

Abstract
Allosterism is an essential biological regulatory mechanism. In enzymes, allosteric regulation results in an activation or inhibition of catalytic turnover. The mechanisms by which this is accomplished are unclear and vary significantly depending on the enzyme. It is commonly the case that a metabolite binds to the enzyme at a site distant from the catalytic site yet its binding is coupled to and sensed by the active site. This coupling can manifest in changes in structure, dynamics, or both at the active site. These interactions between allosteric and active site, which are often quite distant from one another involve numerous atoms as well as complex conformational rearrangements of the protein secondary and tertiary structure. Interrogation of this complex biological phenomenon necessitates multiple experimental approaches. In this article, we outline a combined solution NMR spectroscopic and computational approach using molecular dynamics and network models to uncover mechanistic aspects of allostery in the enzyme imidazole glycerol phosphate synthase.
Keywords: Molecular dynamics, relaxation dispersion, spin-relaxation, network analysis, methyl dynamics
A. Introduction
Allostery is an essential regulatory mechanism in biology whereby enzyme catalytic activity or protein ligand affinity can be modulated in response to changes in the local environment. Thus, changes in the concentrations of protons 1, metal ions 2, inhibitors 3, 4, or metabolites 5 as well as changes in temperature 6 can manifest as alterations in biological processes. Enzymes that respond to allosteric ligands possess at least two spatially distinct binding sites – the active site for the substrate and the effector site for the allosteric ligand. These two sites are linked both thermodynamically and functionally 7. In the case of an enzyme, the binding of the effector molecule alters the active site such that either the equilibrium of the enzyme/substrate binding event is altered or the effector modulates the properties of the catalytic active site to change the rate of the chemical reaction. These two types of allostery are termed K (for equilibrium) or V (for reaction velocity) type, respectively. Because allostery exerts control over enzyme function, it is of great importance to develop a full understanding of allosteric properties at the atomic level. Moreover, amino acid residues involved in these allosteric pathways present unique locations, distinct from the active site, to focus small molecule or drug design efforts8 to modulate control over a particular biological pathway.
The functional interaction between the allosteric site and active site requires that the two sites be linked, thermodynamically 9, 10, which has been borne out from double mutant cylcle experiments11, 12. The coupling free energy (ΔΔG) between the allosteric site and active site can, of course, result from both structural and dynamical effects13–19 and the standard phenomonological models of allostery16, 20 have provided a framework through which much insight into the workings of allosteric proteins has been gleaned. Continued study of allostery requires atomistic level detail to decipher how ligand-binding information is passed between two distant sites through a dense protein matrix that involves both side chain groups and the polypeptide backbone scaffold21, 22. The resulting complexity of the allosteric phenomenon requires multiple approaches to obtain the necessary detail for a fuller understanding. In collaboration with the Batista group at Yale, we have combined solution NMR relaxation experiments with molecular dynamics (MD) simulations and network models to address the allostery in the V-type enzyme, imidazole glycerol phosphate synthase (IGPS). In this protein system discussed below, significant evidence has accumulated suggesting a major role for protein motions in allostery. Below, we discuss the methods used and the insight obtained through a combined application of solution NMR spectroscopy and computational methods.
B. Experimental
Solution NMR spectroscopy is an ideal experimental technique for obtaining atomic detail of dynamic processes in biological macromolecules. Solution NMR experiments can be performed under physiological conditions of solvent, pH, and temperature, thereby making a more direct correlation to the in vivo function. The incorporation of spin-1/2 nuclei into proteins, in addition to being non-perturbing, is relatively straightforward and bacterial expression systems facilitate production of significant quantities of the desired protein sample. Moreover, NMR spectroscopy is a non-destructive technique; therefore the sample can be recovered for subsequent investigations. Crucially, to further our understanding of allostery, NMR provides atomic resolution and access to site-specific dynamic information ranging in time resolution from the picosecond (ps) to tens of seconds. For the purposes of this work, focus is placed on the characterization of fast (ps – ns) and slower (μs – ms) timescale motions. Fast timescale molecular motions perturb the dipolar and chemical shift anisotropy (CSA) Hamiltonians whereas the chemical shift Hamiltonian is modulated by μs – ms motions. In the description below, we focus on 13C-methyl groups, though the theoretical treatment and experimental measurements of other nuclei share many similarities 23.
Fast timescale motions in the ps – ns regime reflect stochastic, equilibrium fluctuations of individual bond vectors. As such NMR measurements of the amplitudes of these fluctuations have been used as surrogates for conformational entropy 24–26 For example, rigidification of bond vectors upon ligand binding, indicate protein regions that ‘pay an entropy price’ due to the interaction with ligand 27. Because of the connection to thermodynamics that underlie these fast timescale motions, they have been particularly useful when trying to identify amino acid residue networks involved in allostery 24,28. Fast timescale motions of methyl groups (δ1, δ, γ, respectively) in the hydrophobic amino acids, Ile, Leu, and Val (ILV), can be assessed by measurement of the intra-methyl 1H-1H dipolar cross-correlated relaxation rate constant, η, i n which 29,
| (1) |
where rHH is the distance between two methyl protons (1.813 Å), γH is the proton gyromagnetic ratio, the rotational correlation time of the protein is given by τc. P2(x) is the second Legendre polynomial where θaxis,HH is the angle between the 3-fold axis of the methyl group and the vector between two methyl protons and is assumed to be 90°, S2 is the generalized order parameter for the motion of the methyl 3-fold symmetry axis and describes its amplitude of equilibrium fluctuations. and are the relaxation rates for the single quantum 1H coherences and denote the fast and slow relaxing transitions, respectively. As shown in equation (1), determination of the amplitude of methyl group motion amounts to measuring η, which is achieved by comparing peak intensities from a two HMQC-based NMR pulse sequences, which differ by a 1H 90° pulse. In one experiment, the individual methyl peak intensity (Ia) is given by
| (2) |
where T is the relaxation time during which 1H transverse magnetization is allowed to return toward equilibrium. In the second experiment in which the 1H 90° is omitted, the peak intensity Ib is
| (3) |
Thus, it is readily noted that the ratio of Ia and Ib provides the 1H-1H dipolar cross-correlated relaxation rate as shown in equation (4) and as measured for IGPS in Figure 1,
Figure 1.

1H-1H dipolar cross-correlated relaxation data for IGPS. The ratio of peak intensities for I93 in Apo IGPS and V158 in acivicin bound IGPS are presented with the solid line being the fit of equation (5) to the data points. Data were acquired at 14.1 T, pH = 7.3, 303 K.
| (4) |
Equation (4) is valid in the case of isolated methyl group. Under conditions where external protons are in close proximity to the CH3 group, the Ia:Ib ratio becomes
| (5) |
that contains an additional parameter δ, which captures the contribution due to remote dipolar interactions as given by
| (6) |
Deuteration of remote protons can significantly minimize these remote 1H dipolar effects. Nonetheless, measurement of η is a sensitive indicator of changes in fast timescale motions in the protein under study. Figure 2 shows measured η values versus primary sequence in the V-type allosteric enzyme, IGPS.
Figure 2.
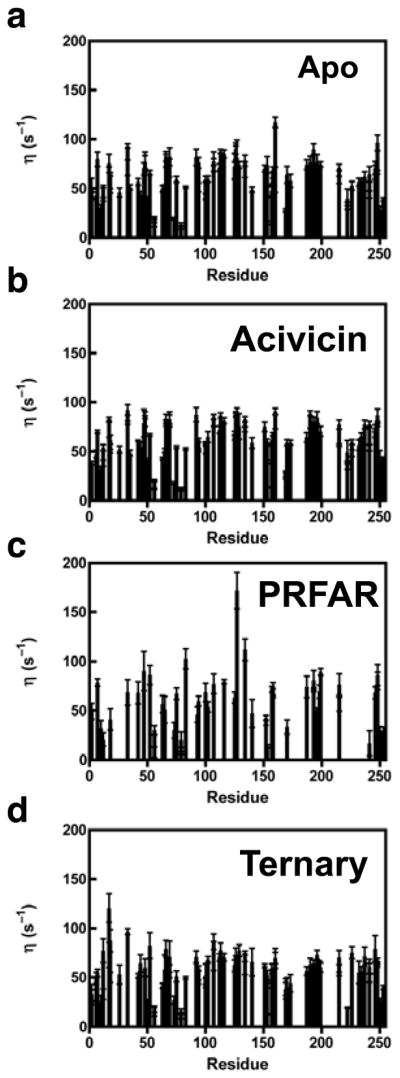
1H-1H dipolar cross-correlated relaxation rate constants (η) determined as shown in (5) for IGPS. Panels a-d show η values for apo, acivicin-bound, PRFAR-bound, and the ternary (acivicin and PRFAR) complex respectively. Data were acquired at 14.1 T, pH = 7.3, 303 K.
In addition to these fast timescale motions it is often the case that allosteric proteins undergo significant conformational changes to convert between active and inactive states. These changes involve barrier-crossing motions, typically involving multiple amino acid residues and therefore the timescale for these motions is in the micro-millisecond (μs – ms) regime. NMR Carr-Purcell-Meiboom-Gill (CPMG) relaxation dispersion experiments30, introduced for the study or 15NH groups in proteins31, and extended and adapted to numerous spin-1/2 pairs in proteins32–37, including methyl groups38–42 represent a powerful set of experiments for characterization of these slower motions. Motions on the μs - ms timescale modulate the isotropic chemical shift of the atoms involved, provided the moving atom experiences unique magnetic environments in, it is assumed, two conformations. This fluctuating magnetic field, whether caused by motion of the site in question or motion of surrounding groups, leads to exchange broadening of the resonance and an increase in the observed transverse relaxation rate (R2). The details of conformational exchange and its effect on relaxation rates have been reviewed previously23. Briefly, the aforementioned relaxation dispersion experiment measures the R2 at varying delays between the 180° CPMG refocusing pulses, which results in modulation of the measured R2 value. For a two-site conformational exchange process of an allosteric enzyme given by
| (7) |
The measured R2 value is a function of the 180° pulse spacing (τcp), the exchange rate constant kex = k1+k−1, the equilibrium populations pA and pB for the active and inactive conformations, respectively, and the chemical shift difference, Δω for the nuclei under observation, in its two conformations. One particularly attractive experiment for characterizing conformational motions in allosteric enzymes, which are often of significant molecular weight, is the multiple quantum (MQ) relaxation dispersion experiment applied to the methyl groups of ILV residues43. The analytical expression for the R2 dependence on conformational exchange processes is given by
| (8) |
These experiments, through non-linear fitting of the relaxation data, can provide detailed information on the timescale of motions, the equilibrium populations for the conformational exchange process, and the chemical shift differences between the two conformations, which can give insight into the structures of the lowly populated, unobservable conformation. Furthermore, careful analysis of relaxation dispersion data can indicate whether the conformational change is due to the concerted motions of many residues or if motions of individual groups are independent and uncorrelated with each other. These MQ relaxation dispersion experiments were applied to IGPS (Figure 3) and demonstrate the ability to monitor ms motions, site-specifically in this large allosteric enzyme. Computation methods provide a powerful complement to these NMR studies by providing access to atomistic details of molecular motions and the correlation of motions between residues.
Figure 3.

Summary of 1H-13C MQ CPMG results for the isoleucine, leucine, and valine methyl groups of IGPS in the binary and ternary complex. Panels a and b show representative multiple quantum CPMG dispersion curves for binary (blue squares) and ternary (red circles). Panel c highlights the residues that undergo millisecond exchange in the both binary and ternary complexes (purple spheres), and the subsets of residues that undergo millisecond motions in only the binary (blue spheres) or ternary (red spheres) complexes. The HisH subunit is shown as blue ribbon and HisF and dark gray. T. maritima IGPS crystal structure (1GPW). The distance between the glutaminase domain (HisH) catalytic residues and the PRFAR binding site in the cyclase domain (HisF) are indicated by the vertical arrow.
C. Computational
Although NMR relaxation experiments provide a wealth of information on molecular motions over a range of time scales, detailed mechanistic information on allostery is not easily obtained from the aforementioned NMR experiments. This mechanistic detail is essential for a fundamental understanding of the allosteric behavior and, additionally, is crucial for aiding in the design of allosteric drugs that interact with the target, blocking the allosteric signal and altering the enzyme response. The atomistic detail on amino acid motions derived from molecular dynamics (MD) simulations is a powerful complement to solution NMR studies to shed further light on allosteric mechanisms.
MD simulations and NMR data can be combined to develop an understanding of how the allosteric signal, initiated by the effector molecule, which is initially confined in the effector binding site and is involved in only a few atomic interactions, can be propagated as large-scale conformational transition that involve hundreds of atoms that span tens of Å. Allosteric transitions of this type often result in conformational changes of significant magnitude that take place in the μs-ms timescale, a time regime that cannot be sampled by means of standard MD techniques. However, the binding of the allosteric effector molecule increases the population of active conformations by modulating protein motions at different time scales and inducing (or accelerating) the allosteric transition. Standard MD simulations can be used to analyze the thermal motion of atomic sites in the ps-ns (up to submicroseconds) time scale. In this time regime, several protein motions take place, including side chain motion of hindered and unhindered surface residues, methyl group rotations, loop motion, collective motion of a few residues, helix-coil transitions and folding of small peptides 44, 45. Modification of these protein motions caused by effector binding can be captured by correlation analysis of motions observed along MD trajectories and combined with observations obtained by solution NMR spectroscopic data.
The cross-correlation analysis of MD trajectories is routinely used for quantifying correlated motion in proteins 46, 47. Cross-correlations between residue motions are obtained from the Pearson correlation coefficient
| (9) |
where xi,j are the fluctuation vectors of atoms i and j, defined as the deviation from the mean, i.e. x= v – <v>, of the atomic position vector v, with < > denoting the ensemble average. The Pearson coefficient depends on the relative orientation of the atomic fluctuations and is exclusively limited to linear correlations, with significant limitations in the quantification of motional correlations. A generalized correlation coefficient based on the measure of mutual information (MI) is able to capture non-linear correlations and it is independent of the relative orientation of the atomic fluctuations 48. The MI for an ensemble of random variables xi (with i=1,.., N) with marginal distribution pi(xi) and joint probability distribution p(x), is defined as
| (10) |
MI gives the ensemble-averaged deviation from the uncorrelated distribution, which vanishes in the case of independent random variables, i.e. MI vanishes for the bivariate case of the fluctuation of the vectors xi and xj for fully uncorrelated motions of the two residues i and j. The mutual information in the case of bivariate random variables is determined from,
| (11) |
where H[xi], H[xj] and H[xi, xj] are the marginal and joint Shannon entropies, for residues i and j respectively. The Shannon entropy H[x] is the value of the information content for the discrete random variable x, which has probability mass function p(x),49
| (12) |
According to Eq.(10), MI values fall in the range [0,..,∞], in contrast with the Pearson coefficient, in which values vary from −1 to 1. For a gaussian multidimensional distribution with unit variance, the generalized correlation coefficient, rMI, defined as
| (13) |
and is used in a manner similar to the Pearson coefficient but instead, obtained from the mutual information, with rMI ranging from zero for fully uncorrelated variables and to 1 for fully correlated variables.
The rMI coefficient, thus, quantifies correlations in an intuitive way and it represents a measure of the mutual information defined by the Shannon entropy, allowing for a direct measure of the entropy redistribution within a protein. Therefore, the generalized correlation coefficient can be used to track the changes in correlated motions induced by binding of the allosteric effector, providing insights into the information flow in an allosteric enzyme. Figure 4, shows how the generalized correlation coefficient, rMI captures the non-linear correlations missed in the Pearson coefficients r[xi, xj], providing a robust measure of protein motional correlations. Changes in correlated protein motions due to the allosteric effector binding may reveal the pathway of allosteric information transfer, suggesting which amino acid residues are integral to the allosteric mechanism.
Figure 4.

Pearson coefficient r[xi, xj] (upper left triangle) and generalized correlation coefficient rMI (lower right triangle) correlation matrices for the case of the apo imidazole glycerol phosphate synthase enzyme. The same color mappings are used for a direct comparison. The rightmost color bar shows the color range for the respective correlation coefficient. Reproduced from Reference 21.
Allosteric enzymes are complex systems in which the signal is transferred for long distances (usually more than 15 Å) utilizing many amino acid residues, involving multiple secondary structures and often, different protein domains. The application of network theory methods for the study of allosteric systems, allows for a simple analysis of the information flow in such complex systems. The protein network of an allosteric enzyme can be defined as the set of amino acid residues, i.e. the network nodes, connected by edges whose length is weighted using the generalized correlation coefficient rMI. In particular, the length (or weight), wij, of the edge that connects nodes i and j, is calculated as
| (14) |
where rij is the corresponding generalized correlation coefficient as defined by Eq.(13). In such a weighted graph, nodes that are closely bound represent residues with highly correlated motions that could exchange a large amount of information, while distant nodes represent pairs of residues that are poorly correlated. Such protein dynamical networks contain information paths and critical nodes that are important for communication within the allosteric complex21, 22, 50.
The protein dynamical network can be partitioned into local substructures that may reveal secondary structures or portions of protein that participate more significantly to the signal transduction and to the overall allosteric mechanism. Using the Girvan-Newmann algorithm51, the weighted graph can be partitioned in “communities”, i.e. groups of nodes within which the network connections are dense but between which they are sparser, using the edge betweenness as the criterion for partitioning. The edge betweenness is defined as the number of shortest pathways that cross an edge and it is a measure of information passing through such an edge within the network. The optimal community structure of the protein complex is obtained by measuring the quality of the community structures obtained during the partitioning procedure. The quality of a given community structure is defined by the “modularity” parameter, i.e. the difference in probability of intra- and inter-community connections. In particular, if eij is the fraction of edges that links nodes in community i to nodes in community j, the modularity, Q, is defined as 52
| (15) |
where ai= Σj eij is the fraction of edges that connect to the nodes in community i. Figure 5 shows a schematic representation of an optimal community structure obtained after the community network analysis of an allosteric protein complex.
Figure 5.
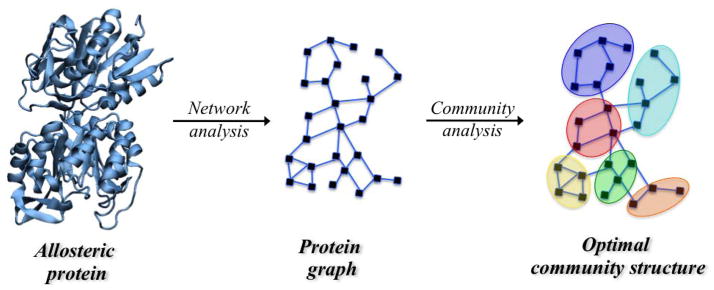
Schematic representation of an optimal community structure obtained by means of the community network analysis of an allosteric protein complex. Black squares (nodes, i.e. amino acid residues) are connected by edges (blue lines) weighted using the generalized correlation coefficient according to Eq. 15.
D. Applications: Imidazole glycerol phosphate synthase
IGPS is a heterodimeric enzyme composed of two catalytic subunits, HisH and HisF (Figure 3). Allostery in IGPS synchronizes ammonia production in HisH via the hydrolysis of glutamine in the HisH active site, with the cleavage and cyclization of N′-[(5′-phosphoribulosyl)formimino]-5-aminoimidazole-4-carboxamide-ribonucleotide (PRFAR) in the active site of HisF to form imidazole glycerol phosphate (IGP) and 5-aminoimidazole-4-carboxamide ribotide (AICAR). IGPS allostery is of the V-type in which catalytic activity, not substrate affinity is altered by the allosteric ligand and vice versa 53, 54. In IGPS, glutamine binding to HisH is unaffected by binding of the allosteric effector (and substrate) PRFAR to HisF. Yet, despite being separated by ~25 Å these two sites are in communication and as a result, PRFAR binding enhances the rate of glutamine hydrolysis by nearly 5000-fold.55 The allostery in IGPS serves to link the chemistry of ammonia production with the binding of PRFAR, which subsequently reacts with the newly generated ammonia. Functionally, allostery in the context of IGPS prevents unnecessary waste of glutamine by HisH if the second substrate, PRFAR is not present in the cell. IGPS is a model for how the information of ligand binding can be transmitted over significant molecular distances to elicit a biochemical response. One aspect of the activation of the HisH active site involves reorienting the ‘oxyanion strand’ residues (h49 –h52)1, which in the apo form of IGPS are positioned such that the C′ of G50 is suboptimally oriented, proximal to the site of the developing negative charge that is characteristic of the tetrahedral intermediate, which forms during the Gln hydrolysis reaction21, 53, 56, 57 (Figure 6). To facilitate the chemistry of NH3 production this strand must rotate nearly 180° such that the position the HN of hV51 now resides in the oxyanion hole site (Figure 6) 58. In the apo enzyme neither HN of hV51 nor C′ of hG50 exhibits crystallographic b-factors greater than those observed for much of the protein. Thus, these residues, which must move for chemistry to occur, do not appear to have an innate tendency to do so in the absence of its allosteric ligand.
Figure 6.
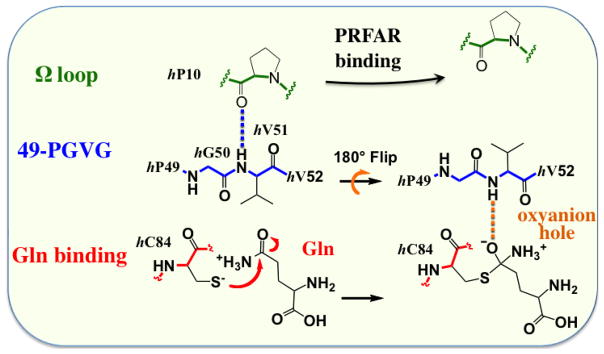
Schematic representation of the proposed conformational changes that are required for proper stabilization of oxyanion formed during the Gln hydrolysis reaction. These changes involve reorientation of the ‘oxyanion strand’ (49-PGVG sequence). Adapted from Reference 21.
The mechanism of allostery in IGPS is not fully understood. Crystal structures of IGPS in binary ligand complexes (either bound to Gln analogs or PRFAR) have provided some insight into the changes that occur upon binding the effector, PRFAR 56, 57, 59, 60. There are no large secondary or tertiary structural changes observed between the crystal structures of apo and PRFAR binary IGPS. Furthermore, the lack of a three-dimensional structure for the ternary complex (PRFAR and Gln (or an analog) leaves a void in the current understanding of allosteric activation in IGPS. Movement of the oxyanion strand is proposed to stabilize the negative charge that develops on the Gln Oε1 substrate atom during hydrolysis 59. While movement of loop regions is proposed to be crucial for successful catalysis, these rearrangements do not appear to come from significant tertiary structure rearrangements or domain reorientation 44.
Fast timescale motions (picosecond-nanosecond) measured by the intra-methyl 1H-1H dipolar cross-correlated relaxation rate constant for apo, binary acivicin, binary PRFAR, and ternary IGPS complexes allow for the assessment of the importance of fast timescale motions of a subset of methyl containing sidechains in allosteric communication. In Figure 1, representative data from intra-methyl 1H-1H dipolar cross-correlated relaxation experiments are shown for I93 and V158. η has a direct relation to the generalized order parameter, S2, and is interpreted as a measure of the methyl group rigidity (Equation 1). Comparison of the η values for each ILV methyl group allows for the comparison of ps-ns motion between the different ligand-bound forms of IGPS (Figure 2). Minimal overall changes in methyl group η values are observed between apo, binary acivicin, binary PRFAR, and ternary IGPS ligand complexes. The lack of a significant difference between the global average η values suggest that ps-ns motions of methyl containing sidechains do not play a significant role in the large, positive entropy change when PRFAR binds 53. However, analysis of site-specific changes in η (Figure 2) show distinct increases and decreases that depend on ligand and furthermore, indicate there is some response of the fast motions particularly due to the binding of the allosteric effector, PRFAR. This analysis provides some insight into regions of IGPS that alter their ps-ns motions in response to binding the allosteric activator. However, the simultaneous enhancement of ms motions upon binding PRFAR results in exchange broadening and loss of signal for several of these ILV sidechain resonances (Figure 2c), precluding further study.
While the structure IGPS is not significantly altered by the presence of the effector or the substrate, the millisecond motions of the ILV side chains as measured by CPMG relaxation dispersion methods change significantly. In apo IGPS, a limited number of ILV side chains experience millisecond exchange motions (Figure 3). There are few alterations in ms apo-like ms motions, when the glutamine mimic, acivicin, is bound to the HisH subunit. However, the addition of PRFAR causes a significant, widespread increase in the number of ILV methyl groups experiencing millisecond motions in HisF. Similarly, in the ternary complex with both acivicin and PRFAR bound, the extent of millisecond exchange is enhanced throughout the HisF domain. The residues affected by PRFAR differ between the binary and ternary complexes. In a comparison between the four IGPS complexes a unique subset of eight sidechains undergoing conformational exchange were identified, which includes V12, V17, V48, L63, I116, V140, V190, and L196 (Figure 3). These residues, many of them conserved, are uniquely flexible in the IGPS ternary complex. In addition, biochemical experiments have identified several of these residues as being functionally important or located adjacent to functionally important residues.
The coupling free energy of allostery is realized in the ternary complex, and comparison of this ternary complex with apo and binary complexes allows for the assessment of the thermodynamics in the allosteric system 9. While acivicin and PRFAR have an effect, small and large respectively, on where and to what extent motions are altered relative to apo-IGPS, the differences caused by the synergistic relationship of acivicin and PRFAR in the ternary complex are the most interesting when assessing allosteric communication (Figure 7). Unique millisecond conformational exchange motions in the ternary complex provide insight into the role allostery plays in regulating the strict one-to-one stoichiometric ratio between the consumption of glutamine and PRFAR. The activation of millisecond exchange of residues unique to the ternary complex by the presence of both substrates shows a contiguous link from the HisF active site to the interface of IGPS providing a network of residues for allosteric communication between active sites. The synergistic effect of acivicin and PRFAR binding loosens the rigid protein structure allowing for a shift to the active state necessary to enhance catalysis and suggests that ms motions are part of the activation process that results in formation of the functional oxyanion hole 53.
Figure 7.

Residues with methyl groups undergoing millisecond exchange are represented as spheres for apo (black), binary acivicin (green), binary PRFAR (blue) and ternary (red) IGPS.
As noted above, NMR studies and computational investigations can be complementary and allow for deeper insight into biophysical processes. MD simulations have been combined with correlation analysis of protein motions based on network theory methods and have been used to obtain fundamental understanding of the IGPS allostery, shedding light on the mechanism of information transfer 21, 22. A comparison of the correlation maps of apo and PRFAR-bound IGPS complexes averaging the generalized correlation coefficient, rMI over four independent MD trajectories for each complex was performed, with the highest rmsd for rMI over all the independent MD simulations of ca. 0.2, and mean rmsd (among all node pairs) being only 0.038 for both apo and PRFAR-bound complexes. Correlation analysis based on the measure of mutual information indicates that effector binding induces changes in correlated motions that are non-uniformly distributed throughout the IGPS complex. On one side of the IGPS complex, where the conserved amino acid residues hW123-fR249 (molecular hinge) and hK181-fD98 (salt-bridge anchor) provide structural stability to the heterodimer (side R), the effector binding induces an increase of correlations at the HisF/HisH interface, indicating that PRFAR binding might further stabilize the heterodimeric form of IGPS during glutamine hydrolysis (Figure 8). At the other side of the complex (sideL), instead, PRFAR binding induces a significant decrease of correlations in the interdomain region, indicating that the entropically driven PRFAR binding 53 promotes the separation of the HisF-HisH domains at sideL. Notably, effector binding in HisF results in a significant reduction of correlations of motions in the HisH active site, including amino acid residues of the oxyanion strand, that is situated over 25 Å away from the effector-binding site.
Figure 8.

Panel a) Changes in the network of ionic interaction at sideR of IGPS, induced by PRFAR binding, involving the fα2, fα3, and hα1 helices in apo (left) and PRFAR bound (right) complexes. The most stable ion-pair interactions, responsible for the increase of correlations at the sideR, are highlighted with solid black lines. (Panel b) Optimal community networks of the apo (left) and PRFAR bound (right) IGPS complexes. The apo communities reported in brackets in the PRFAR network indicate those communities that contribute the most to the newly formed PRFAR communities. Widths of connectivity lines are proportional to the intercommunities total edge betweenness, i.e the sum of the edge betweenness of those edges connecting one community to another one. Adapted from Reference 21.
Community network analysis of the structure of correlations in IGPS has been performed to elucidate the allosteric pathways and to find amino acid residues that are crucial for the signal propagation. As shown in Figure 8, the network analysis indicates that effector binding alters the community structure of interactions in the inactive IGPS and the collective pathways linking the effector and active sites. The number of communities and the distribution of amino acid residues in each community differentiate the optimal community network of apo and PRFAR-bound complexes. The information flow is also affected by effector binding and it reflects the changes in correlated motions, with an increase of information flow (and correlations) on the sideR of the IGPS complex.
The effector binding introduces changes in the apo community network that can be understood in terms of specific hydrophobic, ionic, and hydrogen-bonding interactions associated with the amino acid residues identified by the community network analysis. PRFAR binding causes a stabilization of the flexible two-stranded β-sheet of loop1 and alters hydrophobic interactions at the bottom of the (β/α)8 HisF barrel (Figure 9a). In particular, the formation of the fL50-fI52-fF23 hydrophobic cluster in the fβ2 strand is observed upon effector binding, with disruption of the fV48-fL50 interaction in the ns timescale (Figure 9b). This alteration of hydrophobic interactions is supported by NMR titration experiments (Figure 9c) and it may represent the initial loosening of the protein core that precedes the wholesale enhancement of ms motions observed by solution NMR.
Figure 9.

(Panel a) Hydrogen-bonds in (HisF) loop1 in apo (blue) and PRFAR bound (red) enzymes, showing the extension of the small two-stranded β-sheet upon PRFAR binding. (Panel b) Changes in hydrophobic interactions induced by PRFAR in the (β/α)8 HisF barrel. All highlighted residues but fF23 (in loop1) belong to the fβ2 strand, making a direct link between loop1 and the ammonia tunnel. (panel c) The 1H-13C spectral overlays of the titration of PRFAR into IGPS (0.89 mM) are shown. Spectra were collected for 0 mM (red), 0.56 mM (yellow), 0.74 mM (green), 1.81 mM (blue), and 3.47 mM (purple). Large chemical shift perturbations and significant line broadening are observed for V48 and L50 of HisF. Adapted from Reference 21.
Changes in hydrophobic interactions in the fβ2 strand are coupled with rearrangements of adjacent secondary structures elements, including the fα1 and the fα2 helices. Analysis of the MD trajectories indicate that PRFAR binding alters the network of salt-bridges belonging to the fα2, fα3 and hα1 helices located in the sideR of the IGPS complex (Figure 9a), with charged side chains placed at the protein surface and fα2 packing against the fβ2 hydrophobic region. PRFAR binding induces an increase of correlations in this region, stabilizing specific ionic interactions at the HisF/HisH interface (Figure 9a). The reorganization of ionic interactions at the interdomain interface affects the breathing motion of IGPS, the relative motion between the HisF and HisH domains. We observed that the PRFAR bound complex assumes a conformation that facilitates motions at the interface without damaging the structural stability of the heterodimer but exposing more frequently the interface region to the solvent water molecules and reducing the correlations in motions at the bottom of the HisH domain, suggesting that changes in solvation entropy could contribute to allostery. In this region, the Ω-loop adjacent to the hα1 helix is connected to the oxyanion strand, of the HisH glutaminase active site, through a hydrogen bond between the carbonyl oxygen of hP10 and the NH group of hV51, leading to the “inactive configuration” of IGPS in which the the HN of hV51 does not reside in the oxyanion hole site. The reduction of correlations induced by PRFAR in this region leads to loosening of the hP10-hV51 H-bond, with MD simulations indicating breaking of this H-bond in the tens of ns timescale. Notably, breaking of the hV51-hP10 H-bond upon PRFAR binding results in more rotational degrees of freedom for the oxyanion strand, enabling it to rotate relative to glutaminase binding site (Figure 10). These results support the mechanistic hypothesis that rotation of the conserved oxyanion strand is responsible for the inactive-to-active allosteric transition in IGPS. Single, conserved amino acid residues, such as fL50, fR59, fE91 and hP10 have been identified from this work and shown to play an important role in the allosteric mechanism. These sites are appropriate candidates for future mutagenesis studies.
Figure 10.

MD representative snapshots (at t=50 ns) of apo (left) and PRFAR bound (right) IGPS complexes, showing H-bonds between hP10 (Ω -loop, in green) and hV51 (oxyanion strand, in blue) (panel a) and partial reorientation of the oxyanion strand triggered by PRFAR binding (panel b). Thermal fluctuations of the 49-PGVG backbone are shown (sticks colored from red to blue for 0–50 ns) highlighting the NH amide group of hV51 (larger sticks). Apo (C) and PRFAR bound (D) average structures (colored cartoons) and Gln binding site (hC84, colored sticks) are also shown. Adapted from Reference 21.
These combined studies demonstrate that fast timescale motions identified in NMR experiments show ligand-dependent and residue specific changes in ILV sidechain motions of conserved, functionally important residues. These motions are further explored in the MD simulations to elucidate the allosteric activation process. These multi-nanosecond MD simulations show a variety of correlated motions for a network of residues bridging the PRFAR and glutamine active sites. Our network analysis further demonstrates an alteration in the pathway of information flow between the effector site and catalytic site, yet these simulations do not show an oxyanion strand that fully rotates to the active conformation, suggesting additional changes that take place on an even slower timescale. NMR CPMG relaxation studies characterized these additional motions, which occur on the ms timescale and are required for achieving the optimal HisH catalytic activity and further identified ms fluctuations at the HisH active site as a result of PRFAR binding 53. These ns motions identified in the MD studies therefore likely represent the initial fluctuations that ultimately lead to a catalytically functional glutaminase. Studies such as those described here could be further enhanced by combination with NMR residual dipolar coupling (RDC) measurements. NMR RDCs have been used with much success in characterizing the role of protein domain motions in allostery 61, 62. The work described here highlights the importance of combined computational and NMR techniques for characterizing biological processes.
Biographies
J. Patrick Loria grew up in Fairmont, WV and received his B.Sc. in Chemistry from The George Washington University in 1990. He received his Ph.D. in Biochemistry from the University of Notre Dame under the direction of Professor Thomas Nowak, studying the enzymological and allosteric properties of Yeast Pyruvate Kinase. In 1997, he trained in solution NMR techniques as an NIH Postdoctoral Fellow with Professor Arthur G. Palmer at Columbia University. Since 2001 he has been on the faculty at Yale University in the Department of Chemistry. His research interests are focused on understanding conformational fluctuations in the function of enzymes.
Ivan Rivalta received his PhD in Chemistry at Università della Calabria (Italy) in 2007, working with Prof. Emilia Sicilia and Prof. Nino Russo on computational studies of catalytic systems. In 2009, he was visiting postdoc researcher in Prof. Michele Parrinello’s group at ETH Zurich (Switzerland), working on ab initio (and hybrid QM/MM) molecular dynamics (MD) simulations and accelerated MD methods for studying solvent effects in catalysis. In 2010, he worked with Prof. Victor S. Batista at Yale University (USA) as associate research scientist until 2012, working on biophysical studies of allosteric enzymes and G-protein-coupled receptors and on computational studies of natural and artificial photosynthesis. He recently joined Prof. Marco Garavelli’s group at Università di Bologna (Italy) as senior postdoc, working on structure and dynamics of biomolecules by two-dimensional electronic spectroscopy.
Gregory A. Manley is a Ph.D. candidate in the Chemistry Department at Yale University working with J. Patrick Loria. He received a B.S. Chemistry from Bucknell University. His current research focuses on the role of motions in allostery.
Footnotes
Italic lettering corresponds to the protein in which the amino acid resides, h for HisH and f for HisF.
References
- 1.Carpenter JF, Hand SC. Comparison of Ph-Dependent Allostery and Dissociation for Phosphofructokinases from Artemia Embryos and Rabbit Muscle: Nature of the Enzymes Acylated with Diethylpyrocarbonate. Arch Biochem Biophys. 1986;248:1–9. doi: 10.1016/0003-9861(86)90394-2. [DOI] [PubMed] [Google Scholar]
- 2.Mesecar AD, Nowak T. Metal-Ion-Mediated Allosteric Triggering of Yeast Pyruvate Kinase. 1. A Multidimensional Kinetic Linked-Function Analysis. Biochemistry. 1997;36:6792–6802. doi: 10.1021/bi962869t. [DOI] [PubMed] [Google Scholar]
- 3.Frantom PA, Zhang HM, Emmett MR, Marshall AG, Blanchard JS. Mapping of the Allosteric Network in the Regulation of Alpha-Isopropylmalate Synthase from Mycobacterium Tuberculosis by the Feedback Inhibitor L-Leucine: Solution-Phase H/D Exchange Monitored by Ft-Icr Mass Spectrometry. Biochemistry. 2009;48:7457–7464. doi: 10.1021/bi900851q. [DOI] [PubMed] [Google Scholar]
- 4.Schachman HK. Still Looking for the Ivory Tower. Annu Rev Biochem. 2000;69:1–29. doi: 10.1146/annurev.biochem.69.1.1. [DOI] [PubMed] [Google Scholar]
- 5.Changeux JP, Gerhardt JC, Schachman HK. Allosteric Interaction in Aspartate Transcarbamoylase. I. Binding of Specific Ligands to the Native Enzyme and Its Isolated Subunits. Biochemistry. 1968;7:531–538. doi: 10.1021/bi00842a007. [DOI] [PubMed] [Google Scholar]
- 6.Liu L, Wales ME, Wild JR. Temperature Effects on the Allosteric Responses of Native and Chimeric Aspartate Transcarbamoylases. Journal Of Molecular Biology. 1998;282:891–901. doi: 10.1006/jmbi.1998.2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fenton AW. Allostery: An Illustrated Definition for the ‘Second Secret of Life’. Trends Biochem Sci. 2008;33:420–425. doi: 10.1016/j.tibs.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maksay G. Allostery in Pharmacology: Thermodynamics, Evolution and Design. Progress in biophysics and molecular biology. 2011;106:463–473. doi: 10.1016/j.pbiomolbio.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 9.Reinhart GD. Quantitative Analysis and Interpretation of Allosteric Behavior. Methods Enzymol. 2004;380:187–203. doi: 10.1016/S0076-6879(04)80009-0. [DOI] [PubMed] [Google Scholar]
- 10.Weber G. Ligand Binding and Internal Equilibria in Proteins. Biochemistry. 1972;11:864–878. doi: 10.1021/bi00755a028. [DOI] [PubMed] [Google Scholar]
- 11.Boyer JA, Clay CJ, Luce KS, Edgell MH, Lee AL. Detection of Native-State Nonadditivity in Double Mutant Cycles Via Hydrogen Exchange. J Am Chem Soc. 2010;132:8010–8019. doi: 10.1021/ja1003922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Horovitz A. Double-Mutant Cycles: A Powerful Tool for Analyzing Protein Structure and Function. Fold Des. 1996;1:R121–126. doi: 10.1016/S1359-0278(96)00056-9. [DOI] [PubMed] [Google Scholar]
- 13.Cooper A, Dryden DTF. Allostery without Conformational Change. Eur Biophys J. 1984;11:103–109. doi: 10.1007/BF00276625. [DOI] [PubMed] [Google Scholar]
- 14.Tsai CJ, del Sol A, Nussinov R. Allostery: Absence of a Change in Shape Does Not Imply That Allostery Is Not at Play. Journal Of Molecular Biology. 2008;378:1–11. doi: 10.1016/j.jmb.2008.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koshland DE, Nemethy G, Filmer D. Comparison of Experimental Binding Data and Theoretical Models in Proteins Containing Subunits. Biochemistry. 1966;5:365–385. doi: 10.1021/bi00865a047. [DOI] [PubMed] [Google Scholar]
- 16.Monod J, Wyman J, Changeux JP. On the Nature of Allosteric Transitions: A Plausible Model. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 17.Kimmel JL, Reinhart GD. Reevaluation of the Accepted Allosteric Mechanism of Phosphofructokinase from Bacillus Stearothermophilus. Proc Natl Acad Sci U S A. 2000;97:3844–3849. doi: 10.1073/pnas.050588097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perutz MF. Mechanisms of Cooperativity and Allosteric Regulation in Proteins. Q Rev Biophys. 1989;22:139–237. doi: 10.1017/s0033583500003826. [DOI] [PubMed] [Google Scholar]
- 19.Hilser VJ, Wrabl JO, Motlagh HN. Structural and Energetic Basis of Allostery. Annu Rev Biophys. 2012;41:585–609. doi: 10.1146/annurev-biophys-050511-102319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Monod J, Changeux JP, Jacob F. Allosteric Proteins and Cellular Control Systems. Journal Of Molecular Biology. 1963;6:306–329. doi: 10.1016/s0022-2836(63)80091-1. [DOI] [PubMed] [Google Scholar]
- 21.Rivalta I, Sultan MM, Lee NS, Manley G, Loria JP, Batista VS. Allosteric Pathways in the Imidazole Glycerol Phosphate Synthase. Proc Natl Acad Sci U S A. 2012;109:E1428–E1436. doi: 10.1073/pnas.1120536109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vanwart AT, Eargle J, Luthey-Schulten Z, Amaro RE. Exploring Residue Component Contributions to Dynamical Network Models of Allostery. J Chem Theory Comput. 2012;8:2949–2961. doi: 10.1021/ct300377a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Palmer AG, Kroenke CD, Loria JP. Nuclear Magnetic Resonance Methods for Quantifying Microsecond-to-Millisecond Motions in Biological Macromolecules. Meth Enzymol. 2001;339(Part B):204–238. doi: 10.1016/s0076-6879(01)39315-1. [DOI] [PubMed] [Google Scholar]
- 24.Akke M, Brüschweiler R, Palmer AG. Nmr Order Parameters and Free Energy: An Analytic Approach and Application to Cooperative Ca2+ Binding by Calbindin D9k. J Am Chem Soc. 1993;115:9832–9833. [Google Scholar]
- 25.Yang D, Kay LE. Contributions to Conformational Entropy Arising from Bond Vector Fluctuations Measured from Nmr-Derived Order Parameters: Application to Protein Folding. J Mol Biol. 1996;263:369–382. doi: 10.1006/jmbi.1996.0581. [DOI] [PubMed] [Google Scholar]
- 26.Li Z, Raychaudhuri S, Wand AJ. Insights into the Local Residual Entropy of Proteins Provided by Nmr Relaxation. Protein Sci. 1996;5:2647–2650. doi: 10.1002/pro.5560051228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stivers JT, Abeygunawardana C, Mildvan AS. 15n Nmr Relaxation Studies of Free and Inhibitor-Bound 4-Oxalocrotonate Tautomerase: Backbone Dynamics and Entropy Changes of an Enzyme Upon Inhibitor Binding. Biochemistry. 1996;35:16036–16047. doi: 10.1021/bi961834q. [DOI] [PubMed] [Google Scholar]
- 28.Popovych N, Sun S, Ebright RH, Kalodimos CG. Dynamically Driven Protein Allostery. Nat Struct Mol Biol. 2006;13:831–838. doi: 10.1038/nsmb1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tugarinov V, Sprangers R, Kay LE. Probing Side-Chain Dynamics in the Proteasome by Relaxation Violated Coherence Transfer Nmr Spectroscopy. J Am Chem Soc. 2007;129:1743–1750. doi: 10.1021/ja067827z. [DOI] [PubMed] [Google Scholar]
- 30.Luz Z, Meiboom S. Nuclear Magnetic Resonance Study of the Protolysis of Trimethylammonium Ion in Aqueous Solution—Order of the Reaction with Respect to Solvent. J Chem Phys. 1963;39:366–370. [Google Scholar]
- 31.Loria JP, Rance M, Palmer AG. A Relaxation-Compensated Carr-Purcell-Meiboom-Gill Sequence for Characterizing Chemical Exchange by Nmr Spectroscopy. J Am Chem Soc. 1999;121:2331–2332. [Google Scholar]
- 32.Ishima R, Baber J, Louis JM, Torchia DA. Carbonyl Carbon Transverse Relaxation Dispersion Measurements and Ms-Ms Timescale Motion in a Protein Hydrogen Bond Network. J Biomol NMR. 2004;29:187–198. doi: 10.1023/B:JNMR.0000019249.50306.5d. [DOI] [PubMed] [Google Scholar]
- 33.Ishima R, Torchia D. Extending the Range of Amide Proton Relaxation Dispersion Experiments in Proteins Using a Constant-Time Relaxation Compensated Cpmg Approach. J Biomol NMR. 2003;25:243–248. doi: 10.1023/a:1022851228405. [DOI] [PubMed] [Google Scholar]
- 34.Korzhnev DM, Kloiber K, Kay LE. Multiple-Quantum Relaxation Dispersion Nmr Spectroscopy Probing Millisecond Time-Scale Dynamics in Proteins: Theory and Application. J Am Chem Soc. 2004;126:7320–7329. doi: 10.1021/ja049968b. [DOI] [PubMed] [Google Scholar]
- 35.Lundström P, Vallurupalli P, Religa TL, Dahlquist FW, Kay LE. A Single-Quantum Methyl 13c-Relaxation Dispersion Experiment with Improved Sensitivity. J Biomol NMR. 2007;38:79–88. doi: 10.1007/s10858-007-9149-7. [DOI] [PubMed] [Google Scholar]
- 36.Mulder FA, Skrynnikov NR, Hon B, Dahlquist FW, Kay LE. Measurement of Slow (Micros-Ms) Time Scale Dynamics in Protein Side Chains by (15)N Relaxation Dispersion Nmr Spectroscopy: Application to Asn and Gln Residues in a Cavity Mutant of T4 Lysozyme. J Am Chem Soc. 2001;123:967–975. doi: 10.1021/ja003447g. [DOI] [PubMed] [Google Scholar]
- 37.Hill RB, Bracken C, DeGrado WF, Palmer AG. Molecular Motions and Protein Folding: Characterization of the Backbone Dynamics and Folding Equilibrium of Alpha D-2 Using C-13 Nmr Spin Relaxation. J Am Chem Soc. 2000;122:11610–11619. [Google Scholar]
- 38.Baldwin AJ, Hansen DF, Vallurupalli P, Kay LE. Measurement of Methyl Axis Orientations in Invisible, Excited States of Proteins by Relaxation Dispersion Nmr Spectroscopy. J Am Chem Soc. 2009;131:11939–11948. doi: 10.1021/ja903896p. [DOI] [PubMed] [Google Scholar]
- 39.Baldwin AJ, Religa TL, Hansen DF, Bouvignies G, Kay LE. (13)Chd(2) Methyl Group Probes of Millisecond Time Scale Exchange in Proteins by (1)H Relaxation Dispersion: An Application to Proteasome Gating Residue Dynamics. J Am Chem Soc. 2010;132:10992–10995. doi: 10.1021/ja104578n. [DOI] [PubMed] [Google Scholar]
- 40.Otten R, Villali J, Kern D, Mulder FA. Probing Microsecond Time Scale Dynamics in Proteins by Methyl (1)H Carr-Purcell-Meiboom-Gill Relaxation Dispersion Nmr Measurements. Application to Activation of the Signaling Protein Ntrc(R) J Am Chem Soc. 2010;132:17004–17014. doi: 10.1021/ja107410x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Skrynnikov NR, Mulder FA, Hon B, Dahlquist FW, Kay LE. Probing Slow Time Scale Dynamics at Methyl-Containing Side Chains in Proteins by Relaxation Dispersion Nmr Measurements: Application to Methionine Residues in a Cavity Mutant of T4 Lysozyme. J Am Chem Soc. 2001;123:4556–4566. doi: 10.1021/ja004179p. [DOI] [PubMed] [Google Scholar]
- 42.Tugarinov V, Kay LE. Separating Degenerate (1)H Transitions in Methyl Group Probes for Single-Quantum (1)H-Cpmg Relaxation Dispersion Nmr Spectroscopy. J Am Chem Soc. 2007;129:9514–9521. doi: 10.1021/ja0726456. [DOI] [PubMed] [Google Scholar]
- 43.Korzhnev DM, Kloiber K, Kanelis V, Tugarinov V, Kay LE. Probing Slow Dynamics in High Molecular Weight Proteins by Methyl-Trosy Nmr Spectroscopy: Application to a 723-Residue Enzyme. J Am Chem Soc. 2004;126:3964–3973. doi: 10.1021/ja039587i. [DOI] [PubMed] [Google Scholar]
- 44.Levitt M. Molecular Dynamics of Native Protein. Ii. Analysis and Nature of Motion. Journal Of Molecular Biology. 1983;168:621–657. doi: 10.1016/s0022-2836(83)80306-4. [DOI] [PubMed] [Google Scholar]
- 45.Post CB, Dobson CM, Karplus M. A Molecular Dynamics Analysis of Protein Structural Elements. Proteins. 1989;5:335–354. doi: 10.1002/prot.340050409. [DOI] [PubMed] [Google Scholar]
- 46.Hünenberger PH, Mark AE, van Gunsteren WF. Fluctuation and Cross-Correlation of Protein Motions Observed in Nanosecond Molecular Dynamics Simulations. JMB. 1995;252:492–503. doi: 10.1006/jmbi.1995.0514. [DOI] [PubMed] [Google Scholar]
- 47.Ichiye T, Karplus M. Collective Motions in Proteins: A Covariance Analysis of Atomic Fluctuations in Molecular Dynamics and Normal Mode Simulations. Proteins: Str Funct Gen. 1991;11:205–217. doi: 10.1002/prot.340110305. [DOI] [PubMed] [Google Scholar]
- 48.Lange OF, Grubmuller H. Generalized Correlation for Biomolecular Dynamics. Proteins. 2006;62:1053–1061. doi: 10.1002/prot.20784. [DOI] [PubMed] [Google Scholar]
- 49.Shannon CE. A Mathematical Theory of Communication. Bell System Technical Journal. 1948;27:379–423. 623–656. [Google Scholar]
- 50.Sethi A, Eargle J, Black AA, Luthey-Schulten Z. Dynamical Networks in Trna:Protein Complexes. Proc Natl Acad Sci U S A. 2009;106:6620–6625. doi: 10.1073/pnas.0810961106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Girvan M, Newman ME. Community Structure in Social and Biological Networks. Proc Natl Acad Sci U S A. 2002;99:7821–7826. doi: 10.1073/pnas.122653799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Newman ME, Girvan M. Finding and Evaluating Community Structure in Networks. Phys Rev E. 2004;69:026113. doi: 10.1103/PhysRevE.69.026113. [DOI] [PubMed] [Google Scholar]
- 53.Lipchock JM, Loria JP. Nanometer Propagation of Millisecond Motions in V-Type Allostery. Structure. 2010;18:1596–1607. doi: 10.1016/j.str.2010.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Myers RS, Amaro RE, Luthey-Schulten ZA, Davisson VJ. Reaction Coupling through Interdomain Contacts in Imidazole Glycerol Phosphate Synthase. Biochemistry. 2005;44:11974–11985. doi: 10.1021/bi050706b. [DOI] [PubMed] [Google Scholar]
- 55.Myers RS, Jensen JR, Deras IL, Smith JL, Davisson VJ. Substrate-Induced Changes in the Ammonia Channel for Imidazole Glycerol Phosphate Synthase. Biochemistry. 2003;42:7013–7022. doi: 10.1021/bi034314l. [DOI] [PubMed] [Google Scholar]
- 56.Chaudhuri BN, Lange SC, Myers RS, Chittur SV, Davisson VJ, Smith JL. Crystal Structure of Imidazole Glycerol Phosphate Synthase: A Tunnel through a (Beta/Alpha)8 Barrel Joins Two Active Sites. Structure. 2001;9:987–997. [PubMed] [Google Scholar]
- 57.Douangamath A, Walker M, Beismann-Driemeyer S, Vega-Fernandez MC, Sterner R, Wilmanns M. Structural Evidence for Ammonia Tunneling across the (Beta Alpha)(8) Barrel of the Imidazole Glycerol Phosphate Synthase Bienzyme Complex. Structure. 2002;10:185–193. doi: 10.1016/s0969-2126(02)00702-5. [DOI] [PubMed] [Google Scholar]
- 58.Mouilleron S, Golinelli-Pimpaneau B. Conformational Changes in Ammonia-Channeling Glutamine Amidotransferases. Curr Opin Struct Biol. 2007;17:653–664. doi: 10.1016/j.sbi.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 59.Chaudhuri BN, Lange SC, Myers RS, Davisson VJ, Smith JL. Toward Understanding the Mechanism of the Complex Cyclization Reaction Catalyzed by Imidazole Glycerolphosphate Synthase: Crystal Structures of a Ternary Complex and the Free Enzyme. Biochemistry. 2003;42:7003–7012. doi: 10.1021/bi034320h. [DOI] [PubMed] [Google Scholar]
- 60.Omi R, Mizuguchi H, Goto M, Miyahara I, Hayashi H, Kagamiyama H, Hirotsu K. Structure of Imidazole Glycerol Phosphate Synthase from Thermus Thermophilus Hb8: Open-Closed Conformational Change and Ammonia Tunneling. J Biochem. 2002;132:759–765. doi: 10.1093/oxfordjournals.jbchem.a003284. [DOI] [PubMed] [Google Scholar]
- 61.Bhattacharya A, Kurochkin AV, Yip GNB, Zhang Y, Bertelsen EB, Zuiderweg ERP. Allostery in Hsp70 Chaperones Is Transduced by Subdomain Rotations. Journal Of Molecular Biology. 2009;388:475–490. doi: 10.1016/j.jmb.2009.01.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lukin JA, Kontaxis G, Simplaceanu V, Yuan Y, Bax A, Ho C. Quaternary Structure of Hemoglobin in Solution. Proc Natl Acad Sci U S A. 2003;21:517–520. doi: 10.1073/pnas.232715799. [DOI] [PMC free article] [PubMed] [Google Scholar]