

Abstract
Purpose
A let-7 microRNA-complementary site (LCS6) polymorphism in the 3’UTR of the KRAS gene has been shown to disrupt let-7 binding and upregulate KRAS expression. We evaluated the LCS6 genotype and its association with KRAS mutation status, clinicopathological features, and disease-free survival (DFS) in stage III colon cancer patients enrolled in a phase III clinical trial (NCCTG N0147).
Experimental Design
The LCS6 genotype was assayed by RT-PCR in DNA extracted from whole blood (n=2834) and compared to paired tumor tissue (n=977). Chi-squared and two-sample t tests were used to compare baseline factors and KRAS mutation status between patients defined by LCS6 variant status. Log-rank tests and multivariate Cox models assessed associations between LCS6 status and DFS, respectively.
Results
We identified 432 (15.2%) blood samples and 143 (14.6%) tumor samples heterozygous or homozygous for the LCS6 G-allele, and 2402 of 2834 (84.8%) blood samples and 834 of 977(85.4%) tumor samples homozygous for the LCS6 T-allele. Genotype results were highly concordant (99.8%) in cases with paired blood and tumor tissue (n=977). G-allele carriers were significantly more frequent in Caucasians vs other races (chi-squared test, P <0.0001). The LCS6 genotype was not associated with KRAS mutation status, clinicopathological features (all P > 0.2) or DFS (log-rank P=0.49; HR 0.929; 95% CI: 0.76–1.14), even after combining LCS6 genotype with KRAS mutation status.
Conclusions
In the largest association study investigating the LCS6 polymorphism in colon cancers, the germline LCS6 genotype was not associated with KRAS mutation status or with clinical outcome in patients with stage III tumors.
Keywords: KRAS-LCS6 genotype, let-7, microRNA, colon cancer, single nucleotide polymorphism
Introduction
Colorectal cancer (CRC) is the third most prevalent cancer in men and the second in women throughout the world, with over 1.2 million new cancer cases and 608,700 cancer-related deaths in 2008 [1]. In the United States, the estimated new CRC cases and the estimated deaths in 2012 are 143,460 and 51,690, respectively [2]. While tumor stage remains the most important prognostic factor [3,4], considerable stage-independent variability exists in clinical outcome which underscores the need for the identification and validation of new predictive and prognostic biomarkers to guide therapeutic decision-making for personalized therapy. At present, the only marker that is routinely utilized in clinical practice is the tumor mutation status of the KRAS gene which predicts non-response to anti-EGFR antibodies, including cetuximab, in metastatic CRC patients [5].
MicroRNAs (miRNAs) are endogenous 21- to 22-nucleotide non-coding RNAs [6,7] that target messenger RNAs (mRNAs) and regulate their expression through complementarity to the 3’-UTRs of mRNAs [8,9]. MiRNAs have been shown to play a role in cancer development and progression [10–13]. The lethal-7 (let-7) family is widely viewed as tumor suppressor miRNA and the expression of let-7 family members is down-regulated in cancers of the lung [12], colorectum [14] and breast [15]. The human KRAS oncogene has been shown to contain multiple let-7 complementary sites (LCSs) in its 3’UTR [16] which subjects KRAS to let-7 miRNA-mediated regulation in vitro [14] and in vivo [17].
Recent studies have identified a KRAS 3’UTR polymorphism (rs61764370), aT-to-G nucleotide change in the 6th LCS (LCS6), that was found to increase KRAS expression by altering let-7 binding capability to the KRAS mRNA [18]. Previous association studies have shown potential prognostic value of the LCS6 variant in early stage CRC [19] and in metastatic CRC patients with wild-type (WT) KRAS tumors receiving cetuximab [20]. However, its’ clinical significance and association with KRAS mutation status remains controversial due to conflicting results in studies with limited sample sizes [21–23].
Given this prior evidence, we hypothesized that the LCS6 variant is associated with KRAS mutation status and may be associated with poor prognosis in colon cancers. We secondarily hypothesized that the LCS6 variant is inversely associated with BRAF V600E mutation and deficient DNA mismatch repair (dMMR). To test our hypothesis and further elucidate the significance of the LCS6 variant in a larger patient population, we genotyped the LCS6 variant in a large cohort of stage III colon cancer patients treated in a randomized trial of FOLFOX alone or combined with cetuximab as postoperative adjuvant chemotherapy (NCCTG N0147). In this study, the addition of cetuximab failed to increase disease-free survival (DFS) compared to FOLFOX alone [24].
Materials and Methods
Study population
Patients were obtained from the NCCTG N0147 Trial, a large randomized phase III study in adjuvant colon cancer designed to assess the potential benefit of cetuximab in resected stage III colon cancer. Patients were enrolled in one of the following treatment arms: FOLFOX +/− cetuximab, FOLFIRI +/− cetuximab, 6 cycles of FOLFOX followed by 6 cycles of FOLFIRI ± cetuximab, and treatment per local physician discretion. A total of 3397 patients, of which 2686 patients with KRAS WT were concurrently randomized to primary comparison arms (FOLFOX + cetuximab vs. FOLFOX). The clinical trial obtained Institutional Review Board approval and all patients provided written informed consent before their participation.
Demographic and clinicopathologic data collection was conducted by the Alliance Statistics and Data Center and included the following: N stage (N1 vs. N2), T stage (T1/T2 vs. T3/T4), histologic grade (high [poorly differentiated/undifferentiated] vs. low [well/moderately differentiated]), right (proximal) tumor side (cecum, ascending and transverse colon), or left (distal) tumor side (splenic flexure, descending and sigmoid colon), and body mass index (BMI; BMI<20 vs. 20<BMI<25 vs. 25<BMI<30 vs. BMI>30). In addition, previously reported data on KRAS (c.35 G>C G12A, c.35 G>A G12D, c.34 G>C G12R, c.34 G>T G12C, c.34 G>A G12S, c.35 G>T G12V, and c.38 G>A G13D) and BRAF (c.1799 T>A V600E) mutations and DNA mismatch repair proteins (dMMR vs. pMMR) were also available [24, 25].
KRAS LCS6 genotyping
A total of 2834 Stage III colon cancer patients with available DNA from whole blood (N=2834) and paired formalin-fixed paraffin-embedded (FFPE) tumor specimens (N=977) were utilized for LCS6 genotyping. A previously published probe-based assay (Life Technologies, Grand Island, NY) was used to determine LCS6 variant status [26]. PCR primer and probe sequences were as follows: forward primer: GCCAGGCTGGTCTCGAA, reverse primer: CTGAATAAATGAGTTCTGCAAAACAGGTT, reporter sequence 1: CTCAAGTGATTCACCCAC-VIC, and report sequence 2: CAA GTGATGCACCCAC-FAM. Amplification and variant detection was performed using the LightCycler 480 RT-PCR system (Roche Applied Science, CA). In order to ensure accurate calls, all genotyping plates contained three Coriell DNA samples with known LCS6 variant genotypes (NA12874 – LCS6-GG genotype, NA11831 – LCS6-GT genotype, and NA11892 – LCS6-TT genotype) and one negative control (no genomic DNA). Both genotyping control samples and negative control were duplicated across all plates. In addition, approximately 10% of patient DNA samples (n=280) were randomly selected for duplication across tested DNA plates to ensure consistent calling. Patients with either the GG or GT genotypes were classified as carriers of the LCS6 variant, while patients with the TT genotype were classified as LCS6 wild-type.
Statistical Analysis
All statistical analysis of the LCS6 variant utilized genotype data obtained from whole blood. The primary objective was to assess the prognostic value of LCS6 status in terms of disease-free-survival (DFS) and time to recurrence (TTR). DFS was defined as the time from the date of randomization to the first documented disease recurrence or death from any causes. TTR was defined as time from the date of randomization to the first documented disease recurrence. For patients who died without recurrence, TTR was censored at the last disease evaluation date. Both DFS and TTR were censored at 4 years or last follow-up whichever was earlier. Chi-squared and unequal variance two-sample t-tests were used to compare categorical and continuous baseline factors, respectively, between patients carrying the LCS6 variant (GG or GT) and patients with LCS6 wild-type (TT) [27,28]. Logistic regression was used to assess the association between LCS6 status and clinical outcomes [28]. The method of Kaplan-Meier was used to estimate the distributions of DFS and TTR [29]. Cox model was used to assess the univariate and multivariate associations between LCS6 and clinical outcomes [30]. Unless otherwise specified, all multivariate models were adjusted for age, sex, race, performance score, stratification factors (T/N stage and grade), primary tumor site, treatment, and KRAS, BRAF, and MMR status. The interaction between LCS6 and KRAS, BRAF, and MMR status were assessed by Cox model with corresponding interaction terms. All analyses were performed in SAS v9 and conducted by the Alliance Statistics and Data Center.
Results
LCS6 Genotype in blood DNA and tumor DNA
KRAS LCS6 genotyping was performed on 2834 blood samples with the finding that 432/2834 (15.2%) were heterozygous (GT, 14.6%, n=413) or homozygous (GG, 0.7%, n=19) for the LCS6 G-allele (LCS6 variant), and 2402/2834 (84.8%) were homozygous (TT) for the LCS6 T-allele (LCS6 wild-type). KRAS LCS6 genotyping was also performed in 977 tumor samples (paired with the corresponding blood samples) of which 143/977 (14.6%) were heterozygous (GT, 14.0%, n=137) or homozygous (GG, 0.6%, n=6) for the LCS6 G-allele and 834/977 (85.4%) were homozygous (TT) for the LCS6 T-allele. Results for blood and tumor samples were highly concordant (99.8%) with discrepant results identified in samples from two patients (sample 1: TT/blood and GT/tumor; sample 2: GT/blood and GG/tumor). Repeating the LCS6 genotyping assay for both whole blood and tumor-derived DNA from the two discrepant samples showed identical results.
LCS6 variant, patient demographic and clinicopathological variables
The median age for both LCS6 variant and wild-type carriers was 58 years. Among the study population, 53.2% were male and 87.5% were Caucasian. The frequency of the LCS6 variant was 17.2% in Caucasian, 3.1% in Black or African-American and 0.8% in Asian patients. G-allele carriers were significantly more frequent in Caucasians than in other races (chi-squared test, P<0.0001). No statistically significant differences were found between LCS6 variant carriers and LCS6 wild-type carriers for age, sex or study treatment arm (all P>0.1, Table 1). Additionally, no associations were found between the LCS6 genotype (variant vs. wild-type) and T stage, number of positive lymph node, tumor differentiation, performance status, primary tumor site, bowel obstruction or perforation, or body mass index (all P>0.1, Table 2).
Table 1.
Patient demographics by KRAS-LCS6 status
| Carrier (N=432) |
wild-type (N=2402) |
Total (N=2834) |
p value | |
|---|---|---|---|---|
| Age, years | 0.391 | |||
| N | 432 (15.2) | 2402 (84.8) | 2834 (100.0) | |
| Median | 58.00 | 58.00 | 58.00 | |
| Range | (22.00–85.00) | (19.00–86.00) | (19.00–86.00) | |
| Age, n (%) | 0.182 | |||
| <50 | 89 (13.6) | 566 (86.4) | 655 (23.1) | |
| >=50 | 343 (15.7) | 1836 (84.3) | 2179 (76.9) | |
| Sex, n (%) | 0.912 | |||
| Female | 201 (15.2) | 1125 (84.8) | 1326 (46.8) | |
| Male | 231 (15.3) | 1277 (84.7) | 1508 (53.2) | |
| Race, n (%) | <0.00012 | |||
| Caucasian | 419 (17.2) | 2021 (82.8) | 2440 (87.5) | |
| Black or African-American | 6 (3.1) | 190 (96.9) | 196 (7.0) | |
| Asian | 1 (0.8) | 131 (99.2) | 132 (4.7) | |
| Other | 0 (0.0) | 22 (100.0) | 22 (0.8) | |
| Missing | 6 | 38 | 44 | |
| Treatment Arms, n (%) | 0.782 | |||
| FOLFOX | 177 (15.1) | 997 (84.9) | 1174 (41.4) | |
| FOLFIRI | 15 (17.2) | 72 (82.8) | 87 (3.1) | |
| FOLFOX × 6 → FOLFIRI × 6 | 12 (12.4) | 85 (87.6) | 97 (3.4) | |
| FOLFOX + C225 | 168 (14.9) | 956 (85.1) | 1124 (39.7) | |
| FOLFIRI+ C225 | 3 (10.0) | 27 (90.0) | 30 (1.1) | |
| FOLFOX × 6 → FOLFIRI × 6 + C225 | 6 (19.4) | 25 (80.6) | 31 (1.1) | |
| Treatment per local physician discretion | 51 (17.5) | 240 (82.5) | 291 (10.3) |
Unequal Variance Two Sample T-Test
Chi-Squared Test
Table 2.
Patient clinicopathologic characteristics and genetic biomarkers by LCS6 status
| Carrier (N=432) |
Wildtype (N=2402) |
Total (N=2834) |
p value | |
|---|---|---|---|---|
| T stage, n (%) | 0.221 | |||
| T1 or T2 | 75 (17.2) | 361 (82.8) | 436 (15.4) | |
| T3 or T4 | 357 (14.9) | 2040 (85.1) | 2397 (84.6) | |
| Missing | 0 | 1 | 1 | |
| Number of positive LNs, n (%) | 0.241 | |||
| 1–3 | 246 (14.6) | 1440 (85.4) | 1686 (59.5) | |
| >=4 | 186 (16.2) | 962 (83.8) | 1148 (40.5) | |
| Grade, n (%) | 0.941 | |||
| High | 105 (15.2) | 588 (84.8) | 693 (24.5) | |
| Low | 327 (15.3) | 1814 (84.7) | 2141 (75.5) | |
| PS, n (%) | 0.631 | |||
| PS 0 | 335 (15.4) | 1834 (84.6) | 2169 (76.6) | |
| PS 1 or 2 | 97 (14.7) | 564 (85.3) | 661 (23.4) | |
| Missing | 0 | 4 | 4 | |
| Site of disease, n (%) | 0.211 | |||
| Right | 219 (15.5) | 1197 (84.5) | 1416 (50.2) | |
| Left | 208 (15.2) | 1156 (84.8) | 1364 (48.4) | |
| Both | 2 (5.1) | 37 (94.9) | 39 (1.4) | |
| Missing | 3 | 12 | 15 | |
| Site of disease, n(%) | 0.811 | |||
| Missing | 26 | 149 | 175 | |
| Cecum | 93 (16.2%) | 482 (83.8%) | 575 (21.6%) | |
| Ascending colon | 57 (13.7%) | 359 (86.3%) | 416 (15.6%) | |
| Hepatic flexure | 15 (12.9%) | 101 (87.1%) | 116 (4.4%) | |
| Transverse colon | 36 (16.9%) | 177 (83.1%) | 213 (8.0%) | |
| Splenic flexure | 16 (15.1%) | 90 (84.9%) | 106 (4.0%) | |
| Descending colon | 26 (18.1%) | 118 (81.9%) | 144 (5.4%) | |
| Sigmoid colon | 163 (15.0%) | 926 (85.0%) | 1089 (41.0%) | |
| Bowel obstruction, n (%) | 0.771 | |||
| Yes | 72 (15.7) | 387 (84.3) | 459 (16.2) | |
| No | 360 (15.2) | 2015 (84.8) | 2375 (83.8) | |
| Bowel perforation, n (%) | 0.671 | |||
| Yes | 20 (14.0) | 123 (86.0) | 143 (5.0) | |
| No | 412 (15.3) | 2279 (84.7) | 2691 (95.0) | |
| BMI, n (%) | 0.141 | |||
| Under Weight (BMI<20) | 10 (8.5) | 107 (91.5) | 117 (4.1) | |
| Normal Weight (20<=BMI<25) | 114 (15.6) | 617 (84.4) | 731 (25.9) | |
| Over Weight (25<=BMI<30) | 147 (14.6) | 863 (85.4) | 1010 (35.8) | |
| Obese (BMI>=30) | 158 (16.4) | 806 (83.6) | 964 (34.2) | |
| Missing | 3 | 9 | 12 | |
| KRAS, n(%) | 0.881 | |||
| Missing | 13 | 84 | 97 | |
| Mutant | 150 (15.2) | 839 (84.8) | 989 (36.1) | |
| Wildtype | 269 (15.4) | 1479 (84.6) | 1748 (63.9) | |
| BRAF, n(%) | 0.331 | |||
| Missing | 20 | 139 | 159 | |
| Mutant | 58 (17.2) | 279 (82.8) | 337 (12.6) | |
| Wild type | 354 (15.1) | 1984 (84.9) | 2338 (87.4) | |
| MMR, n(%) | 0.681 | |||
| Missing | 12 | 81 | 93 | |
| pMMR | 375 (15.4) | 2056 (84.6) | 2431 (88.7) | |
| dMMR | 45 (14.5) | 265 (85.5) | 310 (11.3) |
Chi-Squared Test
Association of the LCS6 variant with KRAS, BRAF and MMR status
The overall frequencies of KRAS mutant, BRAF mutant and dMMR tumors were 36.1%, 12.6% and 11.3%, respectively. No statistically significant differences were found between LCS6 variant and wild-type carriers for KRAS, BRAF or MMR status (all P>0.1, Table 2).
Prognostic impact of the LCS6 genotype
The 3-year DFS rate was 74.1% (number of events = 104; 95% CI = 69.5%–78.7%) and 72.5% (number of events = 606; 95% CI = 70.5–74.5%) in LCS6 variant and wild-type carriers, respectively (log-rank test, P=0.49, Figure 1A). The 3 year recurrence-free survival rate was 75.7% (number of events = 93; 95% CI = 71.2%–80.3%) and 74.5% (number of events = 549; 95% CI = 72.6%–76.5%) in LCS6 variant and wild-type carriers, respectively (log-rank test, P =0.43, Figure 1B). Within LCS6 variant and wild-type carriers, no statistically significant differences were found in DFS (HR 0.93, 95% CI: 0.76 to 1.14, Figure 1A) or TTR (HR 0.92, 95% CI: 0.74 to 1.14, Figure 1B). Similar results were obtained after adjusting for age, sex, race, performance score, T/N stage, grade, primary tumor site, KRAS mutation, BRAF mutation, MMR status, and treatment (DFS, HR = 0.885, 95% CI = 0.711 to 1.102, p = 0.2759; TTR, HR = 0.870, 95% CI = 0.689 to 1.097, p = 0.2385). Cox model analysis for the individual LCS6 genotypes (GG vs. GT vs. TT) also showed no significant associations with either DFS (p=0.5738) or TTR (p=0.6713). No significant interaction effect was shown between the LCS6 variant and treatment arm on DFS (p=0.2401) or TTR (p=0.2495). Further analysis within specific treatment arms also showed no statistically significant associations between the LCS6 variant and DFS. In an analysis of the LCS6 genotype in relation to the status of KRAS (Figure 2A), BRAF (Figure 2B), or MMR (Figure 2C), no statistically significant differences in DFS were found (Table 3). In addition, the LCS6 variant showed no significant interaction effect with KRAS mutation status (p=0.42), BRAF mutation status (p=0.16), MMR status (p=0.84), or tumor site (p=0.6616).
Figure 1.
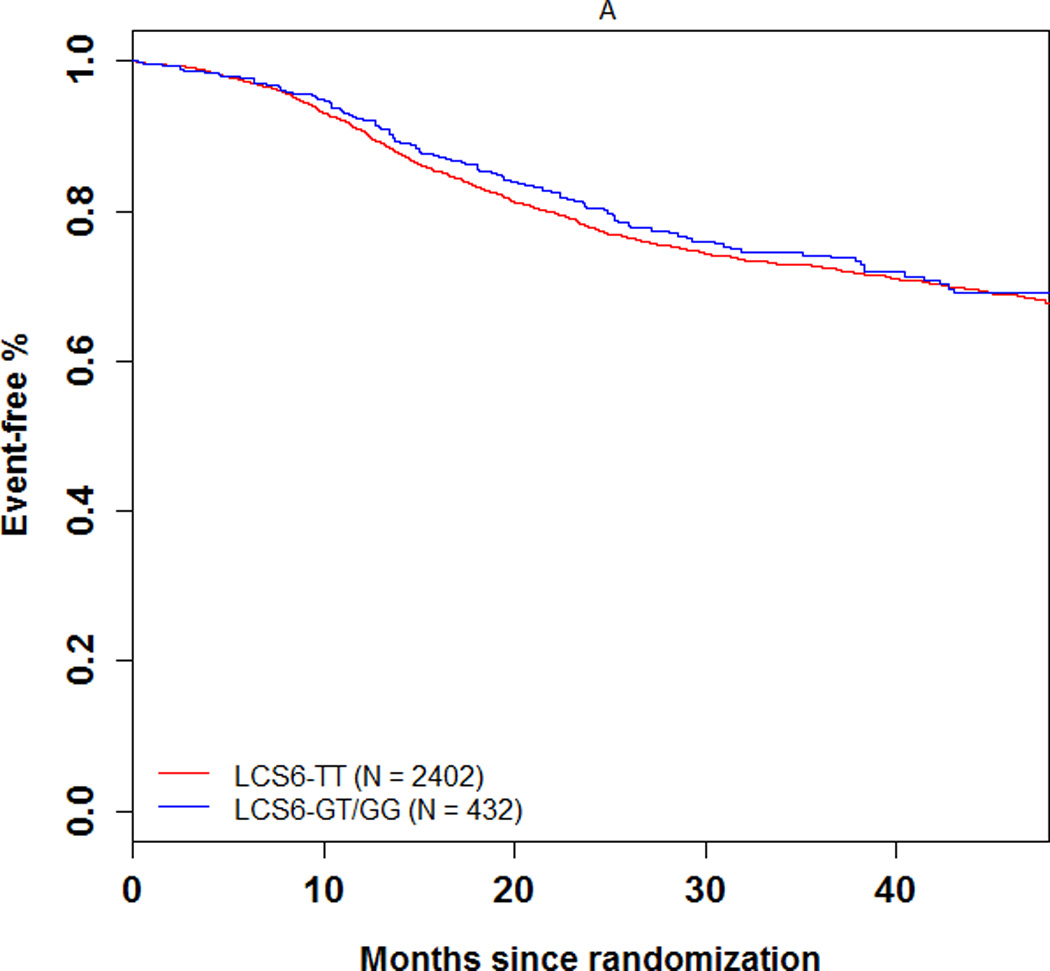
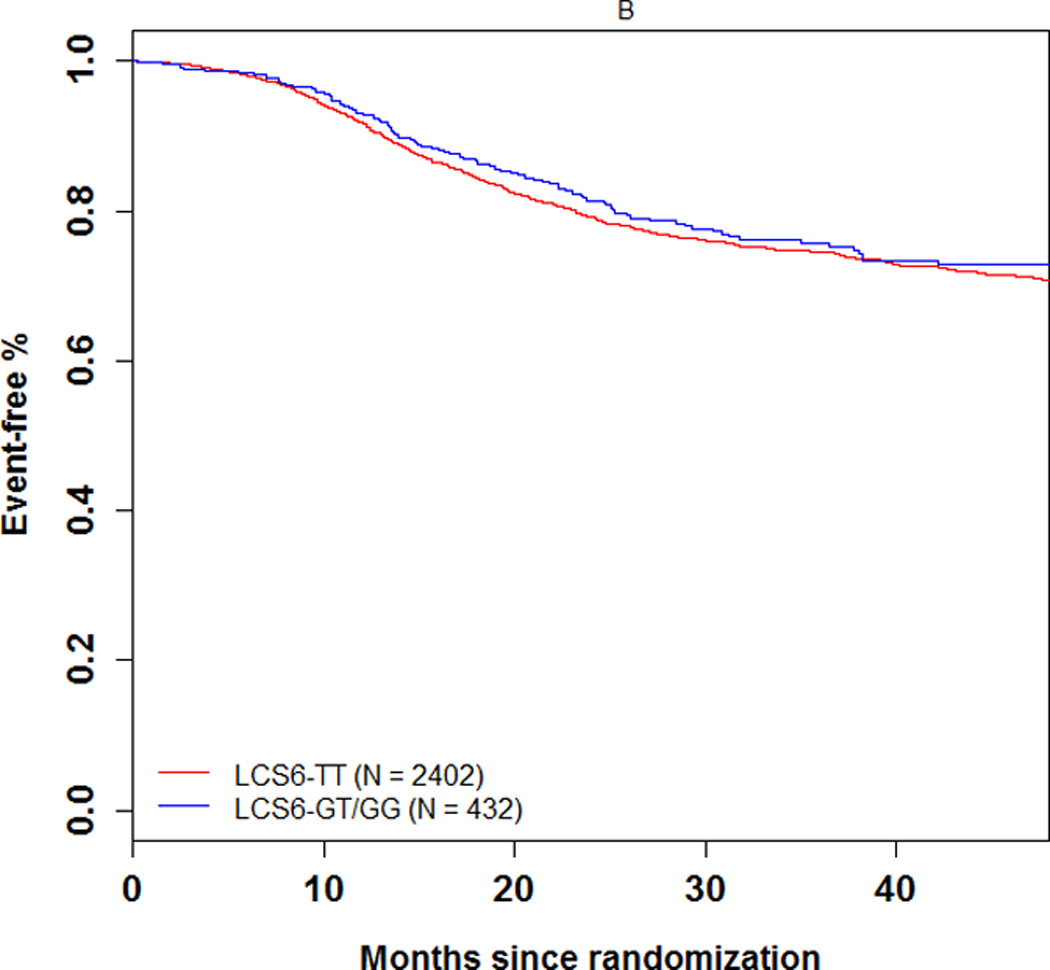
Univariate association of the KRAS-LCS6 variant with (A) DFS and (B) TTR in stage III colon cancer patients. (HR, hazard ratio)
Figure 2.
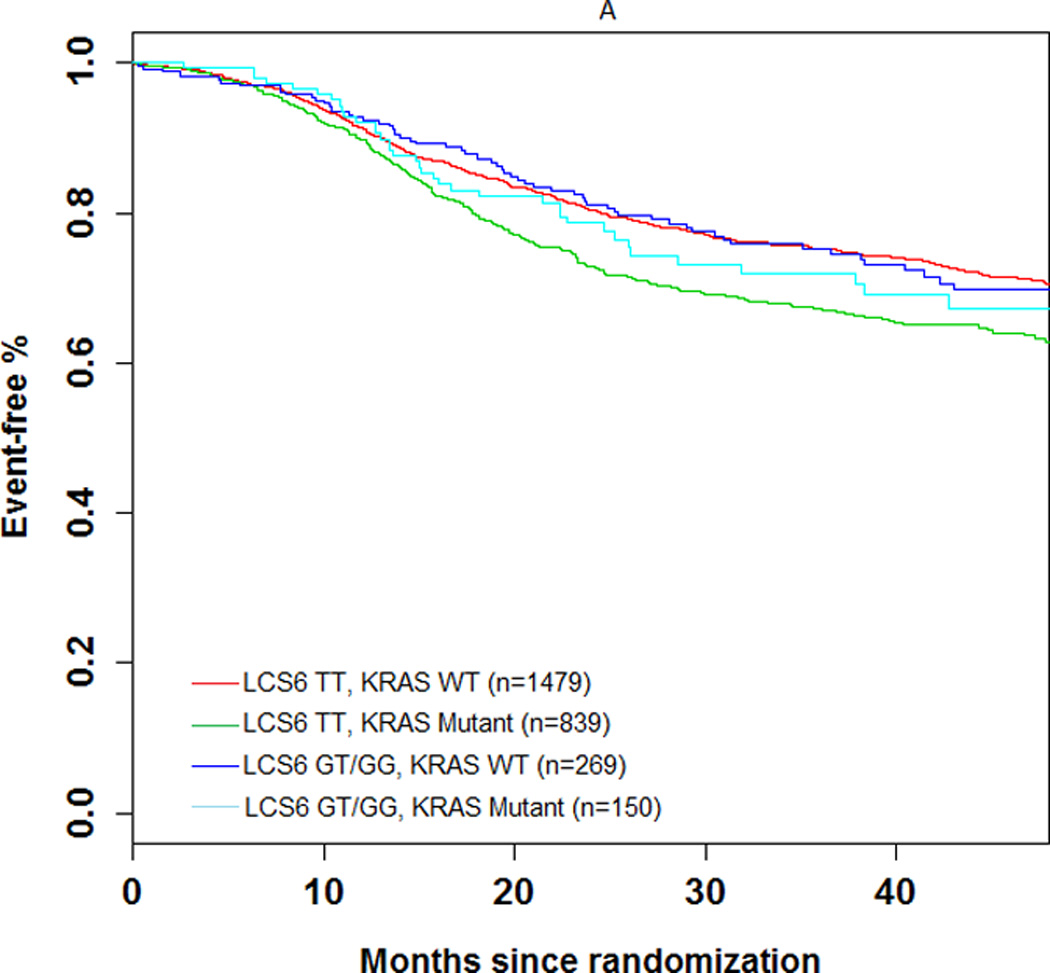

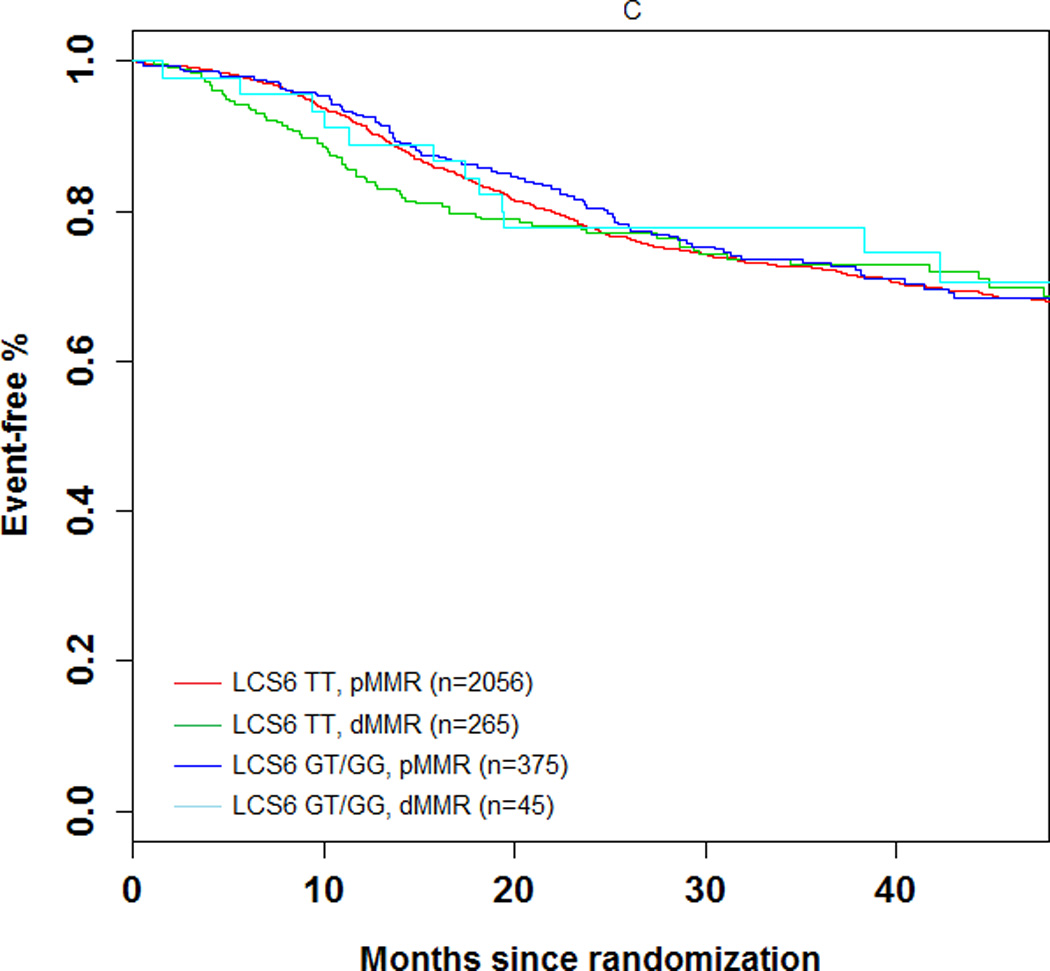
Impact of (A) KRAS mutations, (B) BRAFV600E, and (C) DNA mismatch repair (MMR) status on DFS according to KRAS-LCS6 variant status. (HR, hazard ratio)
Table 3.
Association between the LCS6-variant and DFS stratified by KRAS, BRAF, and MMR status
| HR | 95% CI | P-value | |
|---|---|---|---|
| KRAS MUTATION STATUS | |||
| LCS6 TT, KRAS WT (n=1479) | 0.73 | 0.62 to 0.86 | 0.002 |
| LCS6 TT, KRAS MUTANT (n=839) | Ref | Ref | Ref |
| LCS6 GT/GG, KRAS WT (n=269) | 0.73 | 0.55 to 0.96 | 0.025 |
| LCS6 GT/GG, KRAS MUTANT (n=150) | 0.83 | 0.59 to 1.18 | 0.304 |
| BRAF MUTATION STATUS | |||
| LCS6 TT, BRAF WT (n=1984) | 1.18 | 0.93 to 1.51 | 0.17 |
| LCS6 TT, BRAF MUTANT (n=279) | 1.47 | 1.08 to 2.00 | 0.015 |
| LCS6 GT/GG, BRAF WT (n=354) | Ref | Ref | Ref |
| LCS6 GT/GG, BRAF MUTANT (n=58) | 1.81 | 1.13 to 2.91 | 0.015 |
| MMR STATUS | |||
| LCS6 TT, pMMR (n=2056) | 1.09 | 0.61 to 1.92 | 0.78 |
| LCS6 TT, dMMR (n=265) | 1.13 | 0.61 to 2.08 | 0.70 |
| LCS6 GT/GG, pMMR (n=375) | 1.03 | 0.56 to 1.88 | 0.93 |
| LCS6 GT/GG, dMMR (n=45) | Ref | Ref | Ref |
Discussion
Previous studies have established let-7 as a tumor suppressor miRNA which negatively regulates the RAS pathway [14,16,17]. In 2008, Chin et al. reported on a polymorphism in a let-7 miRNA complementary site 6 in the KRAS 3’UTR (LCS6) that showed a significant association with increased risk for NSCLC among moderate smokers [18]. Since then, the LCS6 polymorphism has been studied extensively in other cancer types, such as oral cavity, ovarian, colorectal and breast [21,26,31–32]. However, the clinical significance of the LCS6 polymorphism in different cancer types and among different stages within CRC has been inconsistent. In order to evaluate the significance of LCS6 variant in colon cancers, we focused on stage III cancer patients from a large, prospectively randomized clinical trial of adjuvant chemotherapy. Our association study indicates that the germline LCS6 genotype was not associated with KRAS mutation status or with clinical outcome in patients with stage III colon cancers.
Our study confirms that the LCS6 variant is a germline polymorphism with genotypes that were highly concordant (99.8%) in paired blood and tumor DNA. Similar to our findings, Sebio et al found a concordance rate of 98%, with two blood DNA samples displaying the LCS6 genotype TG, whereas the two paired tumor DNA samples showed the LCS6 genotype TT [23]. Though a rare occurrence, blood versus tumor DNA discrepancies could result from various events such as loss of heterozygosity in tumor samples, cross-contamination in tissue sampling, DNA fragmentation during the formalin fixation and paraffin embedding processing, or artifactual nucleotide substitutions from problematic PCR amplification [33,34].
Our study identified a significantly higher frequency of the LCS6 G-allele carriers in Caucasians compared to other races which is consistent with the published frequencies Caucasians MAF = 0.086; African MAF = 0.004) [35]. Importantly, racial differences in CRC incidence and mortality exist among Caucasian and African American populations [36] with African Americans being more likely to be diagnosed at a younger age, with late stage disease, proximal tumors, and worse prognosis compared with Caucasians [37]. To date, however, the biological and genetic basis for the existence of a more aggressive CRC phenotype in African Americans awaits further study.
Our analysis showed no associations between the LCS6 variant and either tumor localization, specific tumor subsites, or KRAS somatic mutation status. Tumor location has been shown to display distinct differences in molecular characteristics. Previous studies indicated KRAS-mutated carcinomas were more frequently located in the proximal compared with distal CRC [38]. In addition, cecal cancers have also exhibited the highest frequency of KRAS mutations [39]. In agreement with our findings, previous reports have also shown no correlation between the LCS6 variant and KRAS mutation status in both colon cancer [19] and non-small cell lung cancer [40]. These results suggest that LCS6 and KRAS somatic mutation status are independent events. A possible explanation is that KRAS upregulation accompanying the LCS6 variant does not result in any selective pressure for or against KRAS mutation [40]. However, Graziano et al. reported a conflicting result showing a significantly greater frequency of LCS6 G-allele carriers in the KRAS mutation group compared to the KRAS wild-type group in metastatic CRC patients [21]. It is hypothesized that some clonal selection in tumors may occur, favoring less differentiated and more aggressive clones that harbor both activating KRAS mutations and LCS6. Though the role of LCS6 variant in KRAS mutation remains to be delineated, reported association discrepancies may be explained by the heterogeneity in tumor pathological type and stage, study design, or sample size.
In the current study, we failed to detect any significant association between the LCS6 polymorphism and survival in stage III colon cancer patients, even after combining LCS6 genotype with mutation status of either KRAS or BRAF, or with MMR status. Conflicting data exist regarding this polymorphism in other stages of CRC. In this regard, a significantly better survival was reported in LCS6 G-allele carriers that was enhanced when combined with KRAS mutant status in early stage (stage I and II, n=409), but not in later stage (stage III, n=182 and stage IV, n=69) CRCs [19]. However, Ryan et al. recently showed associations between the LCS6 G allele and reduced risk of mortality in late stage (stage III and IV, n=124), but not in early stage (stage I and II, n=113) CRC patients [22]. Controversy also exists regarding the role of LCS6 polymorphism in prognosis of other solid tumors. A reduced survival was reported in oral cancer patients [26], yet no association between the LCS6 polymorphism and survival was found in NSCLC [40] or ovarian cancer [32]. The conflicting evidence regarding the prognostic value of the LCS6 variant may be attributed to multiple factors: differences in study design, inadequate statistical power, selection bias, and heterogeneity within cancer stages and cancer types.
Our analysis also identified no interaction effect for the LCS6 variant and treatment arm (FOLFOX alone versus FOLFOX and cetuximab) and showed no associations between LCS6 variant status and DFS within the separate treatment groups. Conflicting evidence also exists for the LCS6 variant as a predictive biomarker in KRAS wild-type CRC patients treated with cetuximab. In patients treated with salvage cetuximab-irinotecan therapy, significant associations were found between carriers of the LCS6 G-allele and adverse PFS and overall survival (OS) [21]. However, conflicting results were reported in metastatic CRC patients treated with cetuximab monotherapy with LCS6 wild-type (TT) patients showing a significantly decreased tumor response, but no association between LCS6 genotype and PFS or OS regardless of KRAS status [20]. Most recently, Sebio, et al. identified a significant decrease in tumor response rate in LCS6 G-allele carriers with refractory mCRC; however, there was no significant association between the LCS6 variant and PFS or OS [23]. This association was identified only in patients treated with anti-EGFR-based therapy either alone or in combination, not in patients treated with FOLFIRI alone. While the aforementioned studies were conducted in patients with treatment refractory disease, the Nordic trial was conducted in previously untreated patients with metastatic CRC. In this study, there was no statistically significant effect of the LCS6 variant allele on response rate, PFS or OS in patients treated with FLOX +/− cetuximab [41].
Strengths of our study include the large number of paired blood and tumor specimens that were prospectively collected, analyzed at a single institution and from a clinical trial with meticulous data collection including recurrence and survival. We examined a uniform population of stage III colon cancers as compared to studies that include a mixture of stages with small sample sizes. To our knowledge, our study is the largest conducted to date that examines the LCS6 polymorphism in CRC patients with sufficient statistical power to detect the association between LCS6 variant, KRAS mutation status and disease outcome. However, our study has some limitations. Our trial cohort represents a highly selected group of stage III colon cancer patients through strict inclusion criteria. Thus, bias is unavoidable and geralizability of our findings needs to be proved in colon cancer with other stages (stage I, II and IV) and other cancer types. In addition, KRAS mutation profiling in the N0147 study population remains incomplete. Previous reports have indicated that KRAS mutations in codon 61 and 146 may potentially predict resistance to cetuximab in KRAS codon 12 and 13 wild-type metastatic colorectal cancer [42]. Furthermore, our adjuvant clinical trial population of stage III colon cancer patients is also unable to assess the potential association of the LCS6 variant with tumor response, although recurrence and survival were studied.
In conclusion, we report the largest association study investigating the LCS6 polymorphism and colon cancer outcome. We found that the LCS6 polymorphism is not associated with KRAS mutation status or with disease outcome in stage III colon cancer patients. However, the clinical utility of the LCS6 polymorphism in other stages of colon cancer is poorly understood and awaits further study.
Translational Relevance.
Significant stage-independent variability in clinical outcome of stage III colon cancer continues to be a challenge, highlighting the need for new predictive and prognostic biomarkers. Recent studies have shown potential prognostic value for a KRAS 3’UTR polymorphism in the 6th complimentary site for the miRNA let-7 (KRAS-LCS6); however, the clinical significance of the KRAS-LCS6 remains controversial. In order to determine associations between the KRAS-LCS6 variant and patients’ KRAS mutation status, clinicopathological features, and DFS, our study utilized a total of 2834 stage III colon cancer patients treated with adjuvant FOLFOX or FOLFIRI, alone or combined with cetuximab. To our knowledge, our study examining the KRAS-LCS6 polymorphism is the largest conducted to date. Our results showed no significant association between the KRAS-LCS6 variant and clinical outcomes, indicating limited utility as a prognostic marker in stage III colon cancer.
Acknowledgements
The authors would like to thank Kangsheng Wang for her laboratory work with the Sequenom multiplex assay development, and DNA sample alliqoting. N0147 was supported by the National Cancer Institute, National Institutes of Health (CA25224, CA37404, CA35103, CA63844, CA-35113, CA-3527 2, CA- 114740, CA-32102, CA14028, CA449957, CA21115, CA31946, CA12027, CA37377), Bristol-Myers Squibb, ImClone, Sanofi-Aventis and Pfizer. The study was also supported, in part, by grants from the National Cancer Institute (CA31946) to the Alliance for Clinical Trials in Oncology (Monica M. Bertagnolli, M.D., Chair) and to the Alliance Statistics and Data Center (Daniel J. Sargent, Ph.D., CA33601). Additional funding was also supported, in part, by a National Cancer Institute Senior Scientist Award (K05CA-142885 to F.A.S). The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute.
Funding: N0147 was supported by the National Cancer Institute, National Institutes of Health (CA25224, CA37404, CA35103, CA63844, CA-35113, CA-3527 2, CA- 114740, CA-32102, CA14028, CA449957, CA21115, CA31946, CA12027, CA37377), Bristol-Myers Squibb, ImClone, Sanofi-Aventis and Pfizer. The study was also supported, in part, by grants from the National Cancer Institute (CA31946) to the Alliance for Clinical Trials in Oncology (Monica M. Bertagnolli, M.D., Chair) and to the Alliance Statistics and Data Center (Daniel J. Sargent, Ph.D., CA33601). Additional funding was also supported, in part, by a National Cancer Institute Senior Scientist Award (K05CA-142885 to F.A.S). The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute.
Footnotes
Conflicts of Interest: None
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.A Siegel R, Naishadham D, Jemal A. Cancer statistics 2012. CA Cancer J Clin. 2012;62(1):10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 3.Edge SB, Compton CC. The American Joint Committee on Cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol. 2010;17:1471–1474. doi: 10.1245/s10434-010-0985-4. [DOI] [PubMed] [Google Scholar]
- 4.Hu H, Krasinskas A, Willis J. Perspectives on current tumor-node-metastasis (TNM) staging of cancers of the colon and rectum. Semin Oncol. 2011;38:500–510. doi: 10.1053/j.seminoncol.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 5.Karapetis CS, Khambata-Ford S, Jonker DJ, O'Callaghan CJ, Tu D, Tebbutt NC, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359(17):1757–1765. doi: 10.1056/NEJMoa0804385. [DOI] [PubMed] [Google Scholar]
- 6.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 7.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 8.Lai EC. MicroRNAs are complementary to 3' UTR sequence motifs that mediate negative post-transcriptional regulation. Nat Genet. 2002;30:363–364. doi: 10.1038/ng865. [DOI] [PubMed] [Google Scholar]
- 9.Stark A, Brennecke J, Bushati N, Russell RB, Cohen SM. Animal MicroRNAs confer robustness to gene expression and have a significant impact on 3’UTR evolution. Cell. 2005;123:1133–1146. doi: 10.1016/j.cell.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 10.Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 11.He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–1134. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takamizawa J, Konishi H, Yanagisawa K, Tomida S, Osada H, Endoh H, et al. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. 2004;64:3753–3756. doi: 10.1158/0008-5472.CAN-04-0637. [DOI] [PubMed] [Google Scholar]
- 13.Lu Y, Govindan R, Wang L, Liu PY, Goodgame B, Wen W, et al. MicroRNA profiling and prediction of recurrence/relapse-free survival in stage I lung cancer. Carcinogenesis. 2012;33:1046–1054. doi: 10.1093/carcin/bgs100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akao Y, Nakagawa Y, Naoe T. let-7 microRNA functions as a potential growth suppressor in human colon cancer cells. Biol Pharm Bull. 2006;29:903–906. doi: 10.1248/bpb.29.903. [DOI] [PubMed] [Google Scholar]
- 15.Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65:7065–7070. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- 16.Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, et al. RAS is regulated by the let-7 microRNA family. Cell. 2005;120:635–647. doi: 10.1016/j.cell.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 17.Esquela-Kerscher A, Trang P, Wiggins JF, Patrawala L, Cheng A, Ford L, et al. The let-7 microRNA reduces tumor growth in mouse models of lung cancer. Cell Cycle. 2008;7:759–764. doi: 10.4161/cc.7.6.5834. [DOI] [PubMed] [Google Scholar]
- 18.Chin LJ, Ratner E, Leng S, Zhai R, Nallur S, Babar I, et al. A SNP in a let-7 microRNA complementary site in the KRAS 3' untranslated region increases non-small cell lung cancer risk. Cancer Res. 2008;68:8535–8540. doi: 10.1158/0008-5472.CAN-08-2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smits KM, Paranjape T, Nallur S, Wouters KA, Weijenberg MP, Schouten LJ, et al. A let-7 microRNA SNP in the KRAS 3’UTR is prognostic in early-stage colorectal cancer. Clin Cancer Res. 2011;17:7723–7731. doi: 10.1158/1078-0432.CCR-11-0990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang W, Winder T, Ning Y, Pohl A, Yang D, Kahn M, et al. A let-7 microRNA-binding site polymorphism in 3'-untranslated region of KRAS gene predicts response in wild-type KRAS patients with metastatic colorectal cancer treated with cetuximab monotherapy. Ann Oncol. 2011;22(1):104–109. doi: 10.1093/annonc/mdq315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Graziano F, Canestrari E, Loupakis F, Ruzzo A, Galluccio N, Santini D, et al. Genetic modulation of the Let-7 microRNA binding to KRAS 3'-untranslated region and survival of metastatic colorectal cancer patients treated with salvage cetuximab-irinotecan. Pharmacogenomics J. 2010;10:458–464. doi: 10.1038/tpj.2010.9. [DOI] [PubMed] [Google Scholar]
- 22.Ryan BM, Robles AI, Harris CC. KRAS-LCS6 genotype as a prognostic marker in early-stage CRC-letter. Clin Cancer Res. 2012;18(12):3487–3488. doi: 10.1158/1078-0432.CCR-12-0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sebio A, Pare L, Paez D, Salazar J, González A, Sala N, et al. The LCS6 polymorphism in the binding site of let-7 microRNA to the KRAS 3’untranslated region: its role in the efficacy of anti-EGFR-based therapy in metastatic colorectal cancer patients. Pharmacogenet Genomics. 2013;23(3):142–147. doi: 10.1097/FPC.0b013e32835d9b0b. [DOI] [PubMed] [Google Scholar]
- 24.Alberts SR, Sargent DJ, Nair S, Mahoney MR, Mooney M, Thibodeau SN, et al. Effect of oxaliplatin, fluorouracil, and leucovorin with or without cetuximab on survival among patients with resected stage III colon cancer: a randomized trial. JAMA. 2012;307:1383–1393. doi: 10.1001/jama.2012.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sinicrope FA, Mahoney MR, Smyrk TC, Thibodeau SN, Warren RS, Bertagnolli MM, et al. Prognostic Impact of Deficient DNA Mismatch Repair in Patients With Stage III Colon Cancer From a Randomized Trial of FOLFOX-Based Adjuvant Chemotherapy. J Clin Oncol. 2013;31(29):3664–3672. doi: 10.1200/JCO.2013.48.9591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Christensen BC, Moyer BJ, Avissar M, Ouellet LG, Plaza SL, McClean MD, et al. A let-7 microRNA-binding site polymorphism in the KRAS 3' UTR is associated with reduced survival in oral cancers. Carcinogenesis. 2009;30:1003–1007. doi: 10.1093/carcin/bgp099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Altman DG. “Practical Statistics for Medical Research.”. London: Chapman & Hall; 1991. [Google Scholar]
- 28.Rosner B. Fundamentals of biostatistics. 5th edition. Pacific Grove, CA: Duxbury; 2000. [Google Scholar]
- 29.Kaplan EL, Meier P. Nonparametric Estimation from Incomplete Observations. J AmStat Assoc. 1958;53(282):457–481. [Google Scholar]
- 30.Cox DR. Regression Models and Life-Tables. J Royal Stat Soc Series B (Methodological) 1972;34(2):187–220. [Google Scholar]
- 31.Paranjape T, Heneghan H, Lindner R, Keane FK, Hoffman A, Hollestelle A, et al. A 3'-untranslated region KRAS variant and triple-negative breast cancer: a case-control and genetic analysis. Lancet Oncol. 2011;12(4):377–386. doi: 10.1016/S1470-2045(11)70044-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pharoah PD, Palmieri RT, Ramus SJ, Gayther SA, Andrulis IL, Anton-Culver H, et al. The role of KRAS rs61764370 in invasive epithelial ovarian cancer: implications for clinical testing. Clin Cancer Res. 2011;17:3742–3750. doi: 10.1158/1078-0432.CCR-10-3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lamy A, Blanchard F, Le Pessot F, Sesboüé R, Di Fiore F, Bossut J, et al. Metastatic colorectal cancer KRAS genotyping in routine practice: results and pitfalls. Mod Pathol. 2011;24(8):1090–1100. doi: 10.1038/modpathol.2011.60. [DOI] [PubMed] [Google Scholar]
- 34.Pääbo S, Irwin DM, Wilson AC. DNA damage promotes jumping between templates during enzymatic amplification. J Biol Chem. 1990;265(8):4718–4721. [PubMed] [Google Scholar]
- 35.Database of Single Nucleotide Polymorphisms (dbSNP) Bethesda (MD): National Center for Biotechnology Information, National Library of Medicine; (dbSNP Build ID: human 137). Available from: http://www.ncbi.nlm.nih.gov/SNP/ [Google Scholar]
- 36.Jemal A, Murray T, Ward E, Samuels A, Tiwari RC, Ghafoor A, et al. Cancer statistics, 2005. CA Cancer J Clin. 2005;55(1):10–30. doi: 10.3322/canjclin.55.1.10. [DOI] [PubMed] [Google Scholar]
- 37.Polite BN, Dignam JJ, Olopade OI. Colorectal cancer model of health disparities: understanding mortality differences in minority populations. J Clin Oncol. 2006;24(14):2179–2187. doi: 10.1200/JCO.2005.05.4775. [DOI] [PubMed] [Google Scholar]
- 38.Yamauchi M, Morikawa T, Kuchiba A, Imamura Y, Qian ZR, Nishihara R, et al. Assessment of colorectal cancer molecular features along bowel subsites challenges the conception of distinct dichotomy of proximal versus distal colorectum. Gut. 2012;61(6):847–854. doi: 10.1136/gutjnl-2011-300865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosty C, Young JP, Walsh MD, Clendenning M, Walters RJ, Pearson S, et al. Colorectal carcinomas with KRAS mutation are associated with distinctive morphological and molecular features. Mod Pathol. 2013;26(6):825–834. doi: 10.1038/modpathol.2012.240. [DOI] [PubMed] [Google Scholar]
- 40.Nelson HH, Christensen BC, Plaza SL, Wiencke JK, Marsit CJ, Kelsey KT. KRAS mutation, KRAS-LCS6 polymorphism, and non-small cell lung cancer. Lung Cancer. 2010;69:51–53. doi: 10.1016/j.lungcan.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kjersem JB, Ikdahl T, Guren T, Skovlund E, Sorbye H, Hamfjord J, et al. Let-7 miRNA-binding site polymorphism in the KRAS 3'UTR; colorectal cancer screening population prevalence and influence on clinical outcome in patients with metastatic colorectal cancer treated with 5-fluorouracil and oxaliplatin +/− cetuximab. BMC Cancer. 2012;12:534. doi: 10.1186/1471-2407-12-534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Loupakis F, Ruzzo A, Cremolini C, Vincenzi B, Salvatore L, Santini D, et al. KRAS codon 61, 146 and BRAF mutations predict resistance to cetuximab plus irinotecan in KRAS codon 12 and 13 wild-type metastatic colorectal cancer. Br J Cancer. 2009;101(4):715–721. doi: 10.1038/sj.bjc.6605177. [DOI] [PMC free article] [PubMed] [Google Scholar]