

Abstract
Objective
Protein kinase C (PKC) ζ is a key pathological mediator of endothelial cell apoptosis. p62 is a scaffold protein that regulates several cell signaling pathways by binding to target proteins. Because PKCζ and p62 contain Phox/Bem1p (PB1) modules that mediate protein–protein interactions, we hypothesized that an interaction between p62 and PKCζ is required for tumor necrosis factor α–induced PKCζ signaling in endothelial cells.
Methods and Results
In human umbilical vein endothelial cell, tumor necrosis factor α (10 ng/mL) enhanced the interaction between p62 and PKCζ. Transfection with p62 small interfering RNA reduced the activation of both PKCζ and its downstream targets JNK and caspase 3, suggesting that p62 is necessary for PKCζ signaling. Overexpression of only the PB1 domain of p62 inhibited p62–PKCζ interaction, showing that binding of these 2 proteins is mediated by their PB1 domains. Furthermore, overexpression of the p62 PB1 domain suppressed tumor necrosis factor α–induced PKCζ activation and subsequent activation of JNK and caspase 3. Finally, transfection of either p62 small interfering RNA or the PB1 domain of p62 inhibited human umbilical vein endothelial cell apoptosis.
Conclusion
Our results suggest a novel function of p62 that regulates the activity of PKCζ by binding to PKCζ, thereby activating the PKCζ-JNK-caspase 3 apoptotic pathway in endothelial cells.
Keywords: apoptosis, endothelial cells, p62, Phox/Bem1p domain, protein kinase Cζ
We have previously reported that protein kinase C (PKC) ζ is proapoptotic in endothelial cell (EC).1,2 Tumor necrosis factor (TNF) α activates PKCζ in EC1 and induces its apoptosis,3,4 which contributes to vascular diseases such as atherosclerosis.5 We are, therefore, interested in the mechanism of PKCζ-dependent EC apoptosis in response to TNFα stimulation.
Physiologically, PKCζ is the only PKC isoform that is activated specifically in EC exposed to proapoptotic disturbed flow.6,7 It is likely that PKCζ plays a critical role in EC dysfunction and apoptosis.1,2,8 PKCζ belongs to the atypical PKC family of which binding to other regulatory proteins allosterically regulates their activities.9 PKCζ is among 13 proteins in the mammalian genome that contains a Phox/Bem1p (PB1) domain. PB1 domains are recently recognized protein–protein interaction domains found in the atypical PKC isoenzymes, PKCλ/Ι and PKCζ; members of mitogen-activated protein kinase modules such as mitogen extracellular signal–regulated kinase kinase 5, mitogen extracellular signal–regulated kinase kinase 2, and mitogen extracellular-signal regulated kinase kinase 3; and in several scaffold proteins involved in cellular signaling.10–12 Of great interest is the fact that the PB1 domain–containing protein PKCζ is critical to TNFα signaling in EC.1,6
p62 is a scaffold protein that was initially identified by 2-hybrid screens as a binding partner of atypical PKC family.13,14 p62 has several domains that interact with other proteins, including, in the amino terminus region, a PB1 domain that can interact with other PB1 domain–containing proteins through PB1–PB1 interaction.15 p62 has a proinflammatory role in various cell types. For example, p62 was critical for the sustained activation of nuclear factor (NF)-κB in response to NF-κB ligand activation and mediated the differentiation of osteoclasts.16 p62 also mediated nerve growth factor–induced activation of the NF-κB pathway in PC12 cells.17 In addition, p62 was an important mediator in Th2 cell differentiation18 and tumorigenesis.19 However, the role of p62 in EC remains unclear.
Thus, we investigated the function of p62 by focusing on its interaction with PKCζ in the PKCζ-mediated apoptotic signaling cascade in EC. To study the role of p62 in PKCζ activation, we used human umbilical vein EC (HUVEC) because we showed that these cells recapitulate many of the signaling events that occur in vivo.5,20,21 We used TNFα to stimulate PKCζ because we previously showed that TNFα activates PKCζ, which is required for JNK and caspase 3 activation in EC. We found that the interaction between p62 and PKCζ was induced by TNFα stimulation, and the inhibition of their interaction suppressed PKCζ signaling. Activation of JNK and caspase 3 was diminished, reducing EC apoptosis. Overall, this study defines p62 as an important regulatory molecule in PKCζ activation and EC apoptosis.
Materials and Methods
Cell Culture
HUVEC was isolated from collagenase-digested human umbilical veins and maintained in Medium 200 (Cascade Biologics, Portland, OR) supplemented with low serum growth supplement (Cascade Biologic), 5% fetal bovine serum (Gibco, Carlsbad, CA), 100 U/mL penicillin (Gibco), and 100 μg/mL streptomycin (Gibco). Cells were cultured on 2% gelatin-precoated dishes and used at passages 4 to 5.
Sources of Materials
Antibodies against PKCζ, JNK, caspase 3, p-PKCζ (Thr410), and p-JNK (Thr183/Tyr185) were purchased from Cell Signaling Technology (Beverly, MA); anti-p62 from BD Transduction Laboratories (St. Louis, MO); and anti-green fluorescent protein (GFP) and anti-actin from Santa Cruz Biotechnology Inc (Santa Cruz, CA). The human p62 small interfering RNA (siRNA; J-010230-05 [main figures] and D-010230-03 [Figures in the online-only Data Supplement]) and scrambled siRNA were from Dharmacon RNA Technologies (Lafayette CO). TNFα was purchased from Roche Applied Science (Indianapolis, IN). 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) was purchased from Sigma (St. Louis, MO).
Transfection
For siRNA transfection, HUVEC, at >85% confluence, was transiently transfected with scrambled or p62 siRNA using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. The cells were harvested after 48 hours. For plasmid DNA transfection, HUVEC, at >95% confluence, was transiently transfected with pcDNA3.1 or GFP-tagged PB1 domain of p62 (PB1-GFP) using Lipofectamine 2000 (Invitrogen) and Plus Reagent (Invitrogen) according to the manufacturer’s instructions. DNA (0.3 or 1.5 μg) was used for the transfection of 1 well of a 6-well plate or one 100-mm dish, respectively, and the cells were harvested after 24 hours. In this condition, transfection efficiency measured by the expression of GFP under the Olympus BX51 fluorescent microscope was 28.2±5.7% (Figure I in the online-only Data Supplement). Dr Terje Johansen (University of Tromsø, Norway) kindly provided PB1-GFP.12
Western Blotting and Immunoprecipitation Assay
Cells were harvested and lysed in cell lysis buffer (Cell Signaling Technology) supplemented with protease inhibitor cocktail (Sigma). Cells were harvested and centrifuged at 13 000g for 10 minutes and supernatants collected. Protein concentration was determined by Bradford assay (Bio-Rad, Hercules, CA), and after heating at 95°C for 5 minutes, equivalent amounts of cell lysates were resolved by SDS-PAGE. Proteins were transferred onto nitrocellulose membranes and blocked for 1 hour at room temperature in 5% nonfat milk. Membranes were incubated overnight at 4°C with appropriate primary antibodies, followed by incubation in horseradish peroxidase–conjugated secondary antibody for 1 hour before development using an Immobilon Western Chemiluminescent Horseradish Peroxidase Substrate (Millipore, Billerica, MA). Densitometric analyses of immunoblots were performed with Image J software. For the immunoprecipitation assay, 500 μg of total protein was immunoprecipitated with 1 μg of PKCζ antibody or control IgG at 4°C overnight. Lysates were then incubated with 50 μL of protein A/G agarose beads (Santa Cruz Biotechnology) at 4°C for 2 hours. Immune complexes were collected by centrifugation (3000g for 3 minutes) and washed 5× with the cell lysis buffer. The bound proteins were released by heating in 2× SDS gel sample buffer and were resolved by SDS-PAGE.
MTT Assay
MTT assay was performed as previously described.22 Briefly, cells were treated with TNFα (10 ng/mL)+cyclohexamide (CHX, 10 μg/mL). After 24 hours, MTT (final 1 mg/mL) was added to cells and incubated at 37°C for 4 hours. When the purple precipitate was visible, the medium was removed and dimethylsulfoxide added to solubilize the formazan product. After shaking for 10 minutes to thoroughly mix the formazan with dimethylsulfoxide, the optical density at 590 nm was determined.
Statistics
Data are shown as mean±SD for 3 separate experiments. Analyses for 2 groups were performed using 1-way Student t test. Analyses for 4 groups were performed using repeated-measures 1-way ANOVA test. Values of P<0.05 were considered significant.
Results
TNFα Increases p62 Binding to PKCζ
Because both p62 and PKCζ contain PB1 domains, we first studied their interaction in response to TNFα stimulation. In confluent HUVEC, p62 and PKCζ exhibit a low level of interaction (Figure 1A). TNFα stimulation significantly increased the interaction with a peak at 15 minutes (1.9±0.2-fold compared with no TNFα stimulation; Figure 1B).
Figure 1.
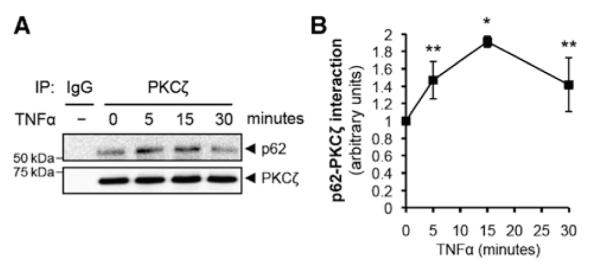
The interaction between p62 and protein kinase C (PKC) ζ is increased by tumor necrosis factor α (TNFα) stimulation. A, Total cell lysates from human umbilical vein endothelial cell that were stimulated by TNFα (10 ng/mL) for the indicated times were immunoprecipitated with PKCζ antibody, and then immunoprecipitates were analyzed by immunoblotting with p62 or PKCζ antibody. B, Quantification of p62 binding to PKCζ. Bound p62 was normalized to the levels of PKCζ immunoprecipitates. *P<0.01 and **P<0.05 compared with no TNFα treatment.
TNFα Activates PKCζ via a p62-Dependent Pathway
After observing an increased interaction between p62 and PKCζ after TNFα stimulation, we hypothesized that this interaction is required for PKCζ activation. To study the role of p62, we depleted p62 using siRNA (J-010230-05) and measured TNFα-induced PKCζ activation. At the optimal concentration of p62 siRNA (80 nmol/L for 48 hours), expression was reduced by 92.8±1.8%, whereas 40 nmol/L siRNA reduced the p62 expression by 83.3±2.7% (Figure 2A and 2B). After depletion of p62, TNFα-induced PKCζ activation was completely inhibited (Figure 2C and 2D). Transfection of HUVEC with another p62 siRNA (D-010230-03) showed a similar inhibitory effect on TNFα-induced PKCζ activation (Figure IIA in the online-only Data Supplement). These data suggest a critical role for p62–PKCζ interaction in TNFα-induced PKCζ signaling.
Figure 2.

Knockdown of p62 suppresses tumor necrosis factor α (TNFα)-induced protein kinase C (PKC) ζ activation. A, Dose-dependent knockdown of p62 expression by small interfering RNA (siRNA) (J-010230-05). Total cell lysates from human umbilical vein endothelial cell (HUVEC) transfected with either scrambled or p62 siRNA for 48 hours were analyzed by immunoblotting. B, Quantification of p62 expression from HUVEC transfected with either scrambled or p62 siRNA. The intensity of p62 expression was normalized to actin expression. *P<0.01. C, HUVEC was transfected with 80 nmol/L of either scrambled or p62 siRNA (J-010230-05) and stimulated with TNFα (10 ng/mL) for the indicated times. Phosphorylation of PKCζ at Thr410 was analyzed by immunoblotting. D, Quantification of PKCζ phosphorylation that was normalized to the expression of total PKCζ. *P<0.01 compared with the respective time control.
Depletion of p62 Suppresses TNFα-Induced PKCζ Signaling Cascade
We previously showed that TNFα-activated PKCζ led to the phosphorylation of JNK.1 We and others have demonstrated that TNFα induced the cleavage of caspase 3 and the subsequent apoptosis of HUVEC when CHX was added together to inhibit protein synthesis.5,22,23 Thus, we investigated the effect of p62 depletion on TNFα-induced JNK activation. As shown in Figure 3A and 3B, TNFα-induced time-dependent phosphorylation of JNK was significantly reduced in p62 siRNA (J-010230-05)–transfected HUVEC compared with scrambled siRNA. Furthermore, the expression of the cleaved form of caspase 3 (17/19 kDa) by TNFα+CHX treatment in p62 siRNA (J-010230-05)–transfected HUVEC was dramatically reduced compared with scrambled siRNA (Figure 3C and 3D). Transfection of HUVEC with another p62 siRNA (D-010230-03) showed a similar inhibitory effect on both JNK activation and caspase 3 activation (Figure IIB and IIC in the online-only Data Supplement). As caspase 8 is required for caspase 3–mediated apoptosis,24 we examined whether caspase 8 is activated by PKCζ pathway. We depleted either PKCζ or p62 with siRNA transfection and stimulated transfected cells with TNFα+CHX for 6 hours. As shown in Figure IIIA to IIIC in the online-only Data Supplement, depletion of either PKCζ or p62 reduced activation of caspase 8 (cleaved form of caspase 8; 41/43 kDa). These data show the important role of p62 in PKCζ signaling as reflected by JNK, caspase 8, and caspase 3 activation.
Figure 3.

Tumor necrosis factor α (TNFα)-induced protein kinase C (PKC) ζ signaling cascade depends on the p62 association with PKCζ. A, Human umbilical vein endothelial cell (HUVEC) was transfected with 80 nmol/L of either scrambled or p62 small interfering RNA (siRNA) (J-010230-05) and stimulated with TNFα (10 ng/mL) for the indicated times. Phosphorylation of JNK at Thr183/Tyr185 was analyzed by immunoblotting. B, Quantification of JNK phosphorylation that was normalized to the expression of total JNK. *P<0.01 compared with the respective time control. C, HUVEC was transfected with 80 nmol/L either scrambled or p62 siRNA (J-010230-05) and stimulated with TNFα (10 ng/mL)+cyclohexamide (CHX) (10 μg/mL) for 6 hours. The cleavage of caspase 3 was analyzed by immunoblotting. D, Quantification of cleaved form of caspase 3 that was normalized to the expression of actin. *P<0.01.
Inhibiting the Interaction Between p62 and PKCxsζ Suppresses TNFα-Induced PKCζ Activation
To provide further support for the role of PB1 domain in mediating the p62–PKCζ interaction, we designed a peptide inhibitor (Figure 4A). Specifically, we thought that exogenous overexpression of the PB1 domain of p62 would compete with endogenous p62 and suppress the binding of p62 to PKCζ by occupying the p62-binding site on PB1 domain of PKCζ (Figure 4A). Consistent with our hypothesis, overexpression of the PB1 domain of p62 that was tagged by GFP (PB1-GFP) suppressed the interaction between p62 and PKCζ (Figure 4B). Furthermore, TNFα-induced PKCζ activation was completely inhibited by the PB1-GFP inhibitor (Figure 4C and 4D). These data suggest that interaction between p62 and PKCζ is critical for the activation of PKCζ by TNFα.
Figure 4.

Interaction between p62 and protein kinase C (PKC) ζ through Phox/Bem1p (PB1) modules is required for tumor necrosis factor (TNFα)-induced PKCζ phosphorylation. A, A model for the inhibitory effect of PB1-GFP on the interaction between p62 and PKCζ. B, Human umbilical vein endothelial cell (HUVEC) was transfected with either pcDNA3.1 or PB1-GFP for 24 hours. Cell lysates were immunoprecipitated with PKCζ antibody, and immunoprecipitates were analyzed by immunoblotting with p62, GFP, or PKCζ antibodies. The transfection of PB1-GFP was analyzed with total cell lysates (TCL) by immunoblotting. C, HUVEC was transfected with either pcDNA3.1 or PB1-GFP and stimulated with TNFα (10 ng/mL) for the indicated times. Phosphorylation of PKCζ at Thr410 was analyzed by immunoblotting. D, Quantification of PKCζ phosphorylation that was normalized to the expression of total PKCζ. *P<0.01 compared with the respective time control.
Inhibiting p62 Binding to PKCζ Suppresses TNFα-Induced PKCζ Signaling Cascade
To study the effect of PB1-GFP on TNFα signaling, we measured the phosphorylation of JNK and cleavage of caspase 3 after overexpression of PB1-GFP. As shown in Figure 5A and 5B, the phosphorylation of JNK induced by TNFα stimulation was significantly reduced in PB1-GFP overexpressing HUVEC. Furthermore, the activation of caspase 8 and caspase 3 was significantly reduced in PB1-GFP overexpressing HUVEC (Figure IIID in the online-only Data Supplement and Figure 5C and 5D). These data suggest that the interaction between the PB1 domains of p62 and PKCζ induced by TNFα stimulation (Figure 1) is important for the PKCζ signaling cascade.
Figure 5.

Tumor necrosis factor (TNF) α-induced protein kinase C (PKC) ζ signaling cascade depends on the interaction between p62 and PKCζ. A, Human umbilical vein endothelial cell (HUVEC) was transfected with either pcDNA3.1 or Phox/Bem1p (PB1)-GFP and stimulated with TNFα (10 ng/mL) for indicated times. Phosphorylation of JNK at Thr183/Tyr185 was analyzed by immunoblotting. B, Quantification of JNK phosphorylation that was normalized to the expression of total JNK. *P<0.01 compared with the respective time control. C, HUVEC was transfected with either pcDNA3.1 or PB1-GFP and stimulated with TNFα (10 ng/mL)+cyclohexamide (CHX) (10 μg/mL) for 6 hours. The cleavage of caspase 3 was analyzed by immunoblotting. D, Quantification of cleaved form of caspase 3 that was normalized to the expression of actin. *P<0.01.
Inhibition of p62 Binding to PKCζ Reduces EC Apoptosis
Activation of PKCζ induces EC apoptosis.2 Thus, as a pathological outcome of p62, we examined whether p62 is required for EC apoptosis. First, we examined whether the inhibition of p62–PKCζ interaction would affect cell viability using an MTT assay. In scrambled siRNA- or pcDNA3.1-transfected control groups, TNFα+CHX treatment induced cell death (Figure 6A and 6D; Figure IVA in the online-only Data Supplement). However, inhibition of p62–PKCζ interaction by either p62 siRNA (J-010230-05; Figure 6A or D-010230-03; Figure IVA in the online-only Data Supplement) or PB1-GFP (Figure 6D) increased cell viability. To show that the cell death was caused by apoptosis, 4′-6-diamidino-2-phenylindole–stained cell nuclei were analyzed, and the percentage of apoptotic cell death was calculated under fluorescence microscopy. TNFα+CHX treatment increased apoptosis in scrambled siRNA- or pcDNA3.1-transfected HUVEC. However, the number of apoptotic cells was significantly reduced in p62 siRNA–transfected (J-010230-05; Figure 6B and 6C or D-010230-03; Figure IVB and IVC in the online-only Data Supplement) or PB1-GFP–transfected HUVEC (Figure 6E and 6F). These results establish the critical role of p62 in PKCζ-mediated EC apoptosis.
Figure 6.

Inhibition of p62 binding to protein kinase C (PKC) ζ reduces endothelial cell (EC) apoptosis. A and D, Human umbilical vein EC (HUVEC) was transfected with either scrambled or p62 small interfering RNA (siRNA) (J-010230-05; A) or either pcDNA3.1 or Phox/Bem1p (PB1)-GFP (D) and then stimulated with tumor necrosis factor (TNF) α (10 ng/mL)+cyclohexamide (CHX) (10 μg/mL). After 24 hours, 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide assay was performed as described in Materials and Methods section. *P<0.01. B, C, E, and F, HUVEC was transfected with either scrambled or p62 siRNA (J-010230-05; B, C) or either pcDNA3.1 or PB1-GFP (D, E) and then stimulated with TNFα (10 ng/mL)+CHX (10 μg/mL). After 24 hours, cells were fixed, stained with 4′-6-diamidino-2-phenylindole (DAPI), and the number of apoptotic cells (indicated by white arrow) was counted under fluorescence microscopy (B, E). Apoptotic nuclear bodies were enumerated and expressed as a percentage of total nuclear counts (C, F). *P<0.01 and **P<0.05.
Discussion
The major finding of this study is that the binding of p62 to PKCζ is critical for the activation of PKCζ, as well as downstream JNK activation, caspase 3 cleavage, and EC apoptosis. Specifically, we showed that binding of p62 to PKCζ occurred via the PB1 domains as shown by inhibition with the specific p62 PB1-GFP peptide. Based on our observations, we propose a new pathway for TNFα-mediated EC apoptosis (Figure V in the online-only Data Supplement). In this model, TNFα increases PB1 domain–dependent interaction between p62 and PKCζ, probably changing the structure of PKCζ (eg, unfolding) and enabling activation of PKCζ. Subsequently, JNK activation and caspase 3 cleavage occur to induce EC apoptosis.
Previous studies have described a canonical mechanism of TNFα-induced apoptosis. Activation of TNFα receptor 1 by TNFα recruits TNFα receptor 1–associated death domain protein, which serves as a platform to subsequently recruit Fas-associated death domain and activate the initiator caspase, caspase 8.24,25 Then executioner caspase, caspase 3 is cleaved, leading to cell apoptosis. Previously, we showed that PKCζ was required for TNFα-mediated EC apoptosis via activation of caspase 3.1,8 There is 1 study which showed that p62 regulated caspase 8 activity.26 However, the present study is the first to show that p62 binding to PKCζ via PB1 domains mediates caspase 8 and caspase 3 activation, leading to EC apoptosis.
Another possible mechanism by which PKCζ could promote EC apoptosis is by inhibiting NF-κB activity, because NF-κB downstream gene expression has been shown to inhibit TNFα-induced apoptosis.27–29 Specifically, it has been reported by the laboratory of Moscat and Rahman that PKCζ is necessary for ligand-mediated phosphorylation of p65 (RelA) on serine 311, which activates NF-κB.30,31 Therefore, we examined the involvement of NF-κB pathway in p62-PKCζ–dependent EC apoptosis. First, we depleted PKCζ and examined the phosphorylation of p65 Ser311. Surprisingly, we found that the depletion of PKCζ enhanced the TNFα-induced phosphorylation of p65 Ser311 (Figure VIA in the online-only Data Supplement). To confirm that NF-κB was activated, we tested the effect of PKCζ depletion on cellular Fas-associated death domain-like interleukin-1β-converting enzyme-like inhibitory protein mRNA expression. As shown in Figure VIB in the online-only Data Supplement, PKCζ-depleted HUVEC showed a higher level of cellular Fas-associated death domain-like interleukin-1β-converting enzyme-like inhibitory protein mRNA in response to TNFα+CHX treatment. Because cellular Fas-associated death domain-like interleukin-1β-converting enzyme-like inhibitory protein expression depends on NF-κB activation,32 these data support the concept that the NF-κB pathway is enhanced in PKCζ-depleted HUVEC. We believe that our results may differ from those reported by Moscat and Rahman as a result of the use of different cell types and stimuli in previous studies (TNFα-stimulated fibroblast30 or lipopolysaccharide-exposed and cigarette smoke–exposed macrophages/monocytes),31 compared with the present study (TNFα-stimulated EC). Consistent with the results of PKCζ depletion, when PKCζ activity was suppressed by either p62 depletion or PB1-GFP transfection, there was a similar increase in p65 Ser311 phosphorylation (Figure VIC and VID in the online-only Data Supplement). These results suggest that inhibition of NF-κB by p62 PB1 domain–mediated PKCζ activation may enhance the TNFα-induced apoptosis. This seems to be another possible mechanism for PKCζ-regulated EC apoptosis and requires more study to fully understand this pathway.
The precise mechanism by which TNFα promotes p62-dependent activation of PKCζ is unknown. Although a direct interaction is possible, a complex that includes TNFα receptor 1 adaptor proteins such as TNF receptor-associated factor 6 may also be necessary.33,34 Our results suggest that the interaction between p62 and PKCζ is direct, because the transfection of GFP-tagged p62 PB1 domain inhibits activation of PKCζ, and there is no PB1 domain in TNF receptor-associated factor 6. Recently, pulmonary arteries from both p62 knockout mice and PKCζ knockout mice showed reduced Ca2+ influx,35 suggesting the similar function of p62 and PKCζ in the modulation of voltage-gated potassium channel function. Furthermore, both p62 knockout mice18 and PKCζ knockout mice36 rarely develop allergic airway inflammation because of reduced production of Th2 cytokine, including interleukin-4 and interleukin-13, which play important roles in airway inflammation such as asthma.37 This supports the concept that p62 regulates PKCζ activity. Interestingly, it was reported that the interaction between PKCΙ and Par6 through a PB1–PB1 interaction is important for PKCΙ-dependent lung cancer cell growth. When their interaction was inhibited, lung cancer cell growth was suppressed because of reduced activation of the PKCΙ signaling cascade, including extracellular signal-regulated kinases-1/2 or Rac1.38–40 These studies show the importance of PB1–PB1 interaction for atypical PKC family activation and strengthen our conclusion that p62 is an important binding partner for the activation of PKCζ in EC.
In combination with previous studies showing the important function of PKCζ in EC dysfunction,1,6,8 our data suggest a key role for p62 in PKCζ-mediated EC apoptosis. Considering the potential proatherogenic function of PKCζ,1,2,6,8 we believe that p62, specifically the PB1 domain, could be a good target for the development of therapy to limit atherosclerosis.
Supplementary Material
Supplemental Figure I. Transfection of PB1-GFP.
After transfection of PB1-GFP, the expression of GFP was analyzed in randomly selected 5 areas under the Olympus BX51 fluorescent microscope. The representative image is shown.
Supplemental Figure II. Inhibitory effect of siRNA-mediated p62 depletion on TNFα-induced PKCζ signaling cascade.
p62 siRNA (Dharmacon RNA Technologies, D-010230-03, 80 nmol/L) was transfected into HUVEC as described in Materials and Methods. The cells were stimulated with TNFα (10 ng/mL) alone or TNFα (10 ng/mL) + CHX (10 μg/mL) for the indicated times and the phosphorylation of PKCζ (A) and JNK (B) and cleavage of caspase 3 (C) were analyzed by western blotting.
Supplemental Figure III. p62-PKCζ pathway regulates caspase 8 activation in response to TNFα + CHX.
Either PKCζ- (Dharmacon RNA Technologies, L-003526-00, 100 nmol/L; A) or p62- (Dharmacon RNA Technologies, J-010230-05, 80 nmol/L; B or D-010230-03, 80 nmol/L; C) depleted HUVEC were stimulated with TNFα (10 ng/mL) + CHX (10 μg/mL) for 6 hours and the cleavage of caspase 8 was analyzed by western blotting. (D) Either pcDNA3.1- or PB1-GFP-transfected HUVEC were stimulated with TNFα (10 ng/mL) + CHX (10 μg/mL) for 6 hours and the cleavage of caspase 8 was analyzed by western blotting.
Supplemental Figure IV. Inhibitory effect of siRNA-mediated p62 depletion on TNFα-induced apoptotic cell death.
p62 siRNA (Dharmacon RNA Technologies, D-010230-03, 80 nmol/L) was transfected into HUVEC as described in Materials and Methods. The cells were stimulated with TNFα (10 ng/mL) + CHX (10 μg/mL) for 24 hours and a MTT assay (A), DAPI staining (B; apoptotic cells were indicated by white arrow) and quantification of apoptotic nuclear body (C) were performed as described in Figure 6.
Supplemental Figure V. A model for the requirement of p62 and PKCζ interaction for the PKCζ signaling cascade that leads to endothelial cell apoptosis.
Supplemental Figure VI. PKCζ negatively regulates TNFα-induced phosphorylation of p65 Ser311.
(A) PKCζ (Dharmacon RNA Technologies, L-003526-00, 100 nmol/L) or scrambled siRNA were transfected into HUVEC which were then stimulated with TNFα (10 ng/mL) for the indicated times and phosphorylation of p65 Ser311 was analyzed by western blotting. (B) PKCζ or scrambled siRNA were transfected into HUVEC which were then stimulated with TNFα (10 ng/mL) + CHX (10 μg/mL) for the indicated times and reverse transcription (RT)-PCR was performed using a reverse transcription kit (Promega, Madison, WI) to measure c-FLIP expression. Following primers were used: forward 5′-TAAAACCACCAGCACCACAA-3′ and reverse 5′-CTACGTGTGGCCCGTATCTT-3′ for c-FLIP; forward 5′-CCAGAAGATGGAGGAAGCTG-3′ and reverse 5′-CGTCTACTGGAGGCTCTTGG-3′ for PKCζ; forward 5′-ACGGATTTGGTCGTATTGGG-3′ and reverse 5′-TGATTTTGGAGGGATCTCGC-3′ for GAPDH. (C) p62 (Dharmacon RNA Technologies, J-010230-05, 80 nmol/L) or scrambled siRNA were transfected into HUVEC which were then stimulated with TNFα (10 ng/mL) for the indicated times and phosphorylation of p65 Ser311 was analyzed by western blotting. (D) Either pcDNA3.1 or PB1-GFP transfected HUVEC were stimulated with TNFα (10 ng/mL) for the indicated times and phosphorylation of p65 Ser311 was analyzed by western blotting. Antibody against p-p65 (Ser311) was purchased from Novus Biologicals (Littleton, CO); anti-p65 from Cell Signaling Technology.
Acknowledgments
We thank Dr Mark Sowden for critical review of the article.
Sources of Funding This study was supported by a grant from National Institutes of Health to Drs Bradford C. Berk (HL-064839), Jun-ichi Abe (HL-064839), and Keigi Fujiwara (HL-064839) and from the American Heart Association to Dr Geun-Young Kim (Postdoctoral Fellowship 12POST9170009).
Footnotes
The online-only Data Supplement is available with this article at http://atvb.ahajournals.org/lookup/suppl/doi: 10.1161/ATVBAHA.112.300054/-/DC1.
Disclosures None.
References
- 1.Garin G, Abe J, Mohan A, Lu W, Yan C, Newby AC, Rhaman A, Berk BC. Flow antagonizes TNF-alpha signaling in endothelial cells by inhibiting caspase-dependent PKC zeta processing. Circ Res. 2007;101:97–105. doi: 10.1161/CIRCRESAHA.107.148270. [DOI] [PubMed] [Google Scholar]
- 2.Heo KS, Lee H, Nigro P, Thomas T, Le NT, Chang E, McClain C, Reinhart-King CA, King MR, Berk BC, Fujiwara K, Woo CH, Abe J. PKCζ mediates disturbed flow-induced endothelial apoptosis via p53 SUMOylation. J Cell Biol. 2011;193:867–884. doi: 10.1083/jcb.201010051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spindel ON, Yan C, Berk BC. Thioredoxin-interacting protein mediates nuclear-to-plasma membrane communication: role in vascular endothelial growth factor 2 signaling. Arterioscler Thromb Vasc Biol. 2012;32:1264–1270. doi: 10.1161/ATVBAHA.111.244681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rennier K, Ji JY. Shear stress regulates expression of death-associated protein kinase in suppressing TNFa-induced endothelial apoptosis. J Cell Physiol. 2012;227:2398–2411. doi: 10.1002/jcp.22975. [DOI] [PubMed] [Google Scholar]
- 5.Nigro P, Satoh K, O′Dell MR, Soe NN, Cui Z, Mohan A, Abe J, Alexis JD, Sparks JD, Berk BC. Cyclophilin A is an inflammatory mediator that promotes atherosclerosis in apolipoprotein E-deficient mice. J Exp Med. 2011;208:53–66. doi: 10.1084/jem.20101174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Magid R, Davies PF. Endothelial protein kinase C isoform identity and differential activity of PKCzeta in an athero-susceptible region of porcine aorta. Circ Res. 2005;97:443–449. doi: 10.1161/01.RES.0000179767.37838.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tricot O, Mallat Z, Heymes C, Belmin J, Lesèche G, Tedgui A. Relation between endothelial cell apoptosis and blood flow direction in human atherosclerotic plaques. Circulation. 2000;101:2450–2453. doi: 10.1161/01.cir.101.21.2450. [DOI] [PubMed] [Google Scholar]
- 8.Nigro P, Abe J, Woo CH, Satoh K, McClain C, O′Dell MR, Lee H, Lim JH, Li JD, Heo KS, Fujiwara K, Berk BC. PKCzeta decreases eNOS protein stability via inhibitory phosphorylation of ERK5. Blood. 2010;116:1971–1979. doi: 10.1182/blood-2010-02-269134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosse C, Linch M, Kermorgant S, Cameron AJ, Boeckeler K, Parker PJ. PKC and the control of localized signal dynamics. Nat Rev Mol Cell Biol. 2010;11:103–112. doi: 10.1038/nrm2847. [DOI] [PubMed] [Google Scholar]
- 10.Hirano Y, Yoshinaga S, Ogura K, Yokochi M, Noda Y, Sumimoto H, Inagaki F. Solution structure of atypical protein kinase C PB1 domain and its mode of interaction with ZIP/p62 and MEK5. J Biol Chem. 2004;279:31883–31890. doi: 10.1074/jbc.M403092200. [DOI] [PubMed] [Google Scholar]
- 11.Nakamura K, Johnson GL. PB1 domains of MEKK2 and MEKK3 interact with the MEK5 PB1 domain for activation of the ERK5 pathway. J Biol Chem. 2003;278:36989–36992. doi: 10.1074/jbc.C300313200. [DOI] [PubMed] [Google Scholar]
- 12.Lamark T, Perander M, Outzen H, Kristiansen K, Øvervatn A, Michaelsen E, Bjørkøy G, Johansen T. Interaction codes within the family of mammalian Phox and Bem1p domain-containing proteins. J Biol Chem. 2003;278:34568–34581. doi: 10.1074/jbc.M303221200. [DOI] [PubMed] [Google Scholar]
- 13.Sanchez P, De Carcer G, Sandoval IV, Moscat J, Diaz-Meco MT. Localization of atypical protein kinase C isoforms into lysosome-targeted endosomes through interaction with p62. Mol Cell Biol. 1998;18:3069–3080. doi: 10.1128/mcb.18.5.3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Puls A, Schmidt S, Grawe F, Stabel S. Interaction of protein kinase C zeta with ZIP, a novel protein kinase C-binding protein. Proc Natl Acad Sci USA. 1997;94:6191–6196. doi: 10.1073/pnas.94.12.6191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moscat J, Diaz-Meco MT, Wooten MW. Of the atypical PKCs, Par-4 and p62: recent understandings of the biology and pathology of a PB1-dominated complex. Cell Death Differ. 2009;16:1426–1437. doi: 10.1038/cdd.2009.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Durán A, Serrano M, Leitges M, Flores JM, Picard S, Brown JP, Moscat J, Diaz-Meco MT. The atypical PKC-interacting protein p62 is an important mediator of RANK-activated osteoclastogenesis. Dev Cell. 2004;6:303–309. doi: 10.1016/s1534-5807(03)00403-9. [DOI] [PubMed] [Google Scholar]
- 17.Wooten MW, Geetha T, Seibenhener ML, Babu JR, Diaz-Meco MT, Moscat J. The p62 scaffold regulates nerve growth factor-induced NF-kappaB activation by influencing TRAF6 polyubiquitination. J Biol Chem. 2005;280:35625–35629. doi: 10.1074/jbc.C500237200. [DOI] [PubMed] [Google Scholar]
- 18.Martin P, Diaz-Meco MT, Moscat J. The signaling adapter p62 is an important mediator of T helper 2 cell function and allergic airway inflammation. EMBO J. 2006;25:3524–3533. doi: 10.1038/sj.emboj.7601250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, Moscat J. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell. 2008;13:343–354. doi: 10.1016/j.ccr.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 20.Wang XQ, Nigro P, World C, Fujiwara K, Yan C, Berk BC. Thioredoxin interacting protein promotes endothelial cell inflammation in response to disturbed flow by increasing leukocyte adhesion and repressing Kruppel-like factor 2. Circ Res. 2012;110:560–568. doi: 10.1161/CIRCRESAHA.111.256362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pang J, Hoefen R, Pryhuber GS, Wang J, Yin G, White RJ, Xu X, O′Dell MR, Mohan A, Michaloski H, Massett MP, Yan C, Berk BC. G-protein-coupled receptor kinase interacting protein-1 is required for pulmonary vascular development. Circulation. 2009;119:1524–1532. doi: 10.1161/CIRCULATIONAHA.108.823997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jin ZG, Lungu AO, Xie L, Wang M, Wong C, Berk BC. Cyclophilin A is a proinflammatory cytokine that activates endothelial cells. Arterioscler Thromb Vasc Biol. 2004;24:1186–1191. doi: 10.1161/01.ATV.0000130664.51010.28. [DOI] [PubMed] [Google Scholar]
- 23.Slowik MR, Min W, Ardito T, Karsan A, Kashgarian M, Pober JS. Evidence that tumor necrosis factor triggers apoptosis in human endothelial cells by interleukin-1-converting enzyme-like protease-dependent and -independent pathways. Lab Invest. 1997;77:257–267. [PubMed] [Google Scholar]
- 24.Guicciardi ME, Gores GJ. Life and death by death receptors. FASEB J. 2009;23:1625–1637. doi: 10.1096/fj.08-111005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thorburn A. Death receptor-induced cell killing. Cell Signal. 2004;16:139–144. doi: 10.1016/j.cellsig.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 26.Jin Z, Li Y, Pitti R, Lawrence D, Pham VC, Lill JR, Ashkenazi A. Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell. 2009;137:721–735. doi: 10.1016/j.cell.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 27.Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 28.Beg AA, Baltimore D. An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 29.Mi J, Zhang X, Liu Y, Reddy SK, Rabbani ZN, Sullenger BA, Clary BM. NF-kappaB inhibition by an adenovirus expressed aptamer sensitizes TNFalpha-induced apoptosis. Biochem Biophys Res Commun. 2007;359:475–480. doi: 10.1016/j.bbrc.2007.05.125. [DOI] [PubMed] [Google Scholar]
- 30.Duran A, Diaz-Meco MT, Moscat J. Essential role of RelA Ser311 phosphorylation by zetaPKC in NF-kappaB transcriptional activation. EMBO J. 2003;22:3910–3918. doi: 10.1093/emboj/cdg370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yao H, Hwang JW, Moscat J, Diaz-Meco MT, Leitges M, Kishore N, Li X, Rahman I. Protein kinase C zeta mediates cigarette smoke/aldehydeand lipopolysaccharide-induced lung inflammation and histone modifications. J Biol Chem. 2010;285:5405–5416. doi: 10.1074/jbc.M109.041418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Benayoun B, Baghdiguian S, Lajmanovich A, Bartoli M, Daniele N, Gicquel E, Bourg N, Raynaud F, Pasquier MA, Suel L, Lochmuller H, Lefranc G, Richard I. NF-kappaB-dependent expression of the antiapoptotic factor c-FLIP is regulated by calpain 3, the protein involved in limb-girdle muscular dystrophy type 2A. FASEB J. 2008;22:1521–1529. doi: 10.1096/fj.07-8701com. [DOI] [PubMed] [Google Scholar]
- 33.Wooten MW, Seibenhener ML, Mamidipudi V, Diaz-Meco MT, Barker PA, Moscat J. The atypical protein kinase C-interacting protein p62 is a scaffold for NF-kappaB activation by nerve growth factor. J Biol Chem. 2001;276:7709–7712. doi: 10.1074/jbc.C000869200. [DOI] [PubMed] [Google Scholar]
- 34.Sanz L, Diaz-Meco MT, Nakano H, Moscat J. The atypical PKC-interacting protein p62 channels NF-kappaB activation by the IL-1-TRAF6 pathway. EMBO J. 2000;19:1576–1586. doi: 10.1093/emboj/19.7.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moreno L, Frazziano G, Cogolludo A, Cobeño L, Tamargo J, Perez-Vizcaino F. Role of protein kinase Czeta and its adaptor protein p62 in voltage-gated potassium channel modulation in pulmonary arteries. Mol Pharmacol. 2007;72:1301–1309. doi: 10.1124/mol.107.037002. [DOI] [PubMed] [Google Scholar]
- 36.Martin P, Villares R, Rodriguez-Mascarenhas S, Zaballos A, Leitges M, Kovac J, Sizing I, Rennert P, Márquez G, Martínez-A C, Diaz-Meco MT, Moscat J. Control of T helper 2 cell function and allergic airway inflammation by PKCzeta. Proc Natl Acad Sci USA. 2005;102:9866–9871. doi: 10.1073/pnas.0501202102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Renauld JC. New insights into the role of cytokines in asthma. J Clin Pathol. 2001;54:577–589. doi: 10.1136/jcp.54.8.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Regala RP, Weems C, Jamieson L, Copland JA, Thompson EA, Fields AP. Atypical protein kinase Ciota plays a critical role in human lung cancer cell growth and tumorigenicity. J Biol Chem. 2005;280:31109–31115. doi: 10.1074/jbc.M505402200. [DOI] [PubMed] [Google Scholar]
- 39.Erdogan E, Lamark T, Stallings-Mann M, Lee Jamieson, Pellecchia M, Pellechia M, Thompson EA, Johansen T, Fields AP. Aurothiomalate inhibits transformed growth by targeting the PB1 domain of protein kinase Ciota. J Biol Chem. 2006;281:28450–28459. doi: 10.1074/jbc.M606054200. [DOI] [PubMed] [Google Scholar]
- 40.Regala RP, Thompson EA, Fields AP. Atypical protein kinase C iota expression and aurothiomalate sensitivity in human lung cancer cells. Cancer Res. 2008;68:5888–5895. doi: 10.1158/0008-5472.CAN-08-0438. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure I. Transfection of PB1-GFP.
After transfection of PB1-GFP, the expression of GFP was analyzed in randomly selected 5 areas under the Olympus BX51 fluorescent microscope. The representative image is shown.
Supplemental Figure II. Inhibitory effect of siRNA-mediated p62 depletion on TNFα-induced PKCζ signaling cascade.
p62 siRNA (Dharmacon RNA Technologies, D-010230-03, 80 nmol/L) was transfected into HUVEC as described in Materials and Methods. The cells were stimulated with TNFα (10 ng/mL) alone or TNFα (10 ng/mL) + CHX (10 μg/mL) for the indicated times and the phosphorylation of PKCζ (A) and JNK (B) and cleavage of caspase 3 (C) were analyzed by western blotting.
Supplemental Figure III. p62-PKCζ pathway regulates caspase 8 activation in response to TNFα + CHX.
Either PKCζ- (Dharmacon RNA Technologies, L-003526-00, 100 nmol/L; A) or p62- (Dharmacon RNA Technologies, J-010230-05, 80 nmol/L; B or D-010230-03, 80 nmol/L; C) depleted HUVEC were stimulated with TNFα (10 ng/mL) + CHX (10 μg/mL) for 6 hours and the cleavage of caspase 8 was analyzed by western blotting. (D) Either pcDNA3.1- or PB1-GFP-transfected HUVEC were stimulated with TNFα (10 ng/mL) + CHX (10 μg/mL) for 6 hours and the cleavage of caspase 8 was analyzed by western blotting.
Supplemental Figure IV. Inhibitory effect of siRNA-mediated p62 depletion on TNFα-induced apoptotic cell death.
p62 siRNA (Dharmacon RNA Technologies, D-010230-03, 80 nmol/L) was transfected into HUVEC as described in Materials and Methods. The cells were stimulated with TNFα (10 ng/mL) + CHX (10 μg/mL) for 24 hours and a MTT assay (A), DAPI staining (B; apoptotic cells were indicated by white arrow) and quantification of apoptotic nuclear body (C) were performed as described in Figure 6.
Supplemental Figure V. A model for the requirement of p62 and PKCζ interaction for the PKCζ signaling cascade that leads to endothelial cell apoptosis.
Supplemental Figure VI. PKCζ negatively regulates TNFα-induced phosphorylation of p65 Ser311.
(A) PKCζ (Dharmacon RNA Technologies, L-003526-00, 100 nmol/L) or scrambled siRNA were transfected into HUVEC which were then stimulated with TNFα (10 ng/mL) for the indicated times and phosphorylation of p65 Ser311 was analyzed by western blotting. (B) PKCζ or scrambled siRNA were transfected into HUVEC which were then stimulated with TNFα (10 ng/mL) + CHX (10 μg/mL) for the indicated times and reverse transcription (RT)-PCR was performed using a reverse transcription kit (Promega, Madison, WI) to measure c-FLIP expression. Following primers were used: forward 5′-TAAAACCACCAGCACCACAA-3′ and reverse 5′-CTACGTGTGGCCCGTATCTT-3′ for c-FLIP; forward 5′-CCAGAAGATGGAGGAAGCTG-3′ and reverse 5′-CGTCTACTGGAGGCTCTTGG-3′ for PKCζ; forward 5′-ACGGATTTGGTCGTATTGGG-3′ and reverse 5′-TGATTTTGGAGGGATCTCGC-3′ for GAPDH. (C) p62 (Dharmacon RNA Technologies, J-010230-05, 80 nmol/L) or scrambled siRNA were transfected into HUVEC which were then stimulated with TNFα (10 ng/mL) for the indicated times and phosphorylation of p65 Ser311 was analyzed by western blotting. (D) Either pcDNA3.1 or PB1-GFP transfected HUVEC were stimulated with TNFα (10 ng/mL) for the indicated times and phosphorylation of p65 Ser311 was analyzed by western blotting. Antibody against p-p65 (Ser311) was purchased from Novus Biologicals (Littleton, CO); anti-p65 from Cell Signaling Technology.