

Abstract
Development of a mild (35°C, no Brönsted acids) tandem Wacker-dehydrogenation of terminal olefins was accomplished using palladium(II) and hypervalent iodine co-catalysis. The reaction affords linear aryl and alkyl α,β- unsaturated ketones directly from readily available terminal olefins in good yields (average 75% per step) with excellent functional group tolerance, chemo- and stereoselectivities. The hypervalent iodine co-catalyst was found to be critical for dehydrogenation but was not effective as a stoichiometric oxidant.
Keywords: Dehydrogenation, Oxidation, C−H oxidation, Palladium, Hypervalent iodine, Tandem catalysis
C—H functionalization reactions,1 due to their capacity for directly installing valuable functionality onto readily available, robust hydrocarbon skeletons, stand to significantly streamline synthetic routes.2 In particular, terminal olefins are appealing starting materials and synthetic intermediates because of their abundance and tolerance of many standard acid-base organic reactions. The ability to selectively transform terminal olefins into ‘traditional’ polar functional groups, especially late in synthetic sequences, continues to be a frontier challenge in reaction development.3 As a classic example, the Wacker oxidation transforms terminal olefins into methyl ketones in complex molecule settings with outstanding selectivities.4 Additionally, Pd/sulfoxide-catalyzed C—H oxidation reactions have emerged that introduce O, N, and C (including C—C double bonds via dehydrogenation) functionality at the allylic position of terminal olefins with high functional group tolerance and chemo- and regioselectivity.5 We herein report a novel olefin/C—H oxidation reaction: Pd(II)/I(III)-catalyzed tandem Wacker oxidation-dehydrogenation that directly and selectively furnishes linear α,β -unsaturated ketones from terminal olefins.
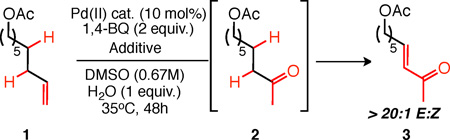
α,β -Unsaturated ketones are a versatile class of synthetic intermediates, readily engaging in Heck reactions, Michael additions, and cycloadditions. Traditionally, these intermediates are prepared via multi-step routes (e.g., sulfoxide or selenoxide elimination,6 Saegusa oxidation7) or from preoxidized starting materials (e.g., carbonyl olefination using stabilized ylides,8 carbonyl dehydrogenation using stoichiometric reagents). 9 Alternatively, we envisioned that a direct route would be possible from terminal olefins via a novel Pd(II)-catalyzed tandem process: Wacker oxidation to form a methyl ketone followed by Pd-catalyzed dehydrogenation (Figure 1).10 Significant progress has been made recently in the development of Pd(II)-catalyzed dehydrogenation reactions of cyclic ketones. 11,12 Despite this, however, no general method has been reported for linear ketone dehydrogenation, due to poor reactivity and competitive overoxidation of aliphatic substrates and their products.11f, 13, 14 To overcome these limitations, we hypothesized that a hypervalent iodine(III) reagent would be capable of generating an iodonium enolate from an intermediate methyl ketone (Figure 1). The iodine(III) reagent, well-precedented to undergo nucleophilic displacement/reduction,15 could serve to activate the ketone for exchange with the palladium (e.g. nucleophilic attack by the Pd(0) byproduct of Pd(II)-catalyzed Wacker oxidation) to provide a Pd(II) enolate, thereby overcoming the low inherent reactivity of linear ketones. Moreover, ketone activation via iodonium enolate formation would hopefully obviate the need for external Brönsted acids/high temperatures, and therefore limit undesired overoxidation. Herein, we report the discovery and development of a Pd(II)/hypervalent iodine-catalyzed tandem Wacker-dehydrogenation reaction of terminal olefins.
Figure 1.

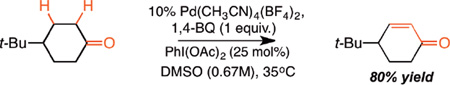
As shown in Table 1, Pd(II)-catalyzed oxidation of aliphatic terminal olefin 1 under mild conditions [Pd(CH3CN)4(BF4)2, 0.67M DMSO, 1,4-benzoquinone, 35°C] led to 68% of the Wacker product (2) with poor conversion to the α,β - unsaturated ketone 3 (13% yield, Entry 1). In accord with our hypothesis, addition of stoichiometric PhI(OAc)2 greatly improved the yield of 3 to synthetically useful levels (56% yield, Entry 2). Unexpectedly, however, lowering the amount of PhI(OAc)2 to substoichiometric levels led to no diminishment in reactivity, with 25 mol% proving optimal (Entries 3–4). Consistent with this, replacing one equivalent of 1,4- benzoquinone with PhI(OAc)2, to test if the I(III) reagent was a competent terminal oxidant for the dehydrogenation step, led to significantly reduced conversion to 3 (Entry 5). Other common aryl iodonium(III) or iodonium(V) reagents led to comparable results (Entry 6, also Supporting Information). Notably, 2-iodoxybenzoic acid (IBX), an iodine(V) reagent known to directly dehydrogenate ketones in DMSO at elevated temperatures,9c is a competent catalytic additive but furnished dehydrogenation product 3 from ketone 2 sluggishly under these mild conditions, with or without palladium (Entries 6–8). Finally, other previously reported palladium(II) dehydrogenation catalysts5g, 11e, 11f as well as other common palladium salts were inferior to Pd(CH3CN)4(BF4)2 for the tandem process (Entries 9–13), while removing the palladium catalyst entirely led to complete elimination of catalysis (Entry 14).
Table 1.
Reaction Development
 | ||||
|---|---|---|---|---|
| entry | SM | catalyst | additive | yield 3a |
| 1 | 1 | Pd(CH3CN)4(BF4)2 | -- | 13% (68% 2)b |
| 2 | 1 | Pd(CH3CN)4(BF4)2 | 100% PhI(OAc)2 | 56% |
| 3 | 1 | Pd(CH3CN)4(BF4)2 | 25% PhI(OAc)2 | 59% (55%c) |
| 4 | 1 | Pd(CH3CN)4(BF4)2 | 10% PhI(OAc)2 | 38% |
| 5 | 1 | Pd(CH3CN)4(BF4)2 | 100% PhI(OAc)2 1 equiv. BQ | 25% |
| 6 | 1 | Pd(CH3CN)4(BF4)2 | 25% IBX | 58% |
| 7 | 2 | Pd(CH3CN)4(BF4)2 | 100% IBX No BQ | 23% |
| 8 | 2 | -- | 100% IBX No BQ | 8% |
| 9 | 1 | Pd(OAc)2 | 25% PhI(OAc)2 | 3% |
| 10 | 1 | Pd(TFA)2 | 25% PhI(OAc)2 | 36% |
| 11 | 1 | Pd(TFA)2 4,5-diazafluorenone | 25% PhI(OAc)2 | 35% |
| 12 | 1 | Pd(OAc)2 1,2-bis(benzylsulfinylethane) | 25% PhI(OAc)2 | trace |
| 13d | 1 | Pd(TFA)2 DMSO | 25% PhI(OAc)2 | -- |
| 14 | 1 | No Pd(II) | 25% PhI(OAc)2 | -- |
Determined by GC, average of two runs at 0.1 mmol, relative to standard curve, external standard: nitrobenzene.
Yield of Wacker product 2 shown in parantheses.
Average isolated yield of 3 shown in parantheses (two runs at 0.3 mmol).
0.67 M in AcOH.
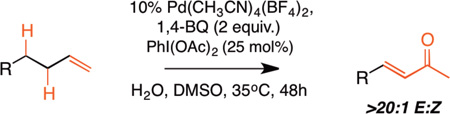
We next examined the scope of the tandem Wacker-dehydrogenation reaction. As shown in Table 2, electron-rich and electron-poor butenylated arenes delivered the desired α,β -unsaturated ketones 4–7 in good yields (Entries 1–4). Ortho- substitution on the arene was tolerated (Entry 5) as was a bioactive chromene functionality in benzopyran 9 (Entry 6). Notably, non-activated, aliphatic substrates also underwent tandem oxidation in good yields under these mild reaction conditions (average 75% yield per step). It is significant to note that these are the highest yields reported to date for direct, catalytic dehydrogenation of this substrate class.14 A range of oxygen and nitrogen functionality are well-tolerated in the α - olefin substrates (i.e., benzoates, phthalimides, amides, esters, benzyl ethers; Entries 7–12). Amides and esters did not undergo competitive dehydrogenation, demonstrating that the dehydrogenation reaction is chemoselective for ketones over other common carbonyl functionality (Entries 10–11). This lack of reactivity has been attributed to the higher pKa value of the α −C—H bonds of esters and amides relative to ketone functionality. 11f A γ-stereocenter did not suffer epimerization in 15 despite its potential lability under enolizable reaction conditions; similarly, 16 retained the trans stereochemistry found within the olefin starting material (Entries 12–13). A disubstituted cyclohexene and an acetate enol ether were both well-tolerated, highlighting the predictable selectivity of the Wacker reaction for terminal olefins (Entries 14–15). Estrone derivative 19 was isolated in 57% yield; the tandem reaction was tolerant of a benzyl ether and an electron-rich aromatic and proceeded next to the hindered D ring of the steroid core (Entry 16). While some steric hindrance is tolerated, β- disubstituted olefins, such as allylcyclohexane, underwent facile Wacker oxidation (85% yield) but failed to undergo significant dehydrogenation with only trace quantities of unsaturated ketone 20 detected (Entry 17). Finally, the dehydrogenation reaction proceeds with comparable efficiency starting from the ketone intermediate and is not limited to linear substrates (eq. 1). Taken together, our results demonstrate that the Pd(II)/PhI(OAc)2-catalyzed tandem Wacker-dehydrogenation readily converts a range of terminal olefins directly into α,β -unsaturated ketones in good yields, with minimal overoxidation and good functional group tolerance.
 |
(1) |
Table 2.
PdII/Phl(OAc)2catalyzed Wacker-dehydrogenation
 | |||
|---|---|---|---|
| entry | unsaturated ketone product |
isolated yielda,b |
|
| 1 | 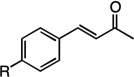 |
R=H,4 | 68% |
| 2 | R=OMe,5 | 69% | |
| 3 | R=Br,6 | 63% | |
| 4 | R=CF3,7 | 61% | |
| 5 | 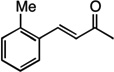 |
8 | 63% |
| 6 | 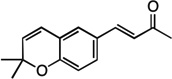 |
9 | 65% |
| 7 | 10 | 61%(58%c) | |
| 8 | 11 | 54% | |
| 9 | 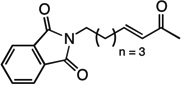 |
12 | 57% |
| 10 | 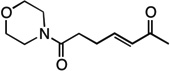 |
13 | 53% |
| 11 | 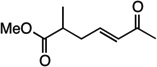 |
14 | 56% |
| 12 | 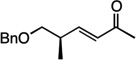 |
15 | 61% |
| 13 | 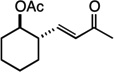 |
16 | 66% |
| 14 | 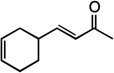 |
17 | 59% |
| 15 | 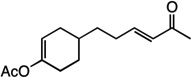 |
18 | 51% |
| 16 | 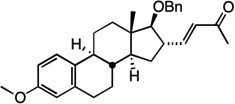 |
19 | 57%d |
| 17 | 20 | tracee | |
Average of two runs, typically at 0.3 mmol.
Unreacted Wacker product typically remained in 10–25% yield, accounting for much of the remaining mass balance.
Isolated yield of 1.0 mmol reaction.
Unreacted Wacker product was recycled 1×.
Wacker product was formed in 85% GC yield.
A critical question raised by these studies relates to the mechanism by which catalytic hypervalent iodine reagents promote the Pd(II)-Wacker/dehydration reaction. To probe this, we monitored the reaction progress of terminal olefin 1 over time using GC analysis. Wacker oxidation occurred rapidly, with full conversion to methyl ketone 2 accomplished within 3 hours, and was not influenced by the addition of PhI(OAc)2. The overall kinetic profile of terminal olefin to α,β-unsaturated ketone was monitored with and without PhI(OAc)2 (Figure 2). A significant increase in the rate of dehydrogenation was observed in the presence of 25 mol% PhI(OAc)2 and did not change significantly upon increasing to stoichiometric PhI(OAc)2 (1 equiv.). Additionally, PhI was never observed by 1H NMR during dehydrogenation.16 Collectively, this data supports the role of PhI(OAc)2 as a dehydrogenation catalyst, not a terminal oxidant and therefore excludes mechanisms that generate PhI (e.g., Figure 1).17 Future studies will seek to better understand the reaction mechanism and the role of the hypervalent iodine catalyst.18
Figure 2.

In summary, we herein report the development of a Pd(II)/hypervalent iodine-catalyzed tandem Wacker-dehydrogenation reaction of terminal olefins. This reaction provides for the expedient and selective synthesis of a broad range of linear aryl and alkyl α,β -unsaturated ketones directly from terminal olefins. Key to this reaction’s broad scope was the discovery of an iodonium(III) co-catalyst that is critical for facilitating dehydrogenation under mild conditions (35°C, no Brönsted acids).
Supplementary Material
ACKNOWLEDGMENT
We gratefully acknowledge Dr. Dustin J. Covell for preliminary investigations of Pd(II)-catalyzed ketone dehydrogenation reactions. We thank Iulia I. Strambeanu for checking the experimental procedure in Table 2, Entry 3.
Funding Sources
Financial support was provided by the NIH/NIGMS (2R01 GM 076153B) and generous gifts from Novartis, Bristol-Myers Squibb and Boehringer Ingelheim. We also thank Johnson Matthey for generous gifts of palladium.
Footnotes
ASSOCIATED CONTENT
Experimental procedures, characterization data, and copies of 1H and 13C NMR spectra for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org
REFERENCES
- 1.(a) Crabtree RH. J. Chem. Soc. Dalton Trans. 2001:2437. [Google Scholar]; (b) Labinger JA, Bercaw JE. Nature. 2002;417:507. doi: 10.1038/417507a. [DOI] [PubMed] [Google Scholar]; (c) Dick AR, Sanford MS. Tetrahedron. 2006;62:2439. [Google Scholar]; (d) Giri R, Shi B-F, Engle KM, Maugel N, Yu J-Q. Chem. Soc. Rev. 2009;38:3242. doi: 10.1039/b816707a. [DOI] [PubMed] [Google Scholar]; (e) White MC. Science. 2012;335:807. doi: 10.1126/science.1207661. [DOI] [PubMed] [Google Scholar]; (f) White MC. Synlett. 2012;23:2746. [Google Scholar]
- 2.For examples of synthetic streamlining using C—H functionalization: Hinman A, Du Bois J. J. Am. Chem. Soc. 2003;125:11510. doi: 10.1021/ja0368305. Wender PA, Hilinski MK, Mayweg AVW. Org. Lett. 2005;7:79. doi: 10.1021/ol047859w. Fraunhoffer KJ, Bachovchin DA, White MC. Org. Lett. 2005;7:223. doi: 10.1021/ol047800p. Covell DJ, Vermeulen NA, Labenz NA, White MC. Angew. Chem. Int. Ed. 2006;45:8217. doi: 10.1002/anie.200603321. Stang EM, White MC. Nat. Chem. 2009;1:547. doi: 10.1038/nchem.351. Vermeulen NA, Delcamp JH, White MC. J. Am. Chem. Soc. 2010;132:11323. doi: 10.1021/ja104826g.
- 3.For recent examples, see: Weiner B, Baeza A, Jerphagnon T, Feringa BL. J. Am. Chem. Soc. 2009;131:9473. doi: 10.1021/ja902591g. Michel BW, McCombs JR, Winkler A, Sigman MS. Angew. Chem. Int. Ed. 2010;131:9473. doi: 10.1002/anie.201004156.
- 4.Keith JA, Henry PM. Angew. Chem. Int. Ed. 2009;48:9038. doi: 10.1002/anie.200902194. [DOI] [PubMed] [Google Scholar]
- 5.(a) Chen MS, White MC. J. Am. Chem. Soc. 2004;126:1346. doi: 10.1021/ja039107n. [DOI] [PubMed] [Google Scholar]; (b) Vermeulen NA, Delcamp JH. J. Am. Chem. Soc. 2010;132:11323. doi: 10.1021/ja104826g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Gormisky PE, White MC. J. Am. Chem. Soc. 2011;133:12584. doi: 10.1021/ja206013j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Fraunhoffer KJ, White MC. J. Am. Chem. Soc. 2007;129:7274. doi: 10.1021/ja071905g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Reed SA, White MC. J. Am. Chem. Soc. 2008;130:3316. doi: 10.1021/ja710206u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Young AJ, White MC. J. Am. Chem. Soc. 2008;130:14090. doi: 10.1021/ja806867p. [DOI] [PubMed] [Google Scholar]; (g) Stang EM, White MC. J. Am. Chem. Soc. 2011;133:14892. doi: 10.1021/ja2059704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Reich HJ, Wollowitz S. Org. React. 1993;44:1. [Google Scholar]; (b) Trost BM. Chem. Rev. 1978;78:363. [Google Scholar]
- 7.Ito Y, Hirao T, Saegusa T. J. Org. Chem. 1978;43:1011. [Google Scholar]; For a catalytic version of this reaction, see: Larock RC, Hightower TR, Kraus GA, Hahn P, Zheng D. Tetrahedron Lett. 1995;36:2423. Yu JQ, Wu HC, Corey EJ. Org. Lett. 2005;7:1415. doi: 10.1021/ol050284y.
- 8.Maryanoff BE, Reitz AB. Chem. Rev. 1989;89:863. [Google Scholar]
- 9.(a) Walker D, Hiebert JD. Chem. Rev. 1967;67:153. doi: 10.1021/cr60246a002. [DOI] [PubMed] [Google Scholar]; (b) Nicolaou KC, Zhong Y-L, Baran PS. J. Am. Chem. Soc. 2000;122:7596. [Google Scholar]; (c) Nicolaou KC, Montagnon T, Baran PS, Zhong Y-L. J. Am. Chem. Soc. 2002;124:2245. doi: 10.1021/ja012127+. [DOI] [PubMed] [Google Scholar]; (d) Nicolaou KC, Montagnon T, Baran PS. Angew. Chem. Int. Ed. 2002;41:993. doi: 10.1002/1521-3773(20020315)41:6<993::aid-anie993>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]; For a catalytic version of this reaction for cyclic ketones using oxone as stoichiometric oxidant, see: Uyanik M, Akakura M, Ishihara K. J. Am. Chem. Soc. 2009;131:251. doi: 10.1021/ja807110n.
- 10.For two reviews of transition metal-catalyzed dehydrogenation of alkanes: Dobereiner GE, Crabtree RH. Chem. Rev. 2010;110:681. doi: 10.1021/cr900202j. Choi J, MacArthur AHR, Brookhart M, Goldman AS. Chem. Rev. 2011;111:1761. doi: 10.1021/cr1003503.
- 11.For a review of Pd(II)-mediated carbonyl dehydrogenation: Muzart J. Eur. J. Org. Chem. 2010:3779. For early examples of this reactivity, see: Theissen RJ. J. Org. Chem. 1971;36:752. Muzart J, Pete JP. J. Mol. Catal. 1982;15:373. For recent examples with greatly improved scope, catalytic efficiency, and selectivity, see: Izawa Y, Pun D, Stahl SS. Science. 2011;333:209. doi: 10.1126/science.1204183. Diao T, Stahl SS. J. Am. Chem. Soc. 2011;133:14566. doi: 10.1021/ja206575j. Diao T, Wadzinski TJ, Stahl SS. Chem. Sci. 2012;3:887. doi: 10.1039/C1SC00724F. Gao W, He Z, Qian Y, Zhao J. Chem. Sci. 2012;3:883. Tokunaga M, Harada S, Iwasawa T, Obora Y, Tsuji Y. Tetrahedron Lett. 2007;48:6860.
- 12.For a rare example of Ir-catalyzed dehydrogenation of cyclic ketones, see: Zhang XW, Wang DY, Emge TJ, Goldman AS. Inorg. Chim. Acta. 2011;369:253.
- 13.For examples of dehydrogenation of linear aliphatic aldehydes: Zhu J, Liu J, Ma RQ, Xie HX, Li J, Jiang HL, Wang W. Adv. Synth. Catal. 2009;351:1229. Liu J, Zhu J, Jiang HL, Wang W, Li J. Chem.— Asian J. 2009;4:1712. doi: 10.1002/asia.200900238.
- 14.The previous highest report described the convesion of 2- octanone into oct-3-en-2-one in 17% yield using a Pd(TFA)2/ 4,5-diazafluorenone catalyst (ref. 11f).
- 15.(a) Zhdankin VV, Stang PJ. Chem. Rev. 2008;108:5299. doi: 10.1021/cr800332c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Moriarty RM, Prakash O. Org. React. 1999;54:273. [Google Scholar]; (c) Koser GF. In: In Hypervalent Iodine Chemistry: Modern Developments in Organic Synthesis. Wirth T, editor. Springer; 2003. p. 137. [Google Scholar]; (d) Merritt EA, Olofsson B. Synthesis. 2011:517. [Google Scholar]
- 16.No products clearly derived from PhI were observed by 1H NMR analysis of the crude reaction mixture (e.g. biaryls, benzene, arylated BQ). If 1,4-benzoquinone were acting to regenerate an I(III) reagent from PhI, thereby explaining our inability to observe PhI, dehydrogenation should proceed similarly beginning from either PhI or PhI(OAc)2 As expected, however, replacing 25 mol% PhI(OAc)2 with 25 mol% PhI and 100% AcOH led to only 15% yield of 3 (see SI).
- 17.Based on the well-precedented ability of hypervalent iodine reagents to oxidize organopalladium(II) intermediates, we considered the possibility of initial generation of a Pd(II) enolate followed by oxidation with the I(III)-based oxidant to provide a higher valent Pd(IV) enolate, with release of PhI (for a review, see: Deprez NR, Sanford MS. Inorg. Chem. 2007;46:1924. doi: 10.1021/ic0620337. )Alternatively, as proposed in Figure 1, an iodonium(III) enolate might undergo reductive displacement with Pd(0), affording a Pd(II) enolate and PhI. In either case, β-hydride elimination from a Pd enolate would release product and the I(III) reagent would function as a terminal oxidant. We disfavor both pathways because we have never observed PhI, the reaction requires stoichiometric 1,4-BQ, and I(III) reagents can be used sub-stoichiometrically.
- 18.A mechanism that cannot be excluded is iodonium(III) enolate formation (Figure 1) followed by a sigma-bond metathesis with Pd(II) to furnish a Pd(II)-enolate with no change in the oxidation state of the palladium or iodine. Examples of sigma bond metathesis have been proposed for silyl enol ethers with I(III) reagents: Chen K, Koser GF. J. Org. Chem. 1991;56:5764.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.