

Abstract
Chiral oxygenated molecules are pervasive in natural products and medicinal agents; however, their chemical syntheses often necessitate numerous, wasteful steps involving functional group and oxidation state manipulations. Herein a strategy for synthesizing a readily diversifiable class of chiral building blocks, allylic alcohols, through sequential asymmetric C—H activation/resolution is evaluated against the state-of-the-art. The C—H oxidation routes’ capacity to strategically introduce oxygen into a sequence and thereby minimize non-productive manipulations is demonstrated to effect significant decreases in overall step-count and increases in yield and synthetic flexibility.
Keywords: C–H oxidation, Allylic oxidation, Allylic alcohols, Enantioselective, Palladium, Sulfoxide
1. Introduction
Polyoxygenation in natural products and medicinal agents is ubiquitous and often confers specific biological function to small molecules. Most traditional organic sequences for assembling such motifs involve acid/base reactions to install the oxygenated functionality as the hydrocarbon core is being constructed. Though reliable, these routes require significant synthetic overhead, commonly in the form of protection/deprotection sequences, oxidation state changes, and functional group manipulations (FGMs). Our group and others have demonstrated that selective hydrocarbon C—H oxidation presents an alternative approach by directly installing oxidation when it is most synthetically appropriate and by significantly reducing the number of reactive functional groups that must be carried through and consequently manipulated in a sequence.1,2,3 Herein, we utilize an asymmetric branched allylic C—H oxidation in combination with enzymatic and small molecule resolution methods to interrogate the streamlining effect of such processes for the construction of chiral polyoxygenated motifs.4
Allylic alcohols such as those shown in Scheme 1 are prevalent, in part due to ease with which they can be further elaborated. Such motifs are particularly challenging to construct asymmetrically when the oxygen atom is remote from other functionality, rendering stereochemical relay approaches unviable.5,6 Most often, these products are obtained through a kinetic resolution of the racemic alcohol obtained by the addition of a vinyl carbanion to an aldehyde.5h-i Additionally, Sharpless asymmetric epoxidation (SAE) of linear allylic alcohols followed by two-step sequences to install the vinyl moiety can be employed.5f-g,7 In general, both of these strategies begin with preoxidized starting materials that are elaborated toward the target through carbanion based reactions that, due to low chemoselectivities, demand lengthy sequences of FGMs.
Scheme 1.
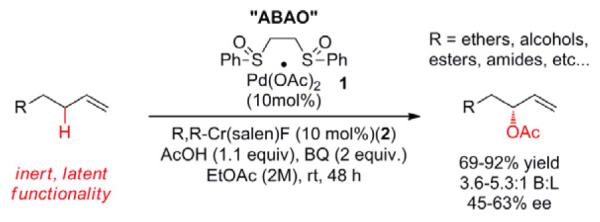
The asymmetric branched allylic oxidation reaction. R,R-Cr(Salen)F = (R,R)-N,N’-Bis(3,5-di-tert-butylsalicylidene)-1,2-cyclohexanediaminochromium (III) fluoride
Palladium (II)/sulfoxide catalysis has proven itself to be a general platform for allylic C—H esterification,8a-d amination,9b,c,e-g,j alkylation,10a-c and dehydrogenation11 of terminal olefins under mildly acidic, oxidative conditions. We previously reported a Pd(II)-bis-sulfoxide 1/chiral Cr(salen)F 2 catalyzed reaction for the asymmetric branched allylic C—H oxidation (ABAO) of terminal olefins (Scheme 1).4 This reaction proceeds under mild conditions with high functional group tolerance and chemoselectivity (primary alcohols and internal olefins are well tolerated) to furnish enantioenriched allylic alcohols in good yields. Although enantioselectivities are modest (43-63% ee) these represent the best to date for C–H activation of this challenging olefin class.12 We hypothesized that combining the asymmetric branched allylic oxidation (ABAO) with enzymatic and/or small-molecule-catalyst based resolutions would enable us to develop streamlined routes to chiral allylic alcohols by circumventing the FGMs of traditional carbanion based approaches. In addition, even the modest enantioenrichment afforded by the ABAO would lead to significant improvements in yield for subsequent resolution steps as compared to racemic approaches (e.g. at 60% ee, 80:20 e.r., only 20% of the material is lost in a highly efficient resolution).
2. Results
We began our explorations by targeting a prototypical bis-oxygenated chiral building block (−)-3, a precursor to the C19 – C26 fragment of the potential cancer therapeutic Bistramide A (Scheme 2).13a In the traditional carbanion based route, diol 4 is selectively protected at one terminus and then oxidized and subjected to a Horner-Wadsworth-Emmons olefination at the other end to generate ester 5.13c-d After reduction of the ester, SAE affords the epoxy alcohol in 93%ee.13e-f The primary alcohol is then converted to a halogen, which is eliminated with zinc to afford the desired allylic alcohol (−)-3 in a total of 7 steps and 34% overall yield.13a,14
Scheme 2.
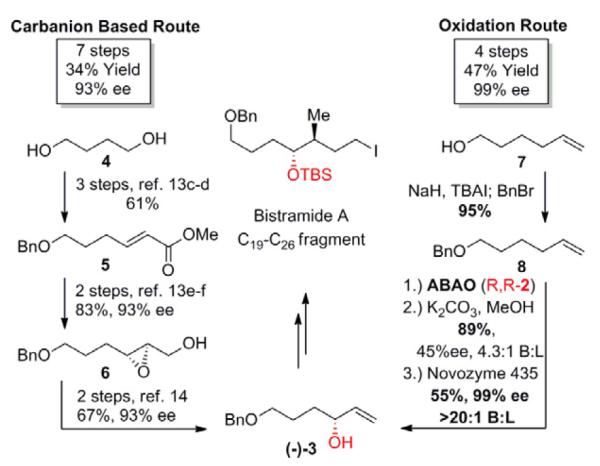
C–H oxidation vs carbanion based route to (−)-3, a precursor of the C19-C26 fragment of Bistramide A
Alternatively, using an oxidation driven route, after simple protection of commercially available 7, allylic C—H oxidation installs the oxygen functionality directly at the desired oxidation state, with significant enrichment toward the desired enantiomer (45% ee, 72:28 e.r.). Subsequent methanolysis and enzymatic resolution via acylation of the minor enantiomer by commercially available Novozyme 435™ gives enantiopure (−)-3 in a total of 4 steps and 47% overall yield. The C–H oxidation route significantly reduces the overall step count and improves the overall yield by reducing FGMs and oxidation state changes (Scheme 2). Additionally, we found that the C–H oxidation sequence requires only two purifications, further enabling rapid access to these chiral building blocks.15
Broad use of an enantioselective transformation requires that either enantiomer of the desired product can be produced with high enantioselectivity. Chiral Cr(salen)F 2 is readily available as either the (R,R)- or (S,S)-enantiomer, and careful selection of commercially available enzymes allows for enrichment of either stereoisomer of the product.16 To demonstrate this, we used Pd(II)/bis-sulfoxide 1/(S,S)-2 and resolution with the commercially available protease Subtilisin Carlsberg to generate (+)-3 in 99%ee and nearly identical overall yield to the route previously described for (−)-3 (Scheme 2, 3). This matches the flexibility of other catalyst-controlled approaches to these compounds, like the SAE which was utilized to make (+)-3 in a total synthesis of the potent biotoxin Azaspiracid A.13,14
Scheme 3.
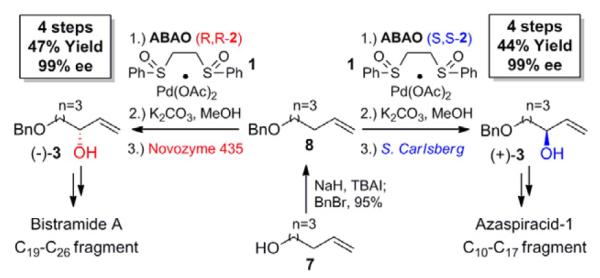
Stereoselective C–H oxidation approach to each enantiomer of alylic alcohol 3
We next sought to compare the asymmetric C–H oxidation route to chiral allylic alcohols to traditional resolution strategies. Ester (−)-9 was synthesized en route to the potent flower inducing factor 9R-KODA (Scheme 4).17 In order to avoid a lengthy sequence of FGMs, the carbanion based route started with ozonolysis of methyl oleate (10) followed by direct vinylation of the resultant aldehyde in the presence of an ester group. While the poor chemoselectivity of this approach resulted in a significant diminishment in overall yield, rapid access to (±)-9 was achieved. Subsequent enzymatic kinetic resolution afforded enantiopure (−)-9 in only three steps, however, with an overall yield of only 9%. Conversely, ABAO of commercially available α-olefin 12 followed by methanolysis and enzymatic resolution yielded (−)-9 with an equivalent step count and enantiopurity, but with a 5-fold increase in total yield (3 steps, 53% yield, 99%ee). High chemoselectivity and functional group tolerance is a general feature of all of the Pd(II)/bis-sulfoxide catalyzed allylic C–H oxidations reported to date and renders them well-suited towards introduction of oxidized functionality late in synthetic sequences.3c-g Importantly, this reaction sequence was run on gram scale with virtually no diminishment in yield (49% versus 53% overall yield).
Scheme 4.
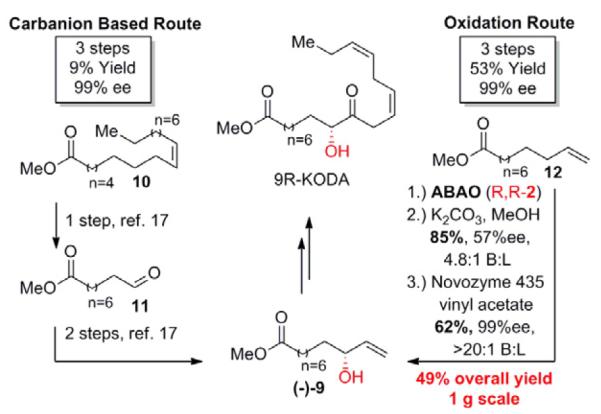
C–H oxidation vs carbanion based route for the synthesis of ester (−)-9
Due to the large number of commercially available α-olefin starting materials, the C–H oxidation sequences presented so far have begun with fully constructed carbon frameworks. We next sought to evaluate cases in which the α-olefin is not available to determine if a C–H oxidation route was still competitive with traditional approaches. Synthesis of allylic alcohol (−)-13 towards (+)-iso-6-cassine began from a commercially available, protected bromide 14 (Scheme 5).18 Formation of a Grignard reagent from bromide 14 followed by its addition into acrolein gave racemic alcohol (±)-13 in one step.18b Again, this carbanion-based carbonyl alkylation reaction proceeds with poor chemoselectivity, giving a mixture of 1,2 and 1,4 acrolein addition products. Enzymatic resolution yielded the desired (−)-13 in two steps and 19% overall yield.18a From the same commercially available bromide 14, a suitable starting material for C–H oxidation can be obtained directly by allylation, a C-C bond forming reaction without the chemoselectivity issues that limited the traditional route. Subjecting the resultant olefin to the allylic C–H oxidation/methanolysis/enzymatic resolution sequence affords the desired enantiopure alcohol (−)-13 in four steps and 46% overall yield, more than doubling the yield of the traditional route. This is enabled by the ease of installing inert α-olefin moieties through powerful allylation methods and the selective nature of these C–H oxidation reactions.
Scheme 5.
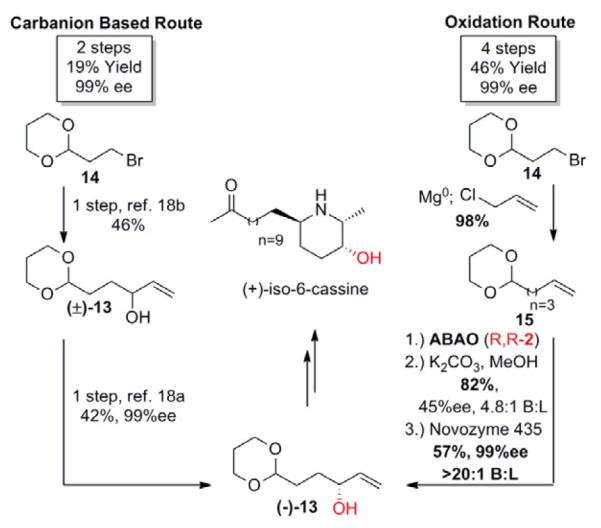
C–H Oxidation vs. carbanion based route for the synthesis of (+)-iso-6-cassine precursor (−)-13
We have demonstrated how the ABAO can be combined with enzymatic resolution to afford enantiopure allylic alcohols rapidly and in good yield. However, powerful, small-molecule catalyst-controlled enantioselective transformations can also be used to enrich the products of this C–H oxidation. We sought to explore this approach to chiral polyol synthesis through construction of the densely functionalized furan core of Goniothalesdiol. Due to its potent activity against mouse leukemia cells, a number of total syntheses of Goniothalesdiol (15) and its epimers have been undertaken.19b-l Generally these routes begin with chiral pool materials; however they lack the synthetic flexibility to rapidly access derivatives of 15. One approach initially developed by Gracza and co-workers to 15 proceeds through the tetrasubstituted furan core 16.19b We recognized that an unprecedented Pd(II)/bis-sulfoxide-catalyzed tandem ABAO/ oxidative Heck sequence could access this core structure rapidly (Scheme 6).20 Subsequent Sharpless asymmetric dihydroxylation (SAD) would generate separable diastereomers, allowing us to obtain enantiopure material for further reaction.21 Significantly, our de novo approach to 16 is quite flexible, allowing us to selectively control the stereochemistry at the 5, 6, and 7 positions of the core furan as well as easily vary the nature of the aryl substituent at position 7. While previous syntheses have relied primarily on C-C bond forming reactions, this plan involves a steady increase in complexity through hydrocarbon oxidations.
Scheme 6.
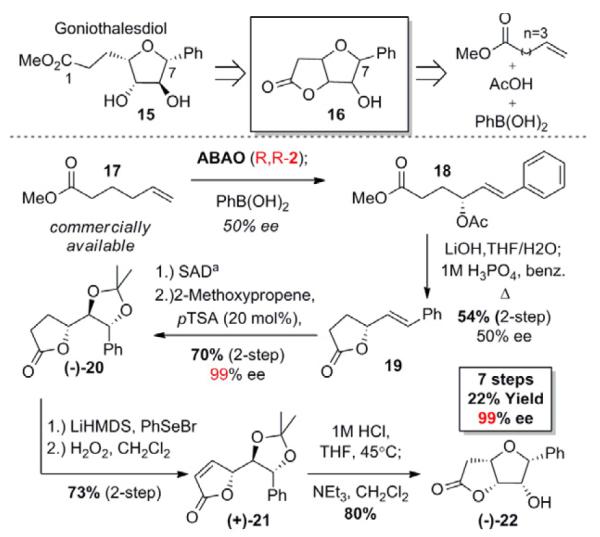
Enantioselective C–H oxidation approach to the core furan precursor (−)-22 of the goniothalesdiol family. aSAD = Sharpless Asymmetric Dihydroxylation. Conditions: K2OsO4-2H2O (1 mol%), (DHQD)2PHAL (5 mol%), K2CO3 (3 equiv.), NaHCO3 (3 equiv.) K3Fe(CN)6 (3 equiv.), MeSO2NH2 (1 equiv.), t-BuOH:H2O:CH2Cl2 10:10:1, 0°C-> rt
We decided upon furan core (−)-22 as an interesting target for this strategy because, to the best of our knowledge, 6-epi-Goniothalesdiol has yet be synthesized or evaluated medicinally. Our route began with the Pd(II)/bis-sulfoxide-catalyzed tandem ABAO/oxidative Heck sequence. Asymmetric branched allylic C—H oxidation (ABAO) reaction on methyl ester 17, followed by the addition of phenyl boronic acid directly furnished ester 18 that was taken on crude through hydrolysis and cyclization to give lactone 19 in 54% yield (two steps) and 50% ee (75:25 e.r.).
Sharpless asymmetric dihydroxylation (SAD) of 19 and subsequent ketal protection of the resultant diol, gave 70% of diastereomerically and enantiomerically pure (−)-20.22 Selenation /dehydroselenation of lactone (−)-20 afforded unsaturated γ-lactone (+)-21 in 73% yield. Deprotection of the acetonide followed by in situ DIPEA assisted cyclization afforded the desired tetrasubstituted pyran (−)-22 in 7 total steps and 22% overall yield. Previous C-C bond forming routes to this core structure proceeded in 8 and 10 steps with 6% and 9% overall yields respectively. This case study demonstrates the streamlining potential of hydrocarbon oxidations for synthesizing densely functionalized fragments.
3. Conclusions
In summary, we have established that the asymmetric branched allylic oxidation reaction4 can be combined with other enantioselective transformations to afford enantiopure, polyoxygenated allylic alcohols rapidly and in good yields. This C–H oxidation approach is demonstrated to be advantageous to commonly used resolution and de novo synthetic strategies that require significant numbers of protection/deprotection steps and functional group manipulations. Strategies such as these, fueled by novel reactions, have the greatest potential to both transform synthetic planning and streamline chemical synthesis.24
Experimental section
4.1 General information
All commercially obtained reagents were used as received; Achiral gas chromatographic (GC) analyses were performed on Agilent Technologies 6890N Series instrument equipped with FID detectors using a HP-5 (5%-Phenyl)-methylpolysiloxane column (30m, 0.32mm, 0.25μm). Chiral gas chromatographic (GC) analyses were performed on an Agilent Technologies 5890A Series instrument equipped with an FID detector using a J&W Scientific β-cyclodextrin column (30m, 0.25mm, 0.25μm). Thin-layer chromatography (TLC) was conducted with E. Merck silica gel 60 F254 precoated plates (0.25 mm) and visualized with UV, potassium permanganate, and ceric ammonium molybdate staining. Flash column chromatography was performed as described by Still et al. using EM reagent silica gel 60 (230-400 mesh).23 1H NMR spectra are reported in ppm using solvent as an internal standard (CDCl3 at 7.26 ppm). Data reported as: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, b = broad; coupling constant(s) in Hz; integration. Proton-decoupled 13C- NMR spectra were reported in ppm using solvent as an internal standard (CDCl3 at 77.0 ppm). IR spectra were recorded as thin films on NaCl plates on a Perkin-Elmer Spectrum BX and are reported in frequency of absorption (cm−1). High-resolution mass spectra were obtained at the University of Illinois Mass Spectrometry Laboratory.
4.2 Experimental section
4.2.1 General procedure for asymmetric branched allylic acetoxylation
A vial (8 mL borosilicate) was charged with the following: 1,2-Bis(phenylsulfinyl)ethane palladium(II) acetate (1) (10 mol%, 0.10 mmol, 50 mg); (1R,2R)-(−)-[1,2-Cyclohexanediamino-N,N’-bis(3,5-di-t-butylsalicylidene)] Chromium(III)F (R,R-2) (10 mol%, 0.10 mmol, 61.6 mg), 1,4-benzoquinone (2 equiv., 2.0 mmol, 216 mg), an activated 4Å MS bead (~30 mg), and a Teflon© stir bar. A separate vial (2 mL, borosilicate) was charged with the following: substrate (1.0 mmol), AcOH (1.1 equiv., 63 μL), and EtOAc (200 μL). The liquids were transferred to the solids via pipette and the vial rinsed with EtOAc (3 × 100 μL). After carefully stirring for 48 hrs at room temperature, the reaction mixture was transferred to a separatory funnel with ~3 mL EtOAc and diluted with hexanes (200 mL). The organic layer was rinsed with sat. aq. NaHSO3 (1 × 50 mL) and 5% aq. K2CO3 (2 × 50 mL). Caution should be taken when combining aqueous layers as carbon dioxide is evolved. The combined aqueous layers were back extracted with hexanes (100 mL). The combined organic layers were dried (MgSO4), filtered, and reduced in vacuo. The resulting oil was re-dissolved in hexanes (50 mL) and extracted again with 5% aq. K2CO3 (3 × 10 mL) to remove residual hydroquinone. The organic layer was again dried (MgSO4), filtered and reduced in vacuo to afford a clean mixture of allylic oxidation products and any unreacted starting material from which the B:L, yield, and conversions were determined (1H NMR). Unless otherwise noted, this material was taken forward without further purification, though each substrate was isolated at least once for characterization purposes. Reported yields and selectivities are an average of at least two runs. Enantiomeric excess was determined by chiral GC (β-cyclodextrin column), as compared to racemic standards generated through our standard branched oxidation chemistry.8a,b Slight variations in B:L ratios and ee’s were noted based on batch of Cr catalyst.
4.2.2 General procedure for cleavage of allylic acetates
To a 25 mL flask containing crude allylic acetate (1 mmol, assumed) was added MeOH (5 mL, 0.2 M) and potassium carbonate (0.276 g, 2 mmol). The reaction was vigorously stirred and monitored via thin layer chromatography (TLC). Upon completion, the reaction was transferred to a separatory funnel with methylene chloride (50 mL). Water (15 mL) was added, and the aqueous layer was extracted with methylene chloride (3 × 50 mL). The combined organics were washed with brine (1 × 10 mL), then dried (MgSO4), filtered, and reduced in vacuo. Products were then purified by standard SiO2 chromatography. While the branched and linear allylic alcohols were commonly separable, it was found that carrying them forward as a mixture had no detrimental effect as the subsequent resolution acylated the linear alcohol rapidly making its separation from branched alcohol trivial. Individual product yields and characterization are reported below.
4.2.2.1 (4R)-1-O-Benzyl-4-acetoxy-5-hexen-1,4-diol 3-OAc
Following the general procedure for the asymmetric branched allylic oxidation afforded: Run 1: 226 mg, 0.910 mmol, 91% yield; [B:L] = 4.7:1, [ee] = 44%. Run 2: 220 mg, 0.886 mmol, 89% yield; [B:L] = 3.8:1, [ee] = 45%. (β-cyclodextrin, 120°C isothermal, tR(R) = 64.98 min., tR(S) = 66.64 min.), [average yield: 90%.]; This material was taken forward without further purification. 1H NMR (500 MHz, CDCl3) δ 7.37-7.27 (m, 5H), 5.77 (ddd, J = 17.2, 10.4, 6.2 Hz, 1H), 5.28-5.15 (m, 3H), 4.50 (bs, 2H), 3.48 (t, J = 6.4 Hz, 2H), 2.06 (s, 3H), 1.75-1.60 (m, 4H); 13C NMR (125 MHz, CDCl3) δ 170.3, 138.4, 136.3, 128.3, 127.6, 127.5, 116.7, 74.5, 72.9, 69.8, 30.8, 25.4, 21.2; IR (neat, cm−1) 3087.8, 3063.9, 3031.2, 2940.3, 2857.9, 1737.7, 1647.0, 1496.0, 1454.1; HRMS (ESI) m/z calculated for C15H20O3Na [M + Na]+: 271.1310; found: 271.1302.
This material was then subjected to the standard procedure for cleavage of the allylic acetate which afforded allylic alcohol (±)-3 ready for subsequent resolution: Run 1: 185 mg, 0.897 mmol, 99% yield, [B:L] = 4.7:1. Run 2: 181 mg, 0.877 mmol, 99% yield; [B:L] = 3.8:1
4.2.2.2 Methyl (9R)-9-acetoxyundec-10-eneoate 9-OAc
The general procedure for the asymmetric branched allylic oxidation afforded: Run 1: 235 mg, 0.915 mmol, 92% yield, [B:L] = 5.1:1, [ee] = 58%. Run 2: 224 mg, 0.876 mmol, 88% yield; [B:L] = 4.3:1, [ee] = 55%. Run 3 (gram scale): 1.09 g, 4.250 mmol, 85% yield; [B:L] = 4.1:1, [ee] = 57%. (β-cyclodextrin, 120°C isothermal, tR(R) = 55.96 min., tR(S) = 57.30 min.), [average yield: 90%]; This material was taken forward without further purification.1H NMR (500 MHz, CDCl3) δ 5.78 (ddd, J = 17.1, 10.5, 6.5 Hz, 1H), 5.24-5.14 (m, 3H), 3.66 (s, 3H), 2.29 (t, J = 8.0 Hz, 2H), 2.05 (s, 3H), 1.62-1.53 (m, 4H), 1.29 (bs, 8H); 13C NMR (125 MHz, CDCl3) δ 174.2, 170.3, 136.5, 116.5, 74.8, 51.4, 34.1, 34.0, 29.1, 29.0, 29.0, 24.9, 24.8, 21.2; IR (neat, cm−1) 3089.1, 2932.9, 2858.2, 1742.0, 1436.3, 1371.5; HRMS (ESI) m/z calculated for C14H24O4Na [M + Na]+: 279.1572; found: 279.1564.
This material was then subjected to the standard procedure for cleavage of the allylic acetate which afforded allylic alcohol (±)-9 ready for subsequent resolution: Run 1: 188 mg, 0.877 mmol, 96% yield, [B:L] = 5.1:1. Run 2: 179 mg, 0.835 mmol, 95% yield; [B:L] = 4.3:1. Run 3 (gram scale): 879 mg, 4.101 mmol, 96% yield; [B:L] = 4.1:1
4.2.2.3 2-((3R)-Pent-3-acetoxy-4-en-1-yl-3-ol)-1,3-dioxane 13-OAc
Following the general procedure for the asymmetric branched allylic oxidation afforded: Run 1: 180 mg, 0.840 mmol, 84% yield; [B:L] = 4.8:1:1, [ee] = 44%. Run 2: 178 mg, 0.831 mmol, 83% yield; [B:L] = 4.3:1, [ee] = 46%. (β-cyclodextrin, 110°C isothermal, tR(R) = 22.21 min., tR(S) = 22.79 min.), [average yield: 84%.]; This material was taken forward without further purification. 1H NMR (500 MHz, CDCl3) δ 5.76 (ddd, J = 6.5, 10.5, 17.3 Hz, 1H), 5.24 (q, J = 6.5 Hz, 1H), 5.23 (dm, J = 17.5 Hz, 1H), 5.16 (dm, J = 10.5 Hz, 1H), 4.53 (t, J = 5.0 Hz, 1H), 4.09 (m, 2H), 3.75 (dt, J = 3.0, 12.5 Hz, 2H), 2.12-2.02 (m, 1H), 2.06 (s, 3H), 1.78-1.58 (m, 4H), 1.33 (dm, J = 13.5 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 170.2, 136.3, 116.7, 101.7, 74.3, 66.9, 30.7, 28.5, 25.8, 21.1; IR (neat, cm−1) 3087.5, 2962.1, 2931.3, 2852.2, 2780.9, 2732.7, 2661.3, 1739.5, 1646.9, 1430.9, 1407.8; HRMS (ESI) m/z calculated for C11H18O4Na [M+Na]+: 237.1103; found 237.1104.
This material was then subjected to the standard procedure for cleavage of the allylic acetate which afforded allylic alcohol (±)-13 ready for subsequent resolution: Run 1: 141 mg, 0.819 mmol, 97% yield, [B:L] = 4.8:1. Run 2: 135 mg, 0.784 mmol, 94% yield; [B:L] = 4.3:1
4.2.2.5 (4R, E)-Methyl 4-acetoxy-6-phenylhex-5-enoate (18)
The general procedure for the asymmetric branched allylic oxidation was modified in the following way to generate allylic acetate (18). A round bottom flask (25 mL ) was charged with the following: 1,2-Bis(phenylsulfinyl)ethane palladium(II) acetate (1) (10 mol%, 0.50 mmol, 250 mg); (1R,2R)-(−)-[1,2-Cyclohexanediamino-N,N’-bis(3,5-di-t-butylsalicylidene)] Chromium(III)F(R,R-2) (10 mol%, 0.50 mmol, 308 mg), 1,4-benzoquinone (2 equiv., 10.0 mmol, 1.08 g), an activated 4Å MS bead (~30 mg), and a Teflon© stir bar. A separate vial (2 mL, borosilicate) was charged with the following: Methyl hexenoate (1.0 equiv, 5.0 mmol, 0.704 mL), AcOH (1.1 equiv., 5.5 mmol, 0.315 mL), and EtOAc (0.50 mL). The liquids were transferred to the solids via pipette and the vial rinsed with EtOAc (4 × 0.50 mL ). After carefully stirring for 48 hrs at room temperature, to the reaction was added phenyl boronic acid (1.5 equiv., 7.5 mmol, 0.914 g), AcOH (1 equiv., 5 mmol, 0.285 mL), and EtOAc (12.5 mL). The reaction was stirred at room temperature until complete by TLC (~4 hr) at which point the reaction mixture was transferred to a separatory funnel with ~5 mL EtOAc and diluted with hexanes (400 mL). The organic layer was rinsed with sat. aq. NaHSO3 (1 × 50 mL) and 5% aq. K2CO3 (2 × 50 mL). Caution should be taken when combining aqueous layers as carbon dioxide is evolved. The combined aqueous layers were back extracted with hexanes (100 mL). The combined organic layers were dried (MgSO4), filtered, and reduced in vacuo. The resulting oil was re-dissolved in hexanes (150 mL) and extracted again with 5% aq. K2CO3 (3 × 25 mL) to remove residual hydroquinone. The organic layer was again dried (MgSO4), filtered and reduced in vacuo This product was generally taken forward without further purification, but was isolated and purified via silica gel chromatography for characterization. [B:L] = >20:1, [ee] = 50%. (Determined on the initial branched acetate product prior to oxidative Heck reaction, β-cyclodextrin, 110°C isothermal, tR(R) = 5.52 min., tR(S) = 5.83 min.), 1H NMR (500 MHz, CDCl3) δ 7.39 (d, J = 7.5 Hz, 2H), 7.34 (t, J = 7.5 Hz, 2H), 7.31-7.25 (m, 1H), 6.64 (d, J = 16.0 Hz, 1H), 6.12 (dd, J = 7.5, 15.8 Hz, 1H), 5.46 (q, J = 6.5 Hz, 1H), 3.68 (s, 3H), 2.42 (dt, J = 2.0, 7.8 Hz, 2H), 2.13-2.07 (m, 2H), 2.10 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 173.2, 170.2, 136.0, 133.0, 128.5, 128.0 126.7, 126.6, 73.7, 51.7, 29.8, 29.5, 21.2; IR (neat, cm−1) 3085.6, 3025.8, 2952.5, 2848.4,v1737.6, 1658.5, 1598.7, 1597.4, 1494.6; HRMS (ESI) m/z calculated for C15H18O4Na [M+Na]+: 285.1103; found 285.1092.
4.2.3 General procedure for resolution with Novozyme 435
To a flame dried round bottom flask containing allylic alcohol to be resolved (1 equiv.) was added vinyl acetate (0.6 M) and Novozyme 435 immobilized on polystyrene beads (33.3 mg/1 mmol). The reaction was stirred vigorously at room temperature for 36 hrs. Upon completion, the solid supported enzyme was removed via filtration. The solid support was rinsed thoroughly with diethyl ether and then the filtrate reduced in vacuo and purified via standard SiO2 chromatography. Enantioselectivities were determined by chiral gas chromatographic analysis on the acetylated derivative of each isolated alcohol. It was found that the recovered solid supported enzyme could be used up to 5 times with little diminishment in activity. Individual yields and selectivities are reported below.
4.2.3.1 (−)-(4R)-1-O-Benzyl-5-hexen-1,4-diol (−)-3
Following the general procedure for Novozyme 435 resolution afforded: Run 1: 105 mg, 0.509 mmol, 57% yield, [B:L] = >20:1, [ee] = 98%. Run 2: 100 mg, 0.485 mmol, 55% yield; [B:L] = >20:1, [ee] = 99%. Enantiomeric access was determined on the acylated derivative of the final product (ee determined on the acylated alcohol, β-cyclodextrin, 120°C isothermal, tR(R) = 65.56 min., tR(S) = 67.14 min), [average yield: 56%.]; [α]26d = −2.86° (c = 2.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.37-7.32 (m, 4H), 7.30-7.25 (m, 1H), 5.87 (ddd, J = 17.0, 10.5, 6.0 Hz, 1H), 5.25 (dt, J = 17.0, 1.5 Hz, 1H), 5.10 (dt, J = 10.5, 1.5 Hz, 1H), 4.52 (s, 2H), 4.13 (m, 1H), 3.52 (t, J = 6.0 Hz, 2H), 2.27 (d, J = 4.5 Hz, 2H), 1.77-1.57 (m, 4H); 13C NMR (125 MHz, CDCl3) δ 141.0, 138.1, 128.3, 127.6, 127.6, 114.4, 72.9, 72.6, 70.2, 34.2, 25.7; IR (neat, cm−1) 3403.8, 3066.3, 3031.6, 2979.5, 2942.9, 2858.0, 2798.2, 1643.1.0, 1496.5, 1454.1; HRMS (ESI) m/z calculated for C14H24O2Na [M+Na]+: 229.1204; found: 229.1204.
4.2.3.2 Methyl (9R)-9-hydroxyundec-10-eneoate (−)-9
Following the general procedure for Novozyme 435 resolution afforded: Run 1: 119 mg, 0.555 mmol, 63% yield, [B:L] = >20:1, [ee] = 99%. Run 2: 109 mg, 0.509 mmol, 61% yield; [B:L] = >20:1, [ee] = 98%, Run 3 (gram scale): 523 mg, 2.441 mmol, 60% yield; [B:L] = >20:1, [ee] = 99%. (ee determined on the acylated alcohol, β-cyclodextrin, 120°C isothermal, tR(R) = 55.92 min.), [average yield: 62%];[α]25d = −5.13° (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 5.86 (ddd, J = 16.9, 10.8, 6.0 Hz, 1H), 5.22 (d, J = 17.0 Hz, 1H), 5.10 (d, J = 10.5 Hz, 1H), 4.09 (p, J = 5.5 Hz, 1H), 3.66 (s, 3H), 2.30 (t, J = 7.5 Hz, 2H), 1.63 – 1.30 (m, 13H); 13C NMR (125 MHz, CDCl3) δ 174.2, 141.3, 114.4, 73.2, 51.4, 37.0, 34.1, 29.3, 29.1, 29.0, 25.2, 24.9; IR (neat, cm−1) 3426.9, 2979.5, 2931.3, 2856.1, 1739.5, 1436.7; HRMS (ESI) m/z calculated for C12H22O3Na [M+Na]+: 237.1467; found: 237.1471.
4.2.3.3 2-((3R)-Pent-4-en-1-yl-3-ol)-1,3-dioxane (−)-13
Following the general procedure for Novozyme 435 resolution afforded: Run 1: 80 mg, 0.464 mmol, 57% yield; [B:L] = >20:1:1, [ee] = 99%. Run 2: 76 mg, 0.441 mmol, 56% yield; [B:L] = >20:1, [ee] = 99%. (ee determined on the acylated alcohol, β-cyclodextrin, 110°C isothermal, tR(R) = 22.31 min., tR(minor) = 22.93 min.), [average yield: 57%.]; [α]24d = −5.01° (c = 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.86 (ddd, J = 5.6, 10.4, 17.3 Hz, 1H), 5.24 (dt, J = 1.6, 17.2 Hz, 1H), 5.10 (dt, J = 1.2, 10.8 Hz, 1H), 4.58 (t, J = 4.4 Hz, 1H), 4.18-4.08 (m, 3H), 3.77 (app t, J = 11.6 Hz, 2H), 2.36 (d, J = 4.0 Hz, 1H), 2.08 (qt, J = 4.0, 12.4, 1H), 1.78-1.58 (m, 4H), 1.35 (d sep, J = 1.2, 13.6 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 141.0, 114.4, 102.1, 72.6, 66.9, 31.2, 31.1, 25.7; IR (neat, cm−1) 3430.8, 3079.8, 2962.1, 2929.4, 2856.1, 2734.6, 1643.1, 1429.0, 1405.9; HRMS (ESI) m/z calculated for C9H16O3Na [M+Na]+: 195.0995; found 195.0997.
4.2.4 General procedure for resolution with Subtilisin. Carlsberg
The active enzyme for resolution was prepared as previously described by Boren and co-workers.15 To a flame dried round bottom flask containing allylic alcohol to be resolved (1 equiv.) was added isoproenyl valerate (1.5 equiv), active S. Carlsberg (36 mg/1 mmol), sodium carbonate (1 equiv.) and THF (0.5 M). The reaction was stirred vigorously at room temperature for 60 hrs. Upon completion, the enzyme was removed via filtration. The enzyme was rinsed thoroughly with diethyl ether and then the filtrate reduced in vacuo and purified via standard SiO2 chromatography. Enantioselectivities were determined by chiral gas chromatographic analysis on the acetylated derivative of each isolated alcohol. Individual yields and selectivities are reported below.
4.2.4.1 (+)-(4S)-1-O-Benzyl-5-hexen-1,4-diol (+)-3
Material for this route was obtained by application of the general ABAO procedure using (S,S)-2 as a chiral catalyst. Subsequent acetate deprotection by the general procedure described afforded: Run 1: 180 mg, 0.873 mmol, 87% yield; [B:L] = 4.4:1, [ee] = 46%. Run 2: 187 mg, 0.906 mmol, 91% yield; [B:L] = 4.1:1, [ee] = 45%. Yields and selectivities are over two-steps. This material was then subjected to the general procedure for resolution with S. Carlsberg to afford chiral allylic alcohol: Run 1: 92 mg, 0.446 mmol, 52% yield, [B:L] = >20:1, [ee] = 99%. Run 2: 97 mg, 0.470 mmol, 54% yield; [B:L] = >20:1, [ee]; Enantiomeric access was determined on the acylated derivative of the final product (ee determined on the acylated alcohol, β-cyclodextrin, 120°C isothermal, tR(S) = 67.03 min) [average yield: 53%.]; [α]26d = +2.85° (c = 2.0, CHCl3).
4.2.5 Synthesis of furan core (−)-22
4.2.5.1 (R, E)-5-styryldihydrofuran-2(3H)-one (19)
To crude 18 (5 mmol, assumed) in a round bottom flask (250 mL ) was added THF (18.75 mL), DI H2O (6.25 mL), and a Teflon© stir bar. The flask was cooled to 0°C and LiOH·H2O (0.623 g, 15 mmol, 3.0 equiv.) was added in one portion. The ice bath was removed after 10 minutes and the reaction monitored via TLC. Upon completion (~2-4 hr) benzene (150 mL) was added and the flask was transferred to a 100°C oil bath and a Dean-Stark trap and a reflux condenser were added. The reaction was brought to a comfortable reflux and then allowed to stir overnight. After removing the flask from the bath and allowing it to cool to room temperature, the contents were transferred to a separatory funnel and the organic layer washed with aq. 1M H3PO4 (3 × 25 mL). The organic layer was then dried (MgSO4), filtered, and reduced in vacuo. The resulting off white solid was purified via silica gel chromatography (10-30% Et2O:Hexanes) to afford a white solid. (0.504 g, 2.678 mmol, 54% (2-step)) 1H NMR (500 MHz, CDCl3) δ 7.40 (d, J = 7.5 Hz, 2H), 7.37-7.32 (m, 2H), 7.30-7.26 (m, 1H), 6.69 (d, J = 16.0 Hz, 1H), 6.21 (dd, J = 6.5, 16.0 Hz, 1H), 5.13 (q, J = 6.5 Hz, 1H), 2.65-2.54 (m, 2H), 2.50 (app sextuplet, J = 9.0 Hz, 1H), 2.11 (ddd, J = 9.0, 12.5, 16.6 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 176.9, 135.6, 132.9, 128.7, 128.4 126.7, 126.4, 80.5, 28.8, 28.5; IR (neat, cm−1) 2989.1, 2950.6, 1762.6, 1722.1, 1454.1, 1415.5; HRMS (ESI) m/z calculated for C12H13O2 [M+H]+: 189.0916; found 189.0918.
4.2.5.2 (R)-5-((4R,5R)-2,2-dimethyl-5-phenyl-1,3-dioxolan-4-yl)dihydrofuran-2(3H)-one ((−)-20)
To a clean, dry 100 mL recovery flask was added sequentially the following: K2OsO4 . 2H2O (0.018 g, 0.05 mmol, 1 mol%), (DHQD)2PHAL (0.199 g, 0.25 mmol, 5 mol%), K3Fe(CN)6 (4.94 g, 15 mmol, 3 equiv.), K2CO3 (2.07 g, 15 mmol, 3 equiv.), NaHCO3 (1.34 g, 15 mmol, 3 equiv.), a Teflon© stir bar, deionized water (24 mL), and tert-butanol (24 mL). The reaction flask was stirred vigorously until both layers became translucent, at which time MeSO2NH2 (0.476 g, 5 mmol, 1 equiv.) was added and the reaction was cooled to 0°C. After the solution became opaque, olefin (19) (0.941 g, 5 mmol, 1 equiv.) was added in one portion. CH2Cl2 (2.4 mL) was added to improve SM solubility and the reaction was stirred vigorously at 0°C for 1 hr, then warmed to room temperature and stirred until completion as indicated by TLC (~5 hr). Upon completion, sodium bisulfite (2 g) was added slowly and the reaction stirred for 1 hour. The reaction mixture was transferred to a separatory funnel and EtOAc (50 ml) was added. The aqueous layer was extracted with additional EtOAc (3 × 50 mL). The combined organic layers were dried (Na2SO4), filtered, and concentrated in vacuo. To the crude diol was added DMF (8.4 mL, 0.6 M) and 2-methoxypropene (4.79 mL). The reaction was cooled to 0°C and p-TsOH·H2O (0.238 g, 1.25 mmol, 0.25 equiv.) was added and the reaction allowed to warm to room temperature while stirring overnight. The reaction mixture was then transferred to a separatory funnel and diluted with Et2O (200 mL). The organic layer was washed with DI H2O (3 × 25mL) and brine (1 × 25 mL). The organic layer was then dried (MgSO4), filtered, and reduced in vacuo. Residual DMF or 2-methoxypropene was removed by addition of benzene and in vacuo concentration. The crude oil was purified by silica gel chromatography in 10-40% Et2O:Hexanes to afford a white solid (0.920 g, 3.52 mmol, 70% yield (2-step), >20:1 dr, 99% ee (Determined on Chiracel AD-RH , 35:75 CH3CN:H2O , tR(major) = 7.3 min., tR(minor) = 6.8 min.) [α]26d = −94.9° (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.39-7.31 (m, 5H), 4.78 (d, J = 8.5 Hz, 1H), 4.60 (ddd, J = 4.0, 6.3, 7.5 Hz, 1H), 4.10 (dd, J = 4.0, 8.3 Hz, 1H), 2.60 (ddd, J = 6.5, 10.0, 18.0 Hz, 1H), 2.51 (ddd, J = 7.5, 9.5, 17.4 Hz, 1H), 2.38-2.24 (m, 2H), 1.56 (s, 3H), 1.52 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 176.5, 137.2, 128.8, 128.7, 126.7 110.2, 83.4, 80.1, 78.7, 28.1, 27.1, 27.0, 22.9; IR (neat, cm−1) 3066.3, 3031.6, 2985.3, 2935.1, 2980.8, 1781.9, 1604.5, 1494.6, 1456.0; HRMS (ESI) m/z calculated for C12H22O4 [M+H]+: 263.1283; found 263.1280. The absolute configuration of this molecule was determined on a crystal grown from benzene of p-bromophenyl-20 synthesized through the same sequence. The cif file for this compound can be found as an additional file of the supporting information.
4.2.5.3 (R)-5-((4R,5R)-2,2-dimethyl-5-phenyl-1,3-dioxolan-4-yl)furan-2(5H)-one ((+)-21)
To a clean, flame dried 25 mL recovery flask charged with a Teflon stir bar and under an argon atmosphere was added THF (5 mL) and hexamethyldisilazane (1.36 mmol, 0.288 mL, 1.1 equiv.). The reaction was cooled to −78°C, and n-Buli (1.30 mmol, 0.813 mL, 1.05 equiv.) was added dropwise via syringe. After stirring for ten minutes, (−)-20 (1.24 mmol, 0.325 g, 1 equiv.) in THF (1 mL, 0.15 mL rinse) was added slowly via cannula. The reaction was stirred a further 25 minutes, and then phenylselenyl bromide (1.24 mmol, 0.293 g, 1 equiv.) in THF (1.15 mL) was added via cannula over ~10 min. The reaction was stirred for an additional 5 minutes and then quenched at −78°C with 1N HCl (5 mL). The reaction mixture was transferred to a separatory funnel and diluted with Et2O (200 ml). The organic layer was washed with sat. aq. NaHCO3 (2 × 10 mL). The organic layer was then dried (Na2SO4), filtered, and concentrated in vacuo. Reproducibility for the elimination step was significantly improved by quickly purifying away fast running selenium containing species by SiO2 chromatography in 5%-10%-20% Et2O:Hexanes. To the mixture of selenides (1.03 mmol, 0.425 g, 1 equiv.) in a clean, dry 100 mL flask was added CH2Cl2 (20.6 mL, 0.05 M) and the reaction flask was cooled to 0°C in an ice bath. Hydrogen peroxide (3.08 mmol, 0.346 mL of 30% solution, 3 equiv.) was then added slowly via syringe. The reaction was stirred at 0°C and conversion monitored by TLC. Upon completion, the reaction mixture was transferred to a separatory funnel and CH2Cl2 was added (200 mL). The organic layer was then washed with DI H2O (2 × 20 mL) and brine (20 mL). The organic layer was then dried (MgSO4), filtered, and reduced in vacuo. The crude oil was purified by silica gel chromatography in 10-40% Et2O:Hexanes to afford a white solid (0.237 g, 0.91 mmol, 73% yield (2-step). [α]25d = 555.6° (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.48 (dd, J = 1.5, 6.0 Hz, 1H), 7.40-7.31 (m, 5H), 6.17 (dd, J = 2.5, 5.8Hz, 1H), 5.16 (dt, J = 2.0, 6.5 Hz, 1H), 5.02 (d, J = 8.0 Hz, 1H), 3.91 (dd, J = 6.5, 7.5 Hz, 1H), 1.56 (s, 3H), 1.54 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 172.2, 153.8, 137.3, 128.6, 128.6, 127.0, 122.6, 110.6, 82.8, 82.7, 80.7, 27.1, 26.8; IR (neat, cm−1) 3089.4, 3033.5, 2989.1, 2935.1, 2894.6, 1783.8, 1758.8, 1602.6, 1496.5, 1456.0; HRMS (ESI) m/z calculated for C15H17O4 [M+H]+: 261.1127; found 263.1123.
4.2.5.4 (3aS,5R,6S,6aS)-6-hydroxy-5-phenyltetrahydrofuro[3,2-b]furan-2(5H)-one((−)-22)
To (+)-21 (0.91 mmol, 0.237 g, 1 equiv.) in a clean, dry round bottom flask (50 mL) with a Teflon© stir bar was added THF (9.1 mL) and 1N HCl (5-10 drops). The reaction mixture was heated to 45°C and monitored via TLC (70% EtOAc:Hex). Deprotection and cyclization would generally proceed to completion under these conditions with prolonged stirring, but could be expedited by the following procedure. After complete acetonide deprotection by TLC, the flask was cooled to 0°C and CH2Cl2 (9.1 mL) and NEt3 was added until a pH of ~10 was obtained. The flask was then allowed to warm to room temperature and monitored via TLC. Upon completion (~4-6 hr), the contents were transferred to a separatory funnel and diluted with further CH2Cl2. The organic layer was then washed with sat. aq. NH4Cl solution (3 × 15 mL). The combined aqueous layers were back extracted with EtOAc (3 × 50 mL) and then the combined organic layers were dried (MgSO4), filtered, and reduced in vacuo. The resulting off white solid was purified via silica gel chromatography (10-50% EtOAc:Hexanes) to afford a white solid. (0.161 g, 0.731 mmol, 80%) [α]26d = −17.1° (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ7.44-7.34 (m, 5H), 5.23 (d, J = 2.5 Hz, 1H), 5.20 (td, J = 1.0, 5.0 Hz, 1H), 5.07 (d, J = 5.0 Hz, 1H), 4.64 (app t, J = 2.0 Hz, 1H), 2.86 (dd, J = 6.0, 18.8 Hz, 1H), 2.79 (d , J = 18.5 Hz, 1H), 1.36 (d, J = 2.5 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 175.4, 134.2, 128.9, 128.6, 126.6, 87.2, 82.8, 77.1, 75.8, 36.0; IR (neat, cm−1) 3948.3, 2975.6, 2948.6, 2923.6, 2858.0, 1766.5, 1496.49, 1454.1; HRMS (ESI) m/z calculated for C12H12O4Na [M+Na]+: 243.0645; found 243.0633.
Supplementary Material
Acknowledgments
Financial support was provided by the NIH/NIGMS (2R01 GM 076153B) and generous gifts from Bristol-Myers Squibb and Boehringer Ingelheim.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material Full experimental details for all compounds synthesized and copies of 1H and 13C NMR spectra as well as chiral GC and HPLC traces for chiral compounds as well as crystallographic data on p-Bromophenyl (−)-20 are available. Supplementary data related to this article can be found at http:
References and notes
- 1(a).Crabtree RH. J. Chem. Soc., Dalton Trans. 2001:2437–2450. [Google Scholar]; (b) Labinger JA, Bercaw JE. Nature. 2002;417:507–514. doi: 10.1038/417507a. [DOI] [PubMed] [Google Scholar]; (c) Dick AR, Sanford MS. Tetrahedron. 2006;62:2439–2463. [Google Scholar]; (d) Giri R, Shi B-F, Engle KM, Maugel N, Yu J-Q. Chem. Soc. Rev. 2009;38:3242–3272. doi: 10.1039/b816707a. [DOI] [PubMed] [Google Scholar]; (e) McMurray L, O’Hara F, Gaunt MJ. Chem. Soc. Rev. 2011;40:1885–1898. doi: 10.1039/c1cs15013h. [DOI] [PubMed] [Google Scholar]; (f) Yamaguchi J, Yamaguchi AD, Itami K. Angew. Chem. Int. Ed. 2012;(51):8960–9009. doi: 10.1002/anie.201201666. [DOI] [PubMed] [Google Scholar]; (g) White MC. Science. 2012;335:807–809. doi: 10.1126/science.1207661. [DOI] [PubMed] [Google Scholar]; (h) White MC. Synlett. 2012;23:2746–2748. [Google Scholar]
- 2(a).For some examples of aliphatic C—H oxidation methods used to install oxygen functionality see: Chen MS, White MC. Science. 2007;318:783–787. doi: 10.1126/science.1148597.; Litvinas ND, Brodsky BH, Du Bois J. Angew. Chem. Int. Ed. 2009;48:4513–4516. doi: 10.1002/anie.200901353.; Chen MS, White MC. Science. 2010;327:566–571. doi: 10.1126/science.1183602.; For directed methods see: Giri R, Liang J, Lei J-G, Li J-J, Wang D-H, Chen X, Naggar IC, Guo C, Foxman BM, Yu J-Q. Angew. Chem. Int. Ed. 2005;44:7420–7424. doi: 10.1002/anie.200502767.; Desai LV, Hull KL, Sanford MS. J. Am. Chem. Soc. 2004;126:9542–9543. doi: 10.1021/ja046831c. Bigi MA, Reed SA, White MC. J. Am. Chem. Soc. 2012;134:9721–9726. doi: 10.1021/ja301685r.; Ren Z, Mo F, Dong G. J. Am. Chem. Soc. 2012;134:16991–16994. doi: 10.1021/ja3082186.
- 3(a).Streamlining synthesis via late-stage C–H oxidation: Nicolaou KC, Yang Z, Liu JJ, Ueno H, Nantermet PG, Guy RK, Claiborne CF, Renaud J, Couladouros EA, Paulvannan K, Sorensen EJ. Nature. 1997;367:630–634. doi: 10.1038/367630a0.; Wender PA, Jesudason CD, Nakahira H, Tamura N, Tebbe AL, Ueno Y. J. Am. Chem. Soc. 1997;119:12976–12977.; Fraunhoffer KJ, Bachovchin DA, White MC. Org. Lett. 2005;7:223–226. doi: 10.1021/ol047800p.; Covell DJ, Vermeulen NA, White MC. Angew. Chem., Int. Ed. 2006;45:8217–8220. doi: 10.1002/anie.200603321.; Stang EM, White MC. Nat. Chem. 2009;1:547–551. doi: 10.1038/nchem.351.; Stang EM, White MC. Angew. Chem. Int. Ed. 2011;50:2094–2097. doi: 10.1002/anie.201007309.; Vermeulen NA, Delcamp JH, White MC. J. Am. Chem. Soc. 2010;132:11323–11328. doi: 10.1021/ja104826g.; For excellent reviews, see: Hoffmann RW. Synthesis. 2006;21:3531–3541.; Hoffmann RW. Elements of Synthesis Planning. Springer; Heidelberg: 2009. For some examples of late stage C–H hydroxylation see: Wender PA, Hilinski MK, Mayweg AVW. Org. Lett. 2005;7:79–82. doi: 10.1021/ol047859w.; (g) Ref. 2a; (h) Ref. 2c (i) Ref. 2f.
- 4.Covell DJ, White MC. Angew. Chem., Int. Ed. 2008;47:6448–6451. doi: 10.1002/anie.200802106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5(a).For examples of asymmetric olefination of aldehydes, see: Oppolzer W, Radinov RN. Tetrahedron Lett. 1991;32:5777–5780.; Brubaker JD, Myers AG. Org. Lett. 2007;9:3523–3525. doi: 10.1021/ol071377d. and references therein; Hargadena GC, Guiry PJ. Adv. Synth. Catal. 2007;349:2407–2424. and references therein; For chiral pool starting material derivatization: Sheddan NA, Mulzer J. Org. Lett. 2006;8:3101–3104. doi: 10.1021/ol061141u.; Ichikawa Y, Yamaoka T, Nakano K, Kotsuki H. Org. Lett. 2007;9:2989–2992. doi: 10.1021/ol0709735.; For asymmetric epoxidation based examples, see: Babu KV, Sharma GVM. Tetrahedron: Asymm. 2008;19:577–583.; Yadav JS, Srihari P. Tetrahedron: Asymm. 2004;15:81–89.; For resolution examples, see: Itoh T, Kudo K, Yokota K, Tanaka N, Hayase S, Renou M. Eur. J. Org. Chem. 2004;2:406–412.; Chênevert R, Gravila S, Bolteb J. Tetrahedron: Asymm. 2005;16:2081–2086.
- 6(a).For elegant metal catalyzed enantioselective routes to chiral allylic esters and alcohols see: Kirsch SF, Overman LE. J. Am. Chem. Soc. 2005;127:2866–2867. doi: 10.1021/ja0425583.; Geurts K, Fletcher SP, Feringa BL. J. Am. Chem. Soc. 2006;128:15572–15573. doi: 10.1021/ja065780b. Evans PA, Grisin A, Lawler MJ. J. Am. Chem. Soc. 2012;134:2856–2859. doi: 10.1021/ja208668u.
- 7(a).For reviews of the Sharpless asymmetric epoxidation (SAE) and its applications see: Johnson RA, Sharpless KB. Comp. Org. Syn. 1991;7:389–436. Johnson RA, Sharpless KB. In: Catalytic Asymmetric Synthesis. 2nd ed Ojima I, editor. Wiley-VCH; New York: 2000. p. 231.
- 8(a).Pd-catalyzed allylic C–H oxidations: Chen MS, White MC. J. Am. Chem. Soc. 2004;126:1346–1347. doi: 10.1021/ja039107n.; Chen MS, Prabagaran N, Labenz NA, White MC. J. Am. Chem. Soc. 2005;127:6970–6971. doi: 10.1021/ja0500198.; Fraunhoffer KJ, Prabagaran N, Sirois LE, White MC. J. Am. Chem. Soc. 2006;128:9032–9033. doi: 10.1021/ja063096r.; Delcamp JH, White MC. J. Am. Chem. Soc. 2006;128:15076–15077. doi: 10.1021/ja066563d.; Mistudome T, Umetani T, Nosaka N, Mori K, Mizugaki T, Ebitani K, Kaneda K. Angew. Chem. Int. Ed. 2006;45:481–485. doi: 10.1002/anie.200502886.; Pilarski LT, Selander N, Bose D, Szabo KJ. Org. Lett. 2009;11:5518–5521. doi: 10.1021/ol9023369.; Lin B-L, Labinger JA, Bercaw JE. Can. J. Chem. 2009;87:264–271.; Thiery E, Aouf C, Belloy J, Harakat D, Le Bras J, Muzart J. J. Org. Chem. 2010;75:1771–1774. doi: 10.1021/jo902587u.; Henderson WH, Check CT, Proust N, Stambuli JP. Org. Lett. 2010;12:824–827. doi: 10.1021/ol902905w.; Campbell AN, White PB, Guzei IA, Stahl SS. J. Am. Chem. Soc. 2010;132:15116–15119. doi: 10.1021/ja105829t.; Check CT, Henderson WH, Wray BB, Eyden MJV, Stambuli JP. J. Am. Chem. Soc. 2011;133:18503–18505. doi: 10.1021/ja2089102.; Alam R, Pilarski LT, Pershagen E, Szabo KJ. J. Am. Chem. Soc. 2012;134:8778–8781. doi: 10.1021/ja302457p.
- 9(a).Pd-catalyzed allylic C–H aminations: Larock RC, Hightower TR, Hasvold LA, Peterson KP. J. Org. Chem. 1996;61:3584–3585. doi: 10.1021/jo952088i.; Fraunhoffer KJ, White MC. J. Am. Chem. Soc. 2007;129:7274–7276. doi: 10.1021/ja071905g.; Reed SA, White MC. J. Am. Chem. Soc. 2008;130:3316–3318. doi: 10.1021/ja710206u.; Liu G, Yin G, Wu L. Angew. Chem., Int. Ed. 2008;47:4733–4736. doi: 10.1002/anie.200801009.; Rice GT, White MC. J. Am. Chem. Soc. 2009;131:11707–11711. doi: 10.1021/ja9054959.; Reed SA, Mazzotti AR, White MC. J. Am. Chem. Soc. 2009;131:11701–11706. doi: 10.1021/ja903939k.; Nahra F, Liron F, Prestat G, Mealli C, Messaoudi A, Poli G. Chem. Eur. J. 2009;15:11078–11082. doi: 10.1002/chem.200901946.; Wu L, Qiu S, Liu G. Org. Lett. 2009;11:2707–2710. doi: 10.1021/ol900941t.; Shimizu Y, Obora Y, Ishii Y. Org. Lett. 2010;12:1372–1374. doi: 10.1021/ol100292g.; Qi X, Rice GT, Lall MS, Plummer MS, White MC. Tetrahedron. 2010;66:4816–4826. doi: 10.1016/j.tet.2010.04.064.
- 10(a).Pd-catalyzed allylic C–H alkylations: Young AJ, White MC. J. Am. Chem. Soc. 2008;130:14090–14091. doi: 10.1021/ja806867p.; Young AJ, White MC. Angew. Chem. Int. Ed. 2011;50:6824–6827. doi: 10.1002/anie.201101654.; Lin S, Song C-X, Cai G-X, Wang W-H, Shi Z-J. J. Am. Chem. Soc. 2008;130:12901–12903. doi: 10.1021/ja803452p.; Trost BM, Thaisrivongs DA, Donckele EJ. Angew. Chem. Int. Ed. 2012;51:1–5. doi: 10.1002/anie.201207870.
- 11.Stang EM, White MC. J. Am. Chem. Soc. 2011;133:14892–14895. doi: 10.1021/ja2059704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12(a).27% ee (15% yield) for octene, 40% ee (28% yield) for allylbenzene, 10 equiv. starting material (SM): Ginotra SK, Singh VK. Tetrahedron. 2006;62:3573–3581.; (b) 30% ee (50% yield) for octene, 36% ee (34% yield) for allylbenzene, 5 equiv. starting material: Andrus MB, Argade AB, Chen X, Pamment MG. Tetrahedron Lett. 1995;36:2945–2948.; (c)15% ee, (98% yield) 2,2-diphenylhex-5-enoic acid (1 equiv.): Takenaka K, Akita M, Tanigaki Y, Takizawa S, Sasai H. Org. Lett. 2011;13:3506–3509. doi: 10.1021/ol201314m.
- 13(a).Yadav JS, Chetia L. Org. Lett. 2007;9:4587–4589. doi: 10.1021/ol702095n. [DOI] [PubMed] [Google Scholar]; (b) Schomaker JM, Reddy PV, Babak B. J. Am. Chem. Soc. 2004;126:13600–13601. doi: 10.1021/ja0469075. [DOI] [PubMed] [Google Scholar]; (c) Garcia C, Martin T, Martin VS. J. Org. Chem. 2001;66:1420–1428. doi: 10.1021/jo0057194. [DOI] [PubMed] [Google Scholar]; (d) Chang MY, Lin JYC, Chen ST, Chang NC. J. Chin. Chem. Soc. 2002;49:1079–1088. [Google Scholar]; (e) Marshall JA, Dehoff BS. J. Org. Chem. 1986;51:863–872. [Google Scholar]; (f) Finan JM, Kishi Y. Tetrahedron Lett. 1982;23:2719–2722. [Google Scholar]
- 14.Yadav JS, Joyasawal S, Duttab SK, Kunwarb AC. Tetrahedron Lett. 2007;48:5335–5340. [Google Scholar]
- 15.Enzymatic acylation not only enhances the enantioenrichment of the branched allylic alcohol, but also rapidly acylates any undesired linear allylic alcohol making final purification simple (See SI for details).
- 16.Boren L, Martin-Matute B, Xu Y, Cordova A, Backvall JE. Chem. Eur. J. 2006;12:225–232. doi: 10.1002/chem.200500758. [DOI] [PubMed] [Google Scholar]
- 17.Yokokawa Y, Kobayashi K, Yokoyama M, Yamamuray S. Chem. Lett. 2003;32:844–845. [Google Scholar]
- 18(a).Singh S, Singh OV, Han H. Tetrahedron Lett. 2007;48:8270–8273. doi: 10.1016/j.tetlet.2007.09.129. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kawashima M, Fujisawa T. Bull. Chem. Soc. Jpn. 1988;61:1, 3377–3379. [Google Scholar]
- 19(a).Isolation: Cao SG, Wu XH, Sim KY, Tan BKH, Pereira JT, Gog S-H. Tetrahedron. 1998;54:2143–2146.; Synthesis: Babjak M, Kapitan P, Gracza T. Tetrahedron Lett. 2002;43:6983–6985.; Yoda H, Nakaseko Y, Takabe K. Synlett. 2002;9:1532–1534.; Murga J, Ruiz P, Falomir E, Carda M, Peris G, Marco JA. J. Org. Chem. 2004;69:1987–1992. doi: 10.1021/jo0356356.; Yadav JS, Raju AR, Rao PP, Rajaiah G. Tetrahedron: Asymm. 2005;16:3283–3290.; Babjak M, Kapitán P, Gracza T. Tetrahedron. 2005;61:2471–2479.; Carreno MC, Hernandez-Torres G, Urbano A, Colobert F. Org. Lett. 2005;7:5517–5520. doi: 10.1021/ol0523603.; Prasad KR, Gholap SL. J. Org. Chem. 2006;71:3643–3645. doi: 10.1021/jo060159f.; Ghosh S, Rao CN, Dutta SK. Synlett. 2007;9:1464–1466.; Kang B, Chang S, Decker S, Britton R. Org. Lett. 2010;12:1716. doi: 10.1021/ol100260z. Brichacek M, Batory LA, Njardarson JT. Angew. Chem. Int. Ed. 2010;49:1648–1651. doi: 10.1002/anie.200906830.; Hernandez-Torres G, Carreco MC, Urbano A, Colobert F. Chem. Eur. J. 2011;17:1283–1293. doi: 10.1002/chem.201002637.
- 20.Although Pd(II)/bis-sulfoxide-catalyzed tandem allylic esterification and aminations/oxidative Heck reactions have been previously demonstrated, this sequence has never been demonstrated for the ABAO, which contains an additional Cr(salen) co-catalyst: (a) Ref. 7d Jiang, Covell DJ, Stepan AF, Plummer MS, White MC. Org. Lett. 2012;14:1386–1389. doi: 10.1021/ol300063t.
- 21.Kolb HC, VanNieuwenhze MS, Sharpless KB. Chem. Rev. 1994;94:2483–2547. [Google Scholar]
- 22.The relative and absolute stereochemistry of this compound was determined by X-ray crystallographic analysis of the diol produced by acetonide deprotection of para-Bromophenyl-(−)-20 (see full SI).
- 23.Still WC, Kahn M, Mitra A. J. Org. Chem. 1978;43:2923–2925. [Google Scholar]
- 24.(a) Wender PA, Croatta MP, Witulski B. Tetrahedron. 2006;62:7505–7511. [Google Scholar]; (b) Wender PA, Verma VA, Paxton TJ, Pillow TH. Acc. Chem. Res. 2008;41:40–49. doi: 10.1021/ar700155p. [DOI] [PubMed] [Google Scholar]; (c) Wender PA, Miller BL. Nature. 2009;460:197–201. doi: 10.1038/460197a. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.