

Abstract
Background
MRI evidence of small vessel disease is common in intracerebral hemorrhage (ICH). We hypothesized that ICH caused by cerebral amyloid angiopathy (CAA) or hypertensive vasculopathy would have different distributions of MRI T2 white matter hyperintensity (WMH) and microbleeds (MB).
Methods
Data were analyzed from 133 consecutive patients with primary supratentorial ICH and adequate MRI sequences. CAA was diagnosed using the Boston criteria. WMH segmentation was performed using a validated semi-automated method. WMH and MB were compared according to site of symptomatic hematoma origin (lobar vs. deep) or by pattern of hemorrhages, including both hematomas and MB, on MRI GRE sequence (grouped as lobar only--probable CAA, lobar only--possible CAA, deep hemispheric only, or mixed lobar and deep hemorrhages).
Results
Lobar and deep hemispheric hematoma patients had similar median nWMH volumes (19.5 cm vs. 19.9 cm3, p=0.74) and prevalence of ≥1 MB (54% vs. 52%, p=0.99). The supratentorial WMH distribution was similar according to hemorrhage location category, however the prevalence of brainstem T2 hyperintensity was lower in lobar hematoma vs. deep hematoma (54% vs. 70%, p=0.004). Mixed ICH was common (23%). Mixed ICH patients had large nWMH volumes and a posterior distribution of cortical hemorrhages similar to that seen in CAA.
Conclusions
WMH distribution is largely similar between CAA-related and non-CAA-related ICH. Mixed lobar and deep hemorrhages are seen on MRI GRE in up to one quarter of patients; in these patients both hypertension and CAA may be contributing to the burden of WMH.
Keywords: intracerebral hemorrhage, MRI, white matter disease, leukoaraiosis, cerebral amyloid angiopathy
INTRODUCTION
Hypertension and cerebral amyloid angiopathy (CAA) account for most primary intracerebral hemorrhage. CAA affects the vessels of the cortex and leptomeninges without significant involvement of the penetrating.1 By contrast, hypertension causes arteriosclerosis in both cortical and subcortical arteries. Patients with multiple strictly lobar hemorrhages, including cerebral microbleeds (MB) on MRI gradient echo sequence (GRE), but no hemorrhages in deep hemispheric brain locations are highly likely to have CAA according to the Boston criteria.2 Some patients with solitary lobar hemorrhage may have hypertension as the cause, however. When patients present with acute or chronic hemorrhages in both lobar and deep hemispheric brain regions it is unclear whether they have severe hypertensive vasculopathy or a combination of hypertensive vasculopathy and CAA.
MRI white matter hyperintensity (WMH) is another manifestation of cerebral small vessel disease. The relationship between WMH distribution and underlying arterial pathology is unclear. Our previous study of WMH in CAA, Alzheimer’s disease and mild cognitive impairment found little difference in distribution.3 WMH anterior to the temporal horn of the lateral ventricles may be relatively specific for CADASIL, however.4, 5
Few studies have compared the severity and distribution of WMH and mircobleeds in CAA and hypertension. Because hypertensive arteriosclerosis and CAA have different vascular distributions we hypothesized that WMH distribution would also differ. We examined a consecutive series of ICH patients to determine whether there might be a more posterior distribution of WMH in lobar ICH caused by CAA than deep ICH caused by hypertension, reflecting the known posterior distribution of CAA pathology,1, 6, 7 and more brainstem WMH in deep ICH, reflecting the paucity of CAA in this vascular territory.1
METHODS
Study population
Study subjects were recruited from an ongoing single-centerprospective longitudinal cohort study of ICH outcome.8 Eligible subjects were consecutive patients age ≥55 years who presented to Massachusetts General Hospital between January 1, 1999 and December 31, 2002 with supratentorial primary ICH, and had MRI within 90 days. Patients with ICH caused malformations or other secondary causes were excluded; ICH associated with anticoagulant use was included. There were 320 consecutive admissions for supratentorial ICH during the study period; 4 patients with multiple simultaneous symptomatic lobar and deep hemispheric ICH were excluded. MRI was performed within 90 days in 141 out of the 316 eligible patients, with adequate FLAIR sequence in 122 (69 lobar, 53 deep hemispheric) and susceptibility sequence in 113 (67 lobar, 46 deep hemispheric). Patients with deep hemispheric ICH, warfarin-related ICH, larger hemorrhages or those who died of ICH were less likely to get MRI (p>0.05, data not shown).
Baseline clinical information was obtained as described.8 APOE genotype was measured8 in 101/141 (72.1%) of subjects who consented to provision of blood for genetic research.
Imaging acquisition and analysis
MR images were acquired on 1.5 Tesla Signa scanners (GE Medical Systems, Milwaukee, WI). Segmentation of white matter T2 hyperintensity, normalized to average subject head size, was performed using the FLAIR sequence as previously described.9, 10 We created a region of interest encompassing the brainstem to test an a priori hypothesis that brainstem T2 hyperintensity would be less frequently present in lobar ICH. White matter hypodensity (WMH) on CT was graded according to the van Swieten scale.11 Cerebral microbleeds (MB) were identified and labeled on the gradient echo MRI sequence by consensus of two experienced raters as previously described.9
Based on MRI and CT data the hemorrhage locations were categorized as lobar origin or deep hemispheric origin (basal ganglia or thalamus). Lobar ICH patients were classified as definite, probable or possible CAA using the previously validated Boston Criteria.2 Patients with probable CAA had either 2 or more strictly lobar hemorrhages. Patients with possible CAA had a single lobar hemorrhage. We additionally defined a category of mixed ICH based on MRI evidence of hemorrhage, either symptomatic hematoma or MB, in both lobar and non-lobar locations. Cerebellar MB were not considered when assigning the mixed ICH category because of pathological evidence that either CAA or hypertensive vasculopathy can cause cerebellar hemorrhage.12 All imaging data was analyzed by raters blinded to clinical and genetic information.
To compare WMH and CMB distribution across subjects, sequences from each subject were anatomically co-registered to sequence data from a single study subject visually selected as having overall brain and ventricular volumes close to the average values for the study population. Following spatial co-registration of the WMH segmentations, maps were created to display the voxel-wise frequency of WMH across the whole brain in the different study groups. Similarly, labels of the MBs and hematoma origins were anatomically co-registered to produce maps of hemorrhage locations in each group of interest. To test the hypothesis that the distribution of lobar hemorrhages along the anterior-posterior (AP) plan would differ among groups, we determined the position coordinate along the AP plane in co-registered space for each hemorrhage, and compared them across groups. Freesurfer software (surfer.nmr.mgh.harvard.edu) was used for these image analyses.13
Statistical Analysis
Univariate comparisons were by Fisher’s exact test, t-test or Pearson correlation coefficient, or Wilcoxon rank-sum test or Spearman correlation coefficient, as appropriate. WMH was log-transformed to a normal distribution for univariate and multivariable analyses as in other studies.9 Logistic or linear regression models, as appropriate, were used to determine the medical and genetic risk factors that were independently associated with the presence of cerebral MB and log-transformed WMH. First, all the variables associated with the outcome in univariate analysis (p≤0.15) were entered into the model, then backward elimination was used to remove non-significant variables (p>0.05). To test hypotheses about hemorrhage location in the AP plane we used linear models to estimate mean hemorrhage location across groups, using a compound symmetry structure for estimation of the covariance matrix. SAS version 9.1.3 (SAS Institute, Cary, NC) was used for the statistical analyses.
RESULTS
Extent of MRI WMH and Microbleeds in Lobar and Deep Hemispheric ICH
Baseline characteristics of the study cohort with adequate MRI data, grouped according to symptomatic ICH location, are shown in table 1. Median nWMH volume was similar in lobar and deep hemispheric ICH (Table 1, 19.5 cm3 vs. 19.9 cm3, p=0.74). Characteristics independently associated with increased nWMH in the whole study cohort, according to a multivariable linear regression model, were increased age (adjusted p=0.03) and hypertension (adjusted p=0.007).
Table 1.
Characteristics associated with lobar and deep intracerebral hemorrhage (ICH), in subjects who had MRI with analyzable sequence (n=133)
| Characteristic | Lobar ICH N=77, % |
Deep ICH N=56, % |
P value |
|---|---|---|---|
| Age (yrs, mean ± SD) | 73.6 ± 8.1 | 73.9 ± 9.5 | 0.86 |
| Male sex | 51 | 57 | 0.49 |
| Hypertension | 61 | 84 | 0.007 |
| Coronary heart disease | 21 | 21 | 0.99 |
| Diabetes mellitus | 10 | 27 | 0.02 |
| Previous ICH | 5 | 0 | 0.14 |
| Warfarin use | 12 | 9 | 0.78 |
| Pre-ICH cognitive impairment | 25 | 14 | 0.19 |
| ≥1 APOE ε2 allele1 | 21 | 3 | 0.01 |
| ≥1 APOE ε4 allele1 | 34 | 19 | 0.16 |
| nWMH2 | 19.5 [8.7, 45.1] | 19.9 [10.0, 31.1] | 0.74 |
| Brainstem T2 hyperintensity | 44 | 70 | 0.004 |
| Proportion with:3 | |||
| Any microbleeds | 54 | 52 | 0.99 |
| Lobar microbleeds | 49 | 30 | 0.05 |
| Deep microbleeds | 18 | 39 | 0.02 |
| Lobar and deep microbleeds | 13 | 20 | 0.44 |
| Cerebellar microbleeds | 12 | 15 | 0.78 |
| Total number of microbleeds | 1 [0, 7] | 1 [0, 3] | 0.53 |
Continuous variables are median [25th percentile, 75th percentile] except as noted.
nWMH, MRI T2 white matter hyperintensity normalized to average subject head size.
Deep hemispheric ICH patients were more likely than lobar ICH patients to have brainstem T2 hyperintensity (Table 1, 70% vs. 44%, p=0.004). Deep hemispheric ICH location was independently associated with the presence of brainstem T2 hyperintensity (adjusted OR 2.79, 95% CI 1.25–6.24, p=0.01) in a multivariable model controlling for age and hypertension.
As with total nWMH volume, there was no difference between lobar ICH and deep ICH in the proportion with ≥1 MB (54% vs. 52%, p=0.99). Lobar ICH was more commonly associated with ≥1 lobar MB (seen in 49% of lobar ICH and 30% of deep ICH, p=0.05), and deep ICH was more commonly associated with deep MB (seen in 18% of lobar ICH and 39% of deep ICH, p=0.02). The median number of MB was also similar in each group (p=0.53, Table 1). Hypertension was the only clinical characteristic associated with the presence of ≥1 MB (48/79 hypertensive patients had ≥1 MB, while 12/34 non-hypertensives had ≥1 MB, p=0.01).
Distribution of MRI WMH and Microbleeds according to Likelihood of Underlying Cerebral Amyloid Angiopathy or Hypertensive Arteriosclerosis
Next, we divided the study cohort according to categories derived from the published Boston criteria for CAA (Table 2).2 We additionally defined a category of mixed ICH consisting of either patients with lobar ICH and deep hemispheric MB (n=12) or deep hemispheric ICH and lobar MB (n=14). Patients with mixed ICH had a similarly high prevalence of hypertension as patients with deep hemispheric ICH, had more microbleeds than deep hemispheric ICH and tended to have high nWMH volumes, suggesting severe small vessel disease (Table 2).
Table 2.
Characteristics according to hemorrhage category by Boston Criteria
| Characteristic | Lobar Only | Mixed n=26, % |
Deep only n=32, % |
P value | |
|---|---|---|---|---|---|
| Probable CAA n=24, % |
Possible CAA n=31, % |
||||
| Age (mean ± SD) | 73.2±9.7 | 74.9±6.3 | 71.0±8.5 | 75.1± 9.9 | 0.27 |
| Male sex | 50 | 52 | 58 | 59 | 0.87 |
| Hypertension | 63 | 45 | 92 | 81 | <0.0011 |
| Coronary heart disease | 29 | 19 | 19 | 22 | 0.81 |
| Diabetes mellitus | 4 | 16 | 23 | 31 | 0.08 |
| Warfarin use | 13 | 6 | 8 | 16 | 0.63 |
| Pre-ICH cognitive impairment | 21 | 23 | 12 | 22 | 0.71 |
| Total nWMH volume | 27.8 [15.2, 52.0] | 11.4 [4.8, 25.7] | 28.6 [20.1, 39.3] | 16.4 [10.9, 22] | 0.0042 |
| Brainstem T2 hyperintensity | 38 | 41 | 67 | 63 | 0.09 |
| Number of microbleeds | 2 [1, 9] | 0 | 7 [2, 10] | 0 [0, 1] | <0.0013 |
| ≥1 cerebellar microbleed | 8 | 0 | 38 | 9 | <0.0014 |
| ≥1 APOE ε2 allele | 12 | 30 | 5 | 5 | 0.055 |
| ≥1 APOE ε4 allele | 24 | 39 | 19 | 26 | 0.48 |
Continuous variables are median [25th percentile, 75th percentile] except as noted.
Statistical testing is for overall comparison across categories by chi-square test or ANOVA (note: nWMH was first log-transformed to a normal distribution). nWMH, MRI white matter hyperintensity normalized to average subject head size.
Significant pairwise comparisons (uncorrected): probable CAA vs. mixed (p=0.02), possible CAA vs. mixed (p<0.001), possible CAA vs. deep (p=0.004).
Significant pairwise comparisons (uncorrected): probable CAA vs. possible CAA (p=0.02), possible CAA vs. mixed (p=0.003).
Significant pairwise comparisons (uncorrected): probable CAA vs. deep (p<0.001), mixed vs. deep (p<0.001).
Significant pairwise comparisons (uncorrected): probable CAA vs. mixed (p=0.02), mixed vs. deep (p=0.01).
Significant pairwise comparisons (uncorrected): possible CAA vs. mixed (p=0.05), possible CAA vs. deep (p=0.05).
Because CAA and hypertension are different arterial pathologies we explored whether the risk factors for WMH and MB were different in possible or probable CAA, compared to the group with deep hemispheric ICH, using interactions terms in fully adjusted models. Mixed ICH patients were excluded because these patients might have both CAA and hypertensive vasculopathy. There was evidence that hypertension (p=0.03) and age (p=0.07) were differently related to nWMH volume. Hypertension was associated with increased nWMH in CAA-related ICH (hypertensives with CAA-related ICH had 191% larger nWMH volume than non-hypertensives, 95% CI 52%–457%, p=0.003, adjusted for other characteristics) but not in deep ICH (p=0.55). Age was correlated with nWMH volume in CAA-related ICH (nWMH volume was 6.0% greater for each additional year of age, 95% CI 1.5%–10.7%, p=0.005) but not in deep ICH (p=0.76). Risk factors for the presence of MB were similar in CAA-related ICH and deep ICH (p>0.20 for comparisons). We also compared the risk factors for nWMH and MB in mixed ICH patients versus all other ICH types, using similar analyses, and failed to find differences.
The voxel-wise frequency of WMH, grouped according to Boston criteria category, is shown in Figure 1. The distribution of WMH appeared similar in each group, despite some differences in overall extent (Table 2). This was confirmed by voxel-wise statistical tests that failed to show major clusters of voxels where the WMH frequency was different between groups (data not shown).
Figure 1.
Distribution of MRI white matter hyperintensity (WMH) at three levels (rows) according to hemorrhage pattern type (columns). Voxels are color-coded according to the frequency of WMH at that location; only voxels with >5% frequency are coded. CAA, cerebral amyloid angiopathy.
To determine the comparability of MRI and CT measures of WMH, we compared total WMH and distribution (anterior vs. posterior grade) among groups using a commonly employed CT visual rating scale in the 110 with available CT. Similar to the MRI findings, we did not find significant group differences in either total CT-WMH grade (p>0.20 for comparisons) or the within-subject difference between posterior and anterior grade (p>0.20 for comparisons, data not shown). We did confirm a previous finding14 that CT-WMH was of higher grade in the posterior than anterior region in patients with probable or possible CAA, however (p=0.01 by Wilcoxon signed rank test). This posterior predominance of CT-WMH was not detected in the groups with mixed ICH (p=0.99) or deep ICH only (p=0.27).
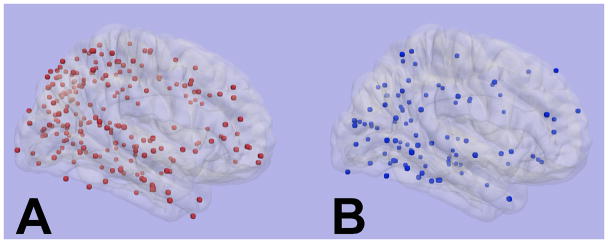
Inspection of the cumulative distribution of hematomas and MBs showed a posterior predominance of the lobar hemorrhages in mixed ICH as well as CAA-associated ICH (Figure 2). Mean hemorrhage location in the anterior-posterior plane was similar between the groups (p=0.99). A single patient with mixed ICH and history of hypertension died and had autopsy; severe CAA and non-CAA-associated arteriolosclerosis was seen (Figure 3).
Figure 2.

Distribution of lobar hemorrhages, including microbleeds, in: A) probable or possible cerebral amyloid angiopathy (n=55 patients), and B) mixed lobar and deep hemorrhages (n=26 patients). The mean hemorrhage location in the anterior-posterior plane was not significantly different (p=0.99, controlling for clustering of hemorrhages by subject).
Figure 3.

A 76 year man died of lobar intracerebral hemorrhage (arrow). MRI GRE sequence (A and B) shows microbleeds in the thalami, basal ganglia and cortex. Autopsy showed severe cerebral amyloid angiopathy in the cortex (C) staining with Congo red (D), with severe arteriosclerosis in the white matter (E) without Congo red staining (F). (C, D: Luxol Fast Blue/H&E stain; E, F: Congo Red stain).
DISCUSSION
Here we show that the WMH volume and frequency of MB are similar in deep hemispheric ICH and lobar ICH. Contrary to our study hypothesis we did not find major differences in supratentorial WMH distribution (Figure 1). The prevalence of brainstem T2 hyperintensity was lower in lobar ICH compared to deep ICH, however, probably because CAA does not affect the arteries that penetrate into the brainstem. The absence of brainstem T2 hyperintensity was not very specific for CAA-related hemorrhage because a substantial number of subjects with CAA had brainstem T2 hyperintensity, reflecting the high population prevalence of hypertension in the elderly.
The periventricular-predominant distribution of WMH in these vascular pathologies with remarkably dissimilar topographical distributions is striking. We have previously reported a similar distribution of WMH in patients with CAA, mild cognitive impairment or Alzheimer’s disease.3 The relatively conserved spatial distribution of WMH across various neurological diseases and syndromes could be related to the spatial distribution of blood flow or spatial variation in the vulnerability of the white matter to ischemia. The periventricular zone is the most distal perfusion zone of the white matter and experiences the lowest resting blood flow.3 Therefore blood flow in this region could be readily compromised regardless of the distribution of vascular pathology. The observation that white matter lesions in CAA are most frequently periventricular, whereas vascular amyloid deposition is mostly restricted to the pial or cortical course of the arteries,1 suggests to us that white matter injury in CAA occurs by impairment of blood flow, possibly caused by altered vascular reactivity.15
In our study we found that lobar hematoma was more closely associated with lobar MB than deep MB, and that deep hemispheric hematoma was more closely associated with deep MB than lobar MB, as previously observed.16 However we also found a relatively high prevalence of mixed ICH (26/113, 23%). Mixed ICH may result from either hypertensive vasculopathy of both the deep and cortical arteries, or from a combination of hypertensive vasculopathy and CAA in the same patient, which would not be unexpected given the relatively high prevalence of hypertension and CAA in the elderly.17, 18 A previous autopsy series found that 7/40 deep ICH patients also had pathologic evidence of CAA.19 There was some evidence from our data to support either possibility. The prevalence of hypertension and brainstem WMH in the group with mixed ICH was similar to the group with deep hemispheric ICH and higher than the group with probable or possible CAA (Table 2). On the other hand, a cumulative map of the lobar hemorrhages in the group with mixed ICH showed a posterior distribution of hemorrhages, consistent with the distribution of hemorrhages seen in probable or possible CAA (Figure 2). A posterior distribution of hemorrhages in CAA, favoring the occipital, parietal and posterior temporal lobes, has previously been observed7, 20, 21 and likely reflects the distribution of vascular amyloid deposition.1, 6, 7 This evidence is not conclusive, however, because some studies of hypertensives and ischemic stroke patients also suggest a relatively posterior distribution of lobar microbleeds,22, 23 although a contribution from coexistent CAA cannot be ruled out.
In contrast to the posterior predominance of hemorrhages and microbleeds, we did not observe a major posterior predominance of MRI WMH in CAA, based on visual inspection of the WMH frequency maps (Figure 1). Despite this, we did confirm a previous finding that posterior CT-WMH was greater than anterior CT-WMH in CAA patients, but could not demonstrate this posterior predominance in the groups with deep hemispheric ICH or mixed ICH, suggesting that it may be feature of CAA. This discrepancy between MRI and CT measures may reflect differences between the type and severity of white matter injuries detected by these two modalities.
WMH volumes were highest in the groups with probable CAA or mixed ICH, likely reflecting more severe small vessel disease. Age and hypertension were risk factors for WMH in the overall study cohort, while hypertension was a risk factor for the presence of MB. Hypertension was associated with WMH in probable or possible CAA but not in deep hemispheric ICH, and was associated with MB in both groups, suggesting that hypertension may add to vascular pathology even in the presence of CAA. The finding of higher WMH volumes in patients with mixed ICH also supports an additive effect of both pathologies. The most likely reason we could not detect a relationship between WMH and hypertension in deep ICH is that few deep hemispheric ICH patients were non-hypertensive, and some may have had unrecognized hypertension before ICH. There was a trend toward a stronger relationship between age and WMH in the group with CAA (p=0.07).
There are several limitations to this study. MRI was only performed in a subset of patients with less severe hemorrhagic stroke severity. The sample size is small and the findings reported here should be confirmed in subsequent larger studies. This is a hospital-based study therefore pre-ICH characteristics were by necessity assessed in retrospect.
These findings have implications for the pathophysiology, diagnosis and management of cerebral small vessel disease. We found that a similar pattern of WMH distribution was present in CAA and hypertensive hemorrhage, supporting the concept that anatomic location is a more important determinant of white matter vulnerability to WMH than the specific small vessel pathology.3 WMH volume was greater in CAA patients who additionally had hypertension. Because WMH have previously been associated with cognitive impairment in lobar ICH patients,14 this finding raises the possibility that strict control of hypertension might reduce disability from CAA. Many consecutive ICH patients with MRI had evidence of mixed lobar and deep hemorrhages on the MRI GRE sequence. These patients would not be diagnosed with CAA based on the Boston criteria, which require that all hemorrhages be lobar in origin, but our data suggest that some of these patients may have CAA. Therefore while the Boston criteria for CAA are highly specific, and are useful for research by allowing selection of a population that is very likely to have CAA, it must be recognized that the true overall burden of CAA-related disease could be underestimated by the Boston criteria. Newer approaches to the diagnosis of CAA, including molecular imaging of beta-amyloid,6 may improve our ability to define the true prevalence of this condition.
Acknowledgments
FUNDING
This study was funded by grants from the National Institute of Neurological Disorders and Stroke (K23NS046327 and K24 NS056207) and National Institute on Aging (R01AG026484).
Footnotes
CONFLICTS OF INTEREST/DISCLOSURES
The authors report no relevant relationships with pharmaceutical or other companies or corporations.
References
- 1.Vinters HV, Gilbert JJ. Cerebral amyloid angiopathy: Incidence and complications in the aging brain. Ii. The distribution of amyloid vascular changes. Stroke. 1983;14:924–928. doi: 10.1161/01.str.14.6.924. [DOI] [PubMed] [Google Scholar]
- 2.Knudsen KA, Rosand J, Karluk D, Greenberg SM. Clinical diagnosis of cerebral amyloid angiopathy: Validation of the boston criteria. Neurology. 2001;56:537–539. doi: 10.1212/wnl.56.4.537. [DOI] [PubMed] [Google Scholar]
- 3.Holland CM, Smith EE, Csapo I, Gurol ME, Brylka DA, Killiany RJ, Blacker D, Albert MS, Guttmann CR, Greenberg SM. Spatial distribution of white-matter hyperintensities in Alzheimer disease, cerebral amyloid angiopathy, and healthy aging. Stroke. 2008;39:1127–1133. doi: 10.1161/STROKEAHA.107.497438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O’Sullivan M, Jarosz JM, Martin RJ, Deasy N, Powell JF, Markus HS. Mri hyperintensities of the temporal lobe and external capsule in patients with CADASIL. Neurology. 2001;56:628–634. doi: 10.1212/wnl.56.5.628. [DOI] [PubMed] [Google Scholar]
- 5.Auer DP, Putz B, Gossl C, Elbel G, Gasser T, Dichgans M. Differential lesion patterns in CADASIL and sporadic subcortical arteriosclerotic encephalopathy: MR imaging study with statistical parametric group comparison. Radiology. 2001;218:443–451. doi: 10.1148/radiology.218.2.r01fe24443. [DOI] [PubMed] [Google Scholar]
- 6.Johnson KA, Gregas M, Becker JA, Kinnecom C, Salat DH, Moran EK, Smith EE, Rosand J, Rentz DM, Klunk WE, Mathis CA, Price JC, Dekosky ST, Fischman AJ, Greenberg SM. Imaging of amyloid burden and distribution in cerebral amyloid angiopathy. Ann Neurol. 2007;62:229–234. doi: 10.1002/ana.21164. [DOI] [PubMed] [Google Scholar]
- 7.Jellinger KA, Lauda F, Attems J. Sporadic cerebral amyloid angiopathy is not a frequent cause of spontaneous brain hemorrhage. European Journal of Neurology. 2007;14:923–928. doi: 10.1111/j.1468-1331.2007.01880.x. [DOI] [PubMed] [Google Scholar]
- 8.O’Donnell HC, Rosand J, Knudsen KA, Furie KL, Segal AZ, Chiu RI, Ikeda D, Greenberg SM. Apolipoprotein e genotype and the risk of recurrent lobar intracerebral hemorrhage. N Engl J Med. 2000;342:240–245. doi: 10.1056/NEJM200001273420403. [DOI] [PubMed] [Google Scholar]
- 9.Gurol ME, Irizarry MC, Smith EE, Raju S, Diaz-Arrastia R, Bottiglieri T, Rosand J, Growdon JH, Greenberg SM. Plasma beta-amyloid and white matter lesions in AD, MCI, and cerebral amyloid angiopathy. Neurology. 2006;66:23–29. doi: 10.1212/01.wnl.0000191403.95453.6a. [DOI] [PubMed] [Google Scholar]
- 10.Nandigam RN, Chen YW, Gurol ME, Rosand J, Greenberg SM, Smith EE. Validation of intracranial area as a surrogate measure of intracranial volume when using clinical mri. J Neuroimaging. 2007;17:74–77. doi: 10.1111/j.1552-6569.2006.00069.x. [DOI] [PubMed] [Google Scholar]
- 11.van Swieten JC, Hijdra A, Koudstaal PJ, van Gijn J. Grading white matter lesions on ct and mri: A simple scale. J Neurol Neurosurg Psychiatry. 1990;53:1080–1083. doi: 10.1136/jnnp.53.12.1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Itoh Y, Yamada M, Hayakawa M, Otomo E, Miyatake T. Cerebral amyloid angiopathy: a significant cause of cerebellar as well as lobar cerebral hemorrhage in the elderly. Journal of the Neurological Sciences. 1993;116:135–141. doi: 10.1016/0022-510x(93)90317-r. [DOI] [PubMed] [Google Scholar]
- 13.Fischl B, Salat DH, Busa E, Albert M, Dieterich M, Haselgrove C, van der Kouwe A, Killiany R, Kennedy D, Klaveness S, Montillo A, Makris N, Rosen B, Dale AM. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron. 2002;33:341–355. doi: 10.1016/s0896-6273(02)00569-x. [DOI] [PubMed] [Google Scholar]
- 14.Smith EE, Gurol ME, Eng JA, Engel CR, Nguyen TN, Rosand J, Greenberg SM. White matter lesions, cognition, and recurrent hemorrhage in lobar intracerebral hemorrhage. Neurology. 2004;63:1606–1612. doi: 10.1212/01.wnl.0000142966.22886.20. [DOI] [PubMed] [Google Scholar]
- 15.Smith EE, Vijayappa M, Lima F, Delgado P, Wendell L, Rosand J, Greenberg SM. Impaired visual evoked flow velocity response in cerebral amyloid angiopathy. Neurology. 2008;71:1424–1430. doi: 10.1212/01.wnl.0000327887.64299.a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee SH, Bae HJ, Kwon SJ, Kim H, Kim YH, Yoon BW, Roh JK. Cerebral microbleeds are regionally associated with intracerebral hemorrhage. Neurology. 2004;62:72–76. doi: 10.1212/01.wnl.0000101463.50798.0d. [DOI] [PubMed] [Google Scholar]
- 17.Pathological correlates of late-onset dementia in a multicentre, community-based population in england and wales. Neuropathology group of the medical research council cognitive function and ageing study (mrc cfas) Lancet. 2001;357:169–175. doi: 10.1016/s0140-6736(00)03589-3. [DOI] [PubMed] [Google Scholar]
- 18.Hajjar I, Kotchen TA. Trends in prevalence, awareness, treatment, and control of hypertension in the united states, 1988–2000. JAMA. 2003;290:199–206. doi: 10.1001/jama.290.2.199. [DOI] [PubMed] [Google Scholar]
- 19.Ritter MA, Droste DW, Hegedus K, Szepesi R, Nabavi DG, Csiba L, Ringelstein EB. Role of cerebral amyloid angiopathy in intracerebral hemorrhage in hypertensive patients. Neurology. 2005;64:1233–1237. doi: 10.1212/01.WNL.0000156522.93403.C3. [DOI] [PubMed] [Google Scholar]
- 20.Rosand J, Muzikansky A, Kumar A, Wisco JJ, Smith EE, Betensky RA, Greenberg SM. Spatial clustering of hemorrhages in probable cerebral amyloid angiopathy. Ann Neurol. 2005;58:459–462. doi: 10.1002/ana.20596. [DOI] [PubMed] [Google Scholar]
- 21.Pettersen JA, Sathiyamoorthy G, Gao FQ, Szilagyi G, Nadkarni NK, St George-Hyslop P, Rogaeva E, Black SE. Microbleed topography, leukoaraiosis, and cognition in probable alzheimer disease from the Sunnybrook Dementia Study. Archives of Neurology. 2008;65:790–795. doi: 10.1001/archneur.65.6.790. [DOI] [PubMed] [Google Scholar]
- 22.Lee SH, Kim SM, Kim N, Yoon BW, Roh JK. Cortico-subcortical distribution of microbleeds is different between hypertension and cerebral amyloid angiopathy. Journal of the Neurological Sciences. 2007;258:111–114. doi: 10.1016/j.jns.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 23.Lee SH, Kwon SJ, Kim KS, Yoon BW, Roh JK. Cerebral microbleeds in patients with hypertensive stroke. Topographical distribution in the supratentorial area. J Neurol. 2004;251:1183–1189. doi: 10.1007/s00415-004-0500-6. [DOI] [PubMed] [Google Scholar]