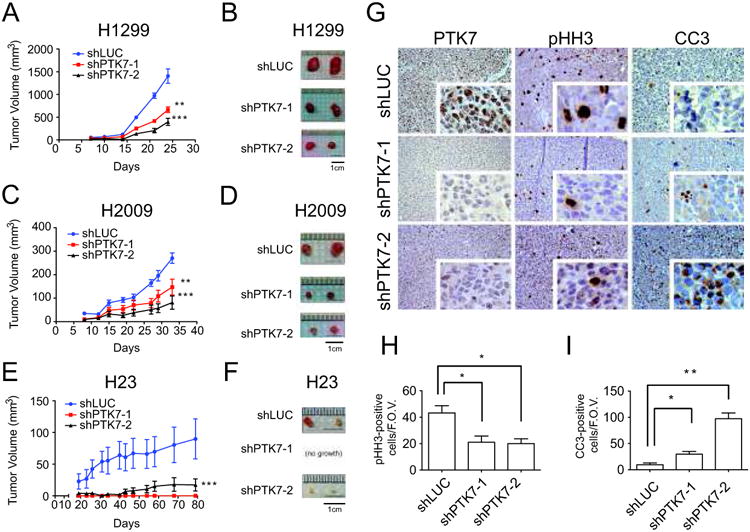
PTK7 depletion in three NSCLC cell lines decreases tumor burden in a xenotransplantation assay. A. Tumor volume over time of xenografted H1299 with control shRNA against luciferase (shLUC), or 2 independent hairpins against PTK7. Error bars represent standard error of the mean (s.e.m) and significant p-values correspond to <0.01 (**) or <0.001 (***) when compared to the shLUC control. Values are mean ± SEM (n = 4-8). B. Representative ex vivo images of H1299 tumors. C and D. Same figures for H2009 cell line. E and F. Same figures for H23 cell line. G. Staining of PTK7, pHH3 and CC3 by IHC in the H1299 xenografts. H. Quantification of pHH3-positive cells per field of view (n=6) I. Quantification of CC3-positive cells per field of view (n=6).
Discussion
The goal of this study was to identify candidate driver genes in lung adenocarcinoma through a meta-analysis of NSCLC gene expression data and to functionally validate their biological significance. The meta-analysis method described here was designed to account for biases inherent in single studies and nominate commonly over-expressed genes. The explosion of large amounts of gene expression data from many different platforms in multiple independent cohorts has enabled the use of in silico tests across numerous patient populations, many more so than a clinical trial could accomplish. With the increasingly widespread availability of gene expression data, a paradigm shift toward inclusion of a “pre-validation” meta-analysis step in biological studies may accelerate translational research.
PTK7 appears to have a context-specific value as a prognostic marker. In gastric cancers it is a favorable marker of differentiated cancers 28, but in AML and esophageal cancer, PTK7 is a poor prognostic marker associated with reduced disease free survival 29, 30. We were unable to find an association between high PTK7 expression and patient survival in NSCLC gene expression datasets with clinical annotations (data not shown), although one report suggested a prognostic significance in NSCLC 48.
Functional studies in vitro identified several NSCLC cell lines that were highly sensitive to depletion of PTK7. Reduced expression of PTK7 led to increased cell death both in vitro and in vivo. This increase in cell death was accompanied by increased signaling through the JNK pathway. However, JNK inhibition alone did not rescue cell viability after PTK7 loss (data not shown), suggesting that PTK7 loss alters other signaling pathways and that JNK activation, while present, is not sufficient to induce apoptosis. Further studies will be needed to identify other signaling pathways downstream of PTK7. Importantly, in NSCLC PTK7 loss did not consistently alter signaling through the ERK, AKT or p38MAPK pathways.
Approximately 10% of the human kinome consists of proteins classified as pseudokinases on the basis of alterations in residues that are thought to be critical to kinase catalytic activity. The pseudokinase PTK7 is most closely related to active tyrosine kinases such as Ros, ALK and LTK 49. Despite the absence of a catalytic domain, increasing evidence suggests a key role for pseudokinases in many biological processes 50. The best-described pseudokinase with an oncogenic role is HER3, which is a member of the EGF receptor family. After ligand binding, HER3 heterodimerizes with HER2, resulting in the interaction of the pseudokinase domain of HER3 and the active kinase domain of HER2, leading to the autophosphorylation of HER2 and activation of downstream PI3K and ERK signaling pathways 51, 52. Pseudokinases can also function as scaffold proteins. For example, KSR proteins interact with protein kinases of the MAPK pathway to localize the signaling components to the membrane 53. In contrast to HER3, no known ligands are known to bind PTK7 in the human setting, and it is unclear whether PTK7 plays a role as a signal modulator, scaffold protein, or another role.
In vertebrates, the PCP pathway regulates convergent extension movements and neural tube closure. PCP also is involved in regulating orientation of stereociliary bundles of sensory hair cells in the inner ear. Studies of Ptk7 C-terminal knock-out mice firmly establish this pseudokinase as a regulator of PCP in mammals 27. The precise signaling role of PTK7 in development is not clear, although the drosophila homologue off-track (otk) may act as a co-receptor for plexins to mediate semaphoring signaling 54. Most recently, PTK7/OTK has been suggested to act by inhibiting canonical Wnt signaling 55.
While loss of PTK7 has been associated with activation of WNT in Xenopus, we did not detect consistent activation of WNT signaling in the absence of PTK7 (data not shown). Thus, the effect of PTK7 loss on lung cancer pathogenesis is likely to be independent of the regulation of WNT signaling. In summary, the data presented here describe PTK7 as a functionally relevant protein in NSCLC. We demonstrate that its disruption in vitro and in vivo decrease tumor cell growth. These observations provide a framework for further studies to characterize PTK7 as a potentially therapeutic target in ADC.
Supplementary Material
Acknowledgments
We thank Julien Sage, Laura Attardi, Steve Artandi, Monte Winslow, and all members of the Sweet-Cordero laboratory for helpful discussions.
Grant Support: R.C. was funded by the Stanford Graduate Fellowship and Cancer Biology Training Grant. P.K., A.B. and E.A.S.C were funded by the US National Cancer Institute (R01 CA138256). P.K.M. and S.V. were funded by the Tobacco-Related Disease Research Program of California (TRDRP). P.K.M was funded by the Stanford University Dean's Fellowship and the Child Health Research Institute and Lucile Packard Foundation for Children's Health Postdoctoral Fellowship. Y.Z. was funded by the American Lung Association.
Footnotes
Declaration of conflict of interest: the authors declare no conflict of interest.
References
- 1.Howlader NNA, Krapcho M, Neyman N, Aminou R, Altekruse SF, Kosary CL, Ruhl J, Tatalovich Z, Cho H, Mariotto A, Eisner MP, Lewis DR, Chen HS, Feuer EJ, Cronin KA, editors. SEER Cancer Statistics Review, 1975-2009 (Vintage 2009 Populations) [accessed August 13, 2012]; http://seer.cancer.gov/csr/1975_2009_pops09/index.html.
- 2.Keedy VL, Temin S, Somerfield MR, et al. American Society of Clinical Oncology provisional clinical opinion: epidermal growth factor receptor (EGFR) Mutation testing for patients with advanced non-small-cell lung cancer considering first-line EGFR tyrosine kinase inhibitor therapy. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2011;29:2121–2127. doi: 10.1200/JCO.2010.31.8923. [DOI] [PubMed] [Google Scholar]
- 3.Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 4.Pao W, Hutchinson KE. Chipping away at the lung cancer genome. Nature medicine. 2012;18:349–351. doi: 10.1038/nm.2697. [DOI] [PubMed] [Google Scholar]
- 5.Sorlie T, Tibshirani R, Parker J, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:8418–8423. doi: 10.1073/pnas.0932692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.West M, Blanchette C, Dressman H, et al. Predicting the clinical status of human breast cancer by using gene expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:11462–11467. doi: 10.1073/pnas.201162998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rhodes DR, Barrette TR, Rubin MA, Ghosh D, Chinnaiyan AM. Meta-analysis of microarrays: interstudy validation of gene expression profiles reveals pathway dysregulation in prostate cancer. Cancer research. 2002;62:4427–4433. [PubMed] [Google Scholar]
- 8.Hoshida Y, Nijman SM, Kobayashi M, et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer research. 2009;69:7385–7392. doi: 10.1158/0008-5472.CAN-09-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shedden K, Taylor JM, Enkemann SA, et al. Gene expression-based survival prediction in lung adenocarcinoma: a multi-site, blinded validation study. Nature medicine. 2008;14:822–827. doi: 10.1038/nm.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nature genetics. 2003;33:49–54. doi: 10.1038/ng1060. [DOI] [PubMed] [Google Scholar]
- 11.Rhodes DR, Yu J, Shanker K, et al. Large-scale meta-analysis of cancer microarray data identifies common transcriptional profiles of neoplastic transformation and progression. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:9309–9314. doi: 10.1073/pnas.0401994101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Segal E, Friedman N, Koller D, Regev A. A module map showing conditional activity of expression modules in cancer. Nature genetics. 2004;36:1090–1098. doi: 10.1038/ng1434. [DOI] [PubMed] [Google Scholar]
- 13.Beer DG, Kardia SL, Huang CC, et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nature medicine. 2002;8:816–824. doi: 10.1038/nm733. [DOI] [PubMed] [Google Scholar]
- 14.Bhattacharjee A, Richards WG, Staunton J, et al. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:13790–13795. doi: 10.1073/pnas.191502998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ding L, Getz G, Wheeler DA, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hou J, Aerts J, den Hamer B, et al. Gene expression-based classification of non-small cell lung carcinomas and survival prediction. PloS one. 2010;5:e10312. doi: 10.1371/journal.pone.0010312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Landi MT, Dracheva T, Rotunno M, et al. Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PloS one. 2008;3:e1651. doi: 10.1371/journal.pone.0001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stearman RS, Dwyer-Nield L, Zerbe L, et al. Analysis of orthologous gene expression between human pulmonary adenocarcinoma and a carcinogen-induced murine model. The American journal of pathology. 2005;167:1763–1775. doi: 10.1016/S0002-9440(10)61257-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Su LJ, Chang CW, Wu YC, et al. Selection of DDX5 as a novel internal control for Q-RTPCR from microarray data using a block bootstrap re-sampling scheme. BMC Genomics. 2007;8:140. doi: 10.1186/1471-2164-8-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takeuchi T, Tomida S, Yatabe Y, et al. Expression profile-defined classification of lung adenocarcinoma shows close relationship with underlying major genetic changes and clinicopathologic behaviors. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2006;24:1679–1688. doi: 10.1200/JCO.2005.03.8224. [DOI] [PubMed] [Google Scholar]
- 21.Lo FY, Chang JW, Chang IS, et al. The database of chromosome imbalance regions and genes resided in lung cancer from Asian and Caucasian identified by array-comparative genomic hybridization. BMC cancer. 2012;12:235. doi: 10.1186/1471-2407-12-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khatri P, Roedder S, Kimura N, et al. A common rejection module (CRM) for acute rejection across multiple organs identifies novel therapeutics for organ transplantation. J Exp Med. 2013;210:2205–2221. doi: 10.1084/jem.20122709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grassot J, Gouy M, Perriere G, Mouchiroud G. Origin and molecular evolution of receptor tyrosine kinases with immunoglobulin-like domains. Molecular biology and evolution. 2006;23:1232–1241. doi: 10.1093/molbev/msk007. [DOI] [PubMed] [Google Scholar]
- 24.Park SK, Lee HS, Lee ST. Characterization of the human full-length PTK7 cDNA encoding a receptor protein tyrosine kinase-like molecule closely related to chick KLG. Journal of biochemistry. 1996;119:235–239. doi: 10.1093/oxfordjournals.jbchem.a021228. [DOI] [PubMed] [Google Scholar]
- 25.Mossie K, Jallal B, Alves F, Sures I, Plowman GD, Ullrich A. Colon carcinoma kinase-4 defines a new subclass of the receptor tyrosine kinase family. Oncogene. 1995;11:2179–2184. [PubMed] [Google Scholar]
- 26.Shnitsar I, Borchers A. PTK7 recruits dsh to regulate neural crest migration. Development. 2008;135:4015–4024. doi: 10.1242/dev.023556. [DOI] [PubMed] [Google Scholar]
- 27.Lu X, Borchers AG, Jolicoeur C, Rayburn H, Baker JC, Tessier-Lavigne M. PTK7/CCK-4 is a novel regulator of planar cell polarity in vertebrates. Nature. 2004;430:93–98. doi: 10.1038/nature02677. [DOI] [PubMed] [Google Scholar]
- 28.Lin Y, Zhang LH, Wang XH, et al. PTK7 as a novel marker for favorable gastric cancer patient survival. Journal of surgical oncology. 2012 doi: 10.1002/jso.23154. [DOI] [PubMed] [Google Scholar]
- 29.Shin WS, Kwon J, Lee HW, et al. Oncogenic role of protein tyrosine kinase 7 in esophageal squamous cell carcinoma. Cancer science. 2013 doi: 10.1111/cas.12194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prebet T, Lhoumeau AC, Arnoulet C, et al. The cell polarity PTK7 receptor acts as a modulator of the chemotherapeutic response in acute myeloid leukemia and impairs clinical outcome. Blood. 2010;116:2315–2323. doi: 10.1182/blood-2010-01-262352. [DOI] [PubMed] [Google Scholar]
- 31.Puppo F, Thome V, Lhoumeau AC, et al. Protein tyrosine kinase 7 has a conserved role in Wnt/beta-catenin canonical signalling. EMBO reports. 2011;12:43–49. doi: 10.1038/embor.2010.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meng L, Sefah K, O'Donoghue MB, et al. Silencing of PTK7 in colon cancer cells: caspase-10-dependent apoptosis via mitochondrial pathway. PLoS ONE. 2010;5:e14018. doi: 10.1371/journal.pone.0014018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhijin W, Irizarry RA, Gentleman R, Martinez-Murillo F, Spencer F. A Model-Based Background Adjustment for Oligonucleotide Expression Arrays. Journal of the American Statistical Association. 2004;99 [Google Scholar]
- 34.Storey J. A direct approach to false discovery rates. Journal of the Royal Statistical Society. 2002;64 [Google Scholar]
- 35.Fisher RA. Statistical methods for research workers. Edinburgh London: Oliver and Boyd; 1925. [Google Scholar]
- 36.Mazur PK, Einwachter H, Lee M, et al. Notch2 is required for progression of pancreatic intraepithelial neoplasia and development of pancreatic ductal adenocarcinoma. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:13438–13443. doi: 10.1073/pnas.1002423107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lundberg AS, Randell SH, Stewart SA, et al. Immortalization and transformation of primary human airway epithelial cells by gene transfer. Oncogene. 2002;21:4577–4586. doi: 10.1038/sj.onc.1205550. [DOI] [PubMed] [Google Scholar]
- 38.Rubinson DA, Dillon CP, Kwiatkowski AV, et al. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nature genetics. 2003;33:401–406. doi: 10.1038/ng1117. [DOI] [PubMed] [Google Scholar]
- 39.Tomayko MM, Reynolds CP. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer chemotherapy and pharmacology. 1989;24:148–154. doi: 10.1007/BF00300234. [DOI] [PubMed] [Google Scholar]
- 40.Sato M, Larsen JE, Lee W, et al. Human lung epithelial cells progressed to malignancy through specific oncogenic manipulations. Molecular cancer research: MCR. 2013;11:638–650. doi: 10.1158/1541-7786.MCR-12-0634-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tang H, Xiao G, Behrens C, et al. A 12-gene set predicts survival benefits from adjuvant chemotherapy in non-small cell lung cancer patients. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:1577–1586. doi: 10.1158/1078-0432.CCR-12-2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Botling J, Edlund K, Lohr M, et al. Biomarker discovery in non-small cell lung cancer: integrating gene expression profiling, meta-analysis, and tissue microarray validation. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:194–204. doi: 10.1158/1078-0432.CCR-12-1139. [DOI] [PubMed] [Google Scholar]
- 43.Micke P, Edlund K, Holmberg L, et al. Gene copy number aberrations are associated with survival in histologic subgroups of non-small cell lung cancer. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2011;6:1833–1840. doi: 10.1097/JTO.0b013e3182295917. [DOI] [PubMed] [Google Scholar]
- 44.Weir BA, Woo MS, Getz G, et al. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450:893–898. doi: 10.1038/nature06358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rhodes DR, Kalyana-Sundaram S, Mahavisno V, et al. Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia. 2007;9:166–180. doi: 10.1593/neo.07112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim TM, Yim SH, Lee JS, et al. Genome-wide screening of genomic alterations and their clinicopathologic implications in non-small cell lung cancers. Clinical cancer research : an official journal of the American Association for Cancer Research. 2005;11:8235–8242. doi: 10.1158/1078-0432.CCR-05-1157. [DOI] [PubMed] [Google Scholar]
- 47.Jackson EL, Willis N, Mercer K, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes & development. 2001;15:3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Endoh H, Tomida S, Yatabe Y, et al. Prognostic model of pulmonary adenocarcinoma by expression profiling of eight genes as determined by quantitative real-time reverse transcriptase polymerase chain reaction. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2004;22:811–819. doi: 10.1200/JCO.2004.04.109. [DOI] [PubMed] [Google Scholar]
- 49.Boudeau J, Miranda-Saavedra D, Barton GJ, Alessi DR. Emerging roles of pseudokinases. Trends in cell biology. 2006;16:443–452. doi: 10.1016/j.tcb.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 50.Zeqiraj E, van Aalten DM. Pseudokinases-remnants of evolution or key allosteric regulators? Current opinion in structural biology. 2010;20:772–781. doi: 10.1016/j.sbi.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF, 3rd, Hynes NE. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:8933–8938. doi: 10.1073/pnas.1537685100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dar AC. A pickup in pseudokinase activity. Biochemical Society transactions. 2013;41:987–994. doi: 10.1042/BST20130110. [DOI] [PubMed] [Google Scholar]
- 53.Kolch W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nature reviews Molecular cell biology. 2005;6:827–837. doi: 10.1038/nrm1743. [DOI] [PubMed] [Google Scholar]
- 54.Winberg ML, Tamagnone L, Bai J, Comoglio PM, Montell D, Goodman CS. The transmembrane protein Off-track associates with Plexins and functions downstream of Semaphorin signaling during axon guidance. Neuron. 2001;32:53–62. doi: 10.1016/s0896-6273(01)00446-9. [DOI] [PubMed] [Google Scholar]
- 55.Peradziryi H, Kaplan NA, Podleschny M, et al. PTK7/Otk interacts with Wnts and inhibits canonical Wnt signalling. EMBO J. 2011;30:3729–3740. doi: 10.1038/emboj.2011.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.