

Abstract
The success of non-viral transfection using polymers hinges on efficient nuclear uptake of nucleic acid cargo and overcoming intra- and extracellular barriers. By incorporating PKKKRKV heptapeptide pendent groups as nuclear localization signals (NLS) on a polymer backbone, we demonstrate protein expression levels higher than obtained from JetPEI™ and Lipofectamine™ 2000, the latter being notorious for coupling high transfection efficiency with cytotoxicity. The orientation of the NLS peptide grafts markedly affected transfection performance. Polymers with the sequence attached to the backbone from the valine residue achieved higher nuclear translocation relative to those having the NLS groups attached in the opposite orientation. The differences in nuclear localization and DNA complexation strength between the two orientations correlated with a striking difference in protein expression, both in cell culture and in vivo. Polyplexes formed from these comb polymer structures exhibited transfection efficiencies superior to those of Lipofectamine 2000 but with greatly reduced toxicity. Moreover, these novel polymers enabled high reporter gene expression in mice when administered by intramuscular ultrasound-mediated delivery, demonstrating their therapeutic promise in vivo.
Keywords: Gene therapy, oligopeptide graft copolymer, Nuclear localization sequence (NLS), Ring- opening metathesis polymerization (ROMP), Polylysine (PLL), Sonoporation
1. INTRODUCTION
Polyelectrolytes comprise numerous platforms in materials and biology, including capsules for drug delivery,1 multilayer films for tissue engineering,2,3 and non-viral vectors for gene therapy.4 Cationic polymers represent appealing alternatives to natural viral vectors that threaten immunogenicity and carcinogenicity stemming from insertional mutagenesis into the chromosomes of infected cells.5,6 Synthetic polymers offer excellent structural and chemical versatility within non-immunogenic delivery systems, as well as high therapeutic carrying capacity and long shelf-life.
The success of polymer-based gene therapy relies on the ability of cationic polymers to complex DNA non-covalently in nanoscale ‘polyplexes’ that enter cells, translocate to the nucleus, and release their nucleotide cargo for transcription and therapeutic protein production. Cationic polymers can be designed as various architectures, such as linear, graft (comb), branched, and dendritic, which influence complexation and delivery capabilities of the polymer vehicle.7–12 Novel synthetic approaches allow integration of many features needed for gene therapy into one macromolecular delivery system, including functionality to prevent immunogenicity,13 recognize cell surfaces,14,15 and localize to the nucleus.16–18
We previously compared the transfection performance of amphiphilic graft copolymers with pendent oligolysine groups to those with pendent oligopeptide groups11 identified as nuclear localization sequences (NLS) in the Simian virus SV40 T-antigen.19,20 Though the copolymers having pendent NLS groups gave higher protein expression than those with simple cationic (i.e., oligolysine) grafts, quantitative polymerase chain reaction (qPCR) evidence and binding strength assays suggested that, for these systems, complexation strength was at least as important, if not more, than nuclear localization. In a study on peptide nucleic acid (PNA)-NLS conjugates, Branden and coworkers reported the effect of NLS (PKKKRKV) orientation on nuclear translocation.20 Cy3-labeled DNA complexed with PNA-NLS, having NLS conjugated to PNA at the valine residue, facilitated nuclear translocation in live cells, as determined from fluorescence micrographs. In contrast, conjugates having the NLS groups attached to PNA at the proline residue showed no translocation.20 Inspired by this finding and the potential to improve the comb polymer transfection platform, we investigated the effect of NLS orientation pendent to a synthetic polymer backbone, as shown in Figure 1 for NLS (proline attachment to backbone) and the reverse, rNLS, case (valine attachment to backbone).
Figure 1.
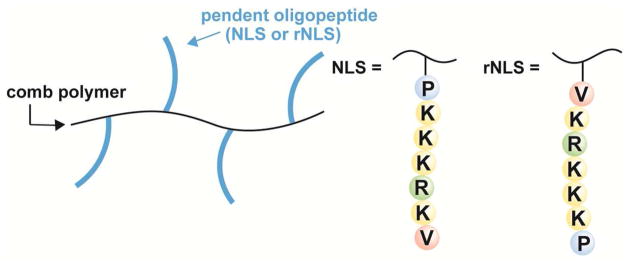
Representation of synthetic polymer transfection reagents with ‘NLS’ and ‘rNLS’ groups pendent to the backbone.
Here we report the preparation of poly(cyclooctene-graft-oligopeptide) comb polymers and their use in complexation of DNA and transfection of SKOV3 (human ovarian adenocarcinoma) and SKVLB1 (multidrug resistant human ovarian adenocarcinoma) cells, and in intramuscular gene delivery in vivo. Higher transfection levels are observed with the rNLS comb structures relative to PLL, JetPEI™, and Lipofectamine™ 2000 in SKOV3 cells, and relative to Lipofectamine 2000 in SKVLB1 cells. As described here, the success of these polymers is attributed to numerous factors, including their biocompatibility, DNA interaction strength, and orientation of the NLS sequence.
2. EXPERIMENTAL SECTION
Materials
2,2,2-Trifluoroethanol (TFE, ≥ 99%), 3-bromopyridine (99%), ethyl vinyl ether (99%), 2nd generation Grubbs catalyst ((1,3-bis(2,4,6-trimethylphenyl)-2-imidazolidinylidene) dichloro(phenylmethylene)(tricyclohexylphosphine) ruthenium), triisopropylsilane (TIPS, 99%), methanol (anhydrous, 99.8 %), N,N-dimethylformamide (DMF, ≥ 99.8%), and N,N-dimethylformamide (anhydrous, 99.8%) were purchased from Aldrich. Fmoc-Val-OH (≥ 98%), Fmoc-Lys(Boc)-OH (≥ 98%), Fmoc-Arg(Pbf)-OH (≥ 98%), Fmoc-Pro-OH (≥ 98%), 1-hydroxybenzotriazole (HOBt, ≥98%) hydrate, O-(benzotriazol-1-yl)-N,N,N′,N′-tetra-methyluronium hexafluorophosphate (HBTU, ≥ 98%), piperidine (≥ 99%), diisopropylethylamine (DIPEA, ≥ 99%), trifluoroacetic acid (TFA, ≥ 99%) and 2-chlorotrityl chloride resin (1.0–1.6 mmol/g, 100–200 mesh) were purchased from Advanced ChemTech. Glacial acetic acid (ACS certified) and dialysis tubing (Spectra/PorR, regenerated cellulose membranes with a molecular weight cut-off of 6000–8000 g/mol) were purchased from Fisher Scientific. 5-Carboxylic acid-1-cyclooctene was prepared from 1,5-cyclooctadiene.21–23 Grubbs Generation III catalyst was prepared according to the literature.24 NLSa-c were prepared as previously reported.11 CH2Cl2 was distilled over CaH2. All other materials were used without purification.
Dulbecco’s Modified Eagles Medium (DMEM), Penicillin and Streptomycin, Lipofectamine™ 2000, phosphate-buffered saline (PBS, 1x, pH 7.4), and the Quant-iT™ PicoGreen® (200 X) double stranded DNA reagent were purchased from Life Technologies. Fetal bovine serum (FBS) was purchased from Atlanta Biologicals. SKOV3 cells and McCoy’s 5A medium were purchased from American Type Culture Collection (ATCC), while SKVLB1 cells were a gift from Dr. Victor Ling. mRFP-IRES-Puro and pEGFP-N1 plasmid DNA were a gift from Dr. Lawrence Schwartz. JetPEI™ (PEI) was purchased from Polyplus Transfection. Heparin, agarose, and DNase I were purchased from Sigma. The CellTiter-Glo Luminescent Cell Viability Assay was purchased from Promega. Optiprep (60% (w/v) Iodixanol) was purchased from Axis-Shield. Brilliant SYBRR Green QPCR Master Mix was purchased from Agilent Technologies. Micromarker microbubbles were purchased from VisualSonics (Toronto, Ontario, Canada).
Polymer characterization
1H and 13C NMR spectra were recorded on a Bruker Spectrospin DPX300 or a Bruker Avance400 spectrometer. Mass spectrometry was performed on a JEOL MStation JMS700 high-resolution two-sector mass spectrometer equipped with fast atom bombardment (FAB) and low resolution electrospray ionization (ESI). Size exclusion chromatography (SEC) on Boc-and Pbf- protected polymers was performed in DMF with 0.01 M LiCl at 50 °C. The system was operated at a flow rate of 1 mL/min with a Sonntek HPLC pump (K-501), one 50 mm × 7.5 mm PL gel mixed guard column, one 300 mm × 7.5 mm PL gel 5 μm mixed C column, one 300 mm × 7.5 mm PL gel 5 μm mixed D column, a Knauer refractive index detector (K-2301), and an Alltech solvent recycler 3000. SEC on deprotected cationic polymers was performed in TFE with 0.02 M sodium trifluoroacetate at 40 °C using an Agilent 1200 system equipped with an isocratic pump operated at 1 mL/min, a degasser, an autosampler, one 50 mm × 8 mm PSS PFG guard column (Polymer Standards Service), three 300 mm × 7.5 mm PSS PFG analytical linear M columns with 7 μm particle size (Polymer Standards Service), and an Agilent 1200 refractive index detector. Both systems were calibrated with PMMA standards.
Synthesis of 5-VK(Boc)R(Pbf)K(Boc)K(Boc)K(Boc)P-1-cyclooctene (rNLS-COE)
5-VK(Boc)R(Pbf)K(Boc)K(Boc)K(Boc)P-1-cyclooctene was prepared similarly to 5-PK(Boc)K(Boc)K(Boc)R(Pbf)K(Boc)V-1-cyclooctene by solid phase peptide by loading the resin with Fmoc-Pro-OH instead of Fmoc-Val-OH and by inverting the order of the sequence of amino acids added to the resin.11 2-Chlorotrityl chloride resin (6.0 g, 1.2 mmol/g, 7.2 mmol of functional groups) was weighed into an oven-dried vessel and swollen in dry CH2Cl2 (60 mL) for 10 min. The solution was filtered, and a solution of Fmoc-Pro-OH (6.5 g, 19.2 mmol), DIPEA (6.7 mL, 38.4 mmol), and dry CH2Cl2 (70 mL) was added to the peptide vessel. The solution was agitated under N2 for 45 min, then filtered and washed with DMF three times. An 80/15/5 anhydrous CH2Cl2/methanol/DIPEA solution (60 mL) was agitated under N2(g) for 10 min (twice), and the resin was washed with DMF three times. The Fmoc protecting groups were removed by agitation under N2(g) with a 25% piperidine solution in DMF (60 mL) for 3 min, followed by filtration and agitation with a fresh piperidine solution for 20 min. The resin was then washed 6 times with DMF, 3 times with CH2Cl2, 3 times with isopropanol, 6 times with hexanes, and once with CH2Cl2. The resin was dried under vacuum. The loading density of amino acid was determined gravimetrically (typically 1.2–1.4 mmol/g of resin).
The remaining amino acids were successively coupled to the proline-functionalized resin, using HBTU and HOBt as coupling agents, in the following order: Fmoc-Lys(Boc)-OH, Fmoc- Lys(Boc)-OH, Fmoc-Lys(Boc)-OH, Fmoc-Arg(Pbf)-OH, Fmoc-Lys(Boc)-OH and Fmoc-Val-OH. For each coupling reaction the amino acid (28.5 mmol), HBTU (10.8 g, 28.5 mmol) and HOBt (4.4 g, 28.5 mmol) were dissolved in anhydrous DMF (80 mL) in an oven-dried 500 mL roundbottom flask. DIPEA (10 mL, 57 mmol) was added to the amino acid solution, which was then added to the peptide vessel and agitated under N2(g) for 1 h. The resin was filtered, and washed 3 times with DMF. The Fmoc protecting groups were removed as described previously using a 25% piperidine solution in DMF. The resin was washed 6 times with DMF. Following the addition of Fmoc-Val-OH, a solution of 5-carboxylic acid-1-cyclooctene (4.4 g, 28 mmol), HOBt (4.37 g, 28.5 mmol) and HBTU (10.8 g, 28.5 mmol) in anhydrous DMF (80 mL) was prepared. DIPEA (10 mL, 57 mmol) was added to the solution, immediately poured into the peptide vessel and agitated under N2(g) for 1 h. The solution was filtered, and the resin was washed 6 times with DMF and 6 times with CH2Cl2. The oligopeptide-functionalized cyclooctene was cleaved from the resin by adding a 4/1 CH2Cl2/TFE solution to the peptide vessel for 45 min (twice). The resin was filtered, washed three times with CH2Cl2, and the filtrates were collected in a clean roundbottom flask. The solution was concentrated by rotary evaporation and precipitated in 2/1 diethyl ether/hexanes. After standing at 4 °C for several hours, the product was isolated by filtration and then dried under vacuum. 6.8 g of a white powder was obtained (57 % yield based upon the estimated loading density). 1H NMR (DMSOd6, 300 MHz): δ =12.44 (br, 1H), 7.51–8.37 (br m, 7H), 5.96–7.13 (br m, 7H), 5.62 (br, 2H), 4.35–4.53 (br m, 1H), 4.12–4.35 (br m, 4H), 3.99–4.12 (br m, 1H), 3.78–3.99 (br m, 1H), 3.60–3.78 (br m, 1H), 3.45–3.60 (br m, 1H), 2.75–3.13 (br, 12 H), 2.34–2.49 (br m, 9H), 1.06–2.19 (br m, 85H), 0.72–0.92 (br m, 6H). 13C NMR (DMSO-d6, 100 MHz): δ = 177.08, 173.27, 173.24, 171.46, 171.29, 171.21, 171.15, 171.01, 169.90, 169.87, 157.45, 156.05, 155.53, 137.29, 134.21, 131.44, 129.96, 129.90, 129.81, 124.29, 116.26, 86.29, 77.35, 59.38, 59.04, 58.54, 58.45, 57.72, 52.55, 52.21, 51.97, 50.26, 46.46, 43.55, 42.50, 32.72, 31.95, 31.80, 31.53, 30.76, 30.16, 29.41, 29.34, 29.25, 28.60, 28.28, 25.30, 24.57, 24.10, 22.61, 22.19, 19.28, 18.97, 18.25, 17.61, 12.29. Low-resolution ESI (m/z): [M+H]+ calculated from C82H138N14O20S, 1672.0; found, 1671.9. High-resolution FAB (m/z): [M+H]+ calculated from C82H138N14O20S, 1672.00; found, 1672.01.
Representative 5-VK(Boc)R(Pbf)K(Boc)K(Boc)K(Boc)P-1-cyclooctene (rNLS-COE) homopolymerization
rNLS-COE (0.2 g, 0.12 mmol) was added to a 20 mL vial and dissolved in TFE (0.4 mL). The monomer solution was degassed using three freeze-pump-thaw cycles. A solution of Grubbs catalyst Generation III in CH2Cl2 (50 mg/mL) was prepared and degassed using three freeze-pump-thaw cycles. Catalyst solution (0.02 mL, 0.001 mg, 0.0011 mmol) was transferred to the monomer solution, and the mixture was stirred for 1 h at room temperature under N2(g). Ethyl vinyl ether (0.1 mL, 1 mmol) was added to terminate the polymerization, and the mixture stirred for 30 min under N2(g). The mixture was precipitated into diethyl ether and stored at 4 °C for 1 h. The precipitate was isolated by filtration and dried under vacuum at room temperature. The product was obtained as a yellow/brown solid (82–93% recovered mass). Table S1 gives reaction conditions and Figure S1 shows GPC traces. 1H NMR (DMSO-d6, 300 MHz): δ =12.45 (br, 1H), 7.52–8.39 (br m, 7H), 5.96–7.49 (br m, 7H), 5.26 (br, 2H), 3.99–4.69 (br m, 7H), 3.77–3.86 (br m, 2H), 3.24–3.76 (br m, 2H), 2.92–3.19 (br m, 4H), 2.86 (br, 8H), 2.33–2.54 (br m, 10H), 0.98–2.18 (br m, 84H), 0.69–0.90 (m, 6H).
Boc- and Pbf-deprotection of poly(cyclooctene-g- VK(Boc)R(Pbf)K(Boc)K(Boc)K(Boc)P)
Poly(cyclooctene-g-VK(Boc)R(Pbf)K(Boc)K (Boc)K(Boc)P) (0.1 g) was dissolved in a TFA/water/TIPS (95/2.5/2.5) solution (5 mL) and stirred 3 h at room temperature. The mixture was precipitated into diethyl ether and stored at 4 °C for 1 h. The precipitate was isolated by filtration, and dissolved in water. The product was dialyzed (MWCO = 6–8 kDa) against deionized water (once), 30% aqueous acetic acid (once) and deionized water (3x) to remove residual macromonomer. The purified polymer was lyophilized to afford a white solid (poly(cyclooctene-g-VKRKKKP)) in 57–69 % yield. Molecular weight characterization of deprotected polymers is given in Table S1, and GPC traces (TFE as mobile phase) are shown in Figure S2. 1H NMR (DMSO-d6, 300 MHz): 5.42 (br, 2H from polymer backbone), 4.13–4.60 (br m, 6H), 3.92–4.13 (br, 1H), 3.36–3.88 (br m, 2H), 3.11–3.30 (br m, 2H from R), 2.90–3.11 (br m, 8H from K), 2.13–2.45 (br m, 2H), 1.08–2.12 (br m, 42 H), 0.76–1.08 (br m, 6H). 13C NMR (D2O, 75 MHz): δ = 181.29, 179.30, 173.55, 173.40, 173.29, 172.27, 171.52, 170.84, 156.69, 130.50, 62.19, 59.45, 53.62, 53.51, 53.26, 53.12, 51.36, 50.97, 47.69, 47.19, 46.61, 46.13, 46.00, 45.87, 42.75, 40.52, 39.10, 32.32, 32.02, 31.88, 31.59, 31.48, 30.50, 29.81, 29.35, 28.33, 26.43, 26.34, 26.25, 25.30, 25.14, 24.61, 24.44, 23.36, 22.11, 21.97, 21.85, 21.73, 18.58, 18.31.
Cell culture, polyplex formation, and transfection
Human ovarian cancer (SKOV3) and multidrug resistant human ovarian cancer (SKVLB1) cells were cultured in McCoy’s 5A medium, containing 10% FBS and Penicillin and Streptomycin (P/S), at 37 °C in a 5% CO2 incubator. Proliferating cells were seeded at approximately 5 × 103 cells per well into black-walled 96 well plates, and transfected when cells reached approximately 40% cell density. The polymers were weighed into low-retention plastic tubes, dissolved in nuclease-free water, and sterilized by filtration. The stock solutions were diluted to the appropriate concentrations to enable complexation with mRFP-IRES-Puro plasmid DNA at various N/P ratios (i.e., the ratio of protonatable nitrogens, N, in the polymer to DNA phosphates, P). DNA solutions (50 μL of 1 μg/50 μL solution, for 4 wells of a 96 well plate) were prepared in 12 × 75 mm polystyrene sterile culture test tubes. Diluted polymer solution (50 μL) was added to the DNA solution, vortexed lightly, and allowed to equilibrate 40 min at room temperature. Prior to transfection, the cells were washed with serum-free media. The polyplexes were then diluted with serum-free media (400 μL), and the diluted polyplex solution (125 μL) was directly added to the cells. A transfection experiment consisted of testing each reagent at N/P ratios from 3–10, with 8 replicates at each N/P ratio. Each experiment was repeated on three different days using a different cell passage and plasmid DNA batch. PLL and commercial reagents, Lipofectamine™ 2000 and JetPEI™, were used according to the manufacturer’s recommended protocol. 6 h post transfection, the media containing transfection reagents was supplemented with 125 μL/well of fresh growth medium containing 20% FBS. 48 h post transfection, the cells were analyzed by fluorescence microscopy (Olympus IX71 fluorescence inverted microscope equipped with an Olympus DP71 digital camera), a plate reader in fluorescence mode (BMG Labtech FLUOstar OPTIMA plate reader), and finally by the CellTiter-Glo Luminescent Cell Viability Assay.
Protein expression and cell viability
mRFP protein expression at different N/P ratios was determined 48 h post transfection by a plate reader in fluorescence mode. Standard error was calculated for each data set. Statistical analysis was conducted using GraphPad Prism 4 for Windows. To determine cell viability 48 h post transfection, Promega’s CellTiter-Glo Luminescent Cell Viability Assay was performed according to the recommended commercial protocol. The CellTiter-Glo buffer and substrate were equilibrated at room temperature, and the buffer was added to the substrate to form the CellTiter-Glo reagent. After equilibrating the 96-well plate at room temperature, the cell media was replaced with serum-free media (50 μL), and then the mixed CellTiter-Glo reagent (50 μL) was added to each well. The plate was mixed on an orbital shaker for 5 min. Luminescence was recorded on a plate reader (BMG Labtech FLUOstar OPTIMA plate reader). The average luminescence values for each condition (i.e., different reagent, N/P ratio) were divided by the average luminescence of the control cells (cells treated with media only) to determine the percent cell viability.
Dynamic light scattering, zeta potential, and serum and salt stability
Polyplex sizes were measured at 633 nm on a Zetasizer Nano ZS instrument (Malvern Instruments). Polyplexes using PLL, NLSc, rNLSa-e and JetPEI™ were formed at N/P = 7 by incubating polymers (500 μL in water) with DNA (500 μL, 0.02 μg/μL in water) for 40 min. Polyplex solution (200 μL) was replaced with 100 mM NaCl (100 μL) and water (100 μL), to bring the final salt concentration in the polyplex solution to 10 mM. The polyplex sizes were then determined at 25 °C, using a detection angle of 173°, and solution pH of 7.1. All measurements were performed in triplicate. The zeta potential measurements were performed on the same samples using the same instrument, employing the Smoluchowski approximation. For serum and salt stability, polyplexes were first incubated with 10% FBS and 150 mM NaCl for either 1 or 60 minutes, then the intensity-average sizes were measured at 37 °C using the procedure described above.
PicoGreen® detection of polyplex formation and quantification of heparin-induced decomplexation
To prepare polyplex solutions, DNA was diluted in nuclease-free water (50 μL, 1 μg/μL). Separately, polymer solutions of various concentrations (corresponding to N/P 1–10) were prepared in 2X PicoGreen® containing nuclease free water. The PicoGreen® dye fluorescence intensity increases significantly upon binding double stranded DNA, allowing the detection and quantification (using fluorescence measured by a plate reader) of plasmid DNA that is either free from, or loosely associated with, the cationic polymer. For polyplex formation assays, polymer solution (50 μL) was added to the DNA solution and PicoGreen® binding with DNA was quantified using a plate reader in fluorescence mode (BMG Labtech FLUOstar OPTIMA plate reader) at 37 °C at several time points up to 16 h. The ratio of fluorescence from DNA incubated with polymers and from naked DNA at each time point gave the relative value of accessible DNA at different N/P ratios. For heparin decomplexation assays, polyplexes formed at N/P 7, by equilibrating polymer and DNA solutions for 40 min, were incubated with 1X PicoGreen® along with 10 units of heparin in 96-well plates at 37 °C for up to 16 h. Heparin, a polyanion, competes with DNA for binding the polycation and displaces DNA, thereby disassembling the polyplex. As a control, naked plasmid DNA was incubated with both PicoGreen® and heparin for the same amount of time. PicoGreen® binding with DNA was similarly quantified using a plate reader in fluorescence mode (BMG Labtech FLUOstar OPTIMA plate reader). The ratio of fluorescence readings for the polyplex and naked DNA at each time point gave the relative value of DNA accessible to PicoGreen® reagent on heparin incubation.
Gel electrophoresis: nuclease protection
Agarose (0.75 g) was added to 1X Tris Acetate EDTA (TAE) buffer (75 mL) to prepare a 1 wt% agarose gel. The solution was heated until boiling and apparent dissolution. After cooling for 5 min, 1 wt% ethidium bromide solution (8 μL) was added to the agarose solution. This mixture was poured into a gel cassette containing a 10-well comb and allowed to set for 30 min. For DNase I protection assays, polymers were incubated with DNase I for 4 h following polyplex formation (see above). After the DNase I treatment, the nuclease was inactivated using the Stop solution (50 mM EDTA) supplied by the manufacturer, and then killed by incubation at 75 °C for 15 min. The DNA was then incubated with heparin (10 units) at 37 °C for 2 h, pelleted by centrifugation at 13000 rpm for 30 min, discarding the supernatant, and then analyzed on a 1 % agarose gel. As a control, plasmid DNA alone was incubated with DNase I, with or without nuclease treatment, and then run on the agarose gel.
Atomic force microscopy (AFM)
Mica substrates were prepared by adhering mica to glass slides using nail polish. Polyplexes were prepared at N/P 7 in filtered, nuclease-free water in 12 × 75 mm polystyrene sterile culture test tubes and allowed to form 40 min at room temperature. Polyplex solution (200 μL) was drop-cast onto freshly cleaved mica and was left on the substrate for 3 minutes before excess solution was removed by tilting the substrate onto filter paper. Filter paper was then held at the edge of the mica to remove any solution between the mica and glass slide. The substrates were subsequently dried with compressed air. Imaging was performed on a DI Dimension3000 instrument in tapping mode at 1 Hz. The imaging area was 5 μm × 5 μm. Line-wise leveling was applied to all images using SPIP 6.0.6 software for Windows.
Nuclei isolation and DNA extraction
The protocol for nuclei isolation and DNA extraction was used as previously reported.11 For whole cell DNA extraction, cells were washed once with sterile PBS, trypsinized for 3 min, centrifuged at 3000 RPM for 3 min, and then lysed using a cell lysis buffer (20 mM EDTA, 10 mM Tris 8.0, 200 mM NaCl, 0.2 % sodium dodecyl sulfate, 100 _g/ml Proteinase K). Total cellular DNA was then isolated by phenol-chloroform extraction followed by ethanol precipitation.
Quantitative real time PCR (qPCR) assay for determining plasmid abundance per cell nucleus and whole cells
The primers used for detection of pEGFP N1 and genomic GAPDH were designed and purchased from Integrated DNA Technologies, and were as follows: a) pEGFP N1: forward primer CATGGTCCTGCTGGAGTTCGTG, reverse primer CGTCGCCGTCCAGCTCGACCAG; and b) mouse genomic GAPDH: forward primer ACCACAGTCCATGCCATCAC, reverse primer TCCACCACCCTGTTGCTGTA. Standard curves of 10 fold serial dilution series with cycle threshold (Ct) plotted against log copy number were made for 1) pEGFP N1 plasmid DNA mixed with a constant amount (10 ng) of SKOV3 genomic DNA and 2) SKOV3 genomic DNA sample; using Brilliant SYBRR Green QPCR Master Mix. Using the Ct values obtained from qPCR for the transfected cell samples, the respective standard curves were used to determine the number of pEGFP N1 plasmid copies per 10 ng sample and the number of haploid genomes present in 10 ng sample DNA, given that there is one GAPDH gene per haploid genome. The number of plasmid copies per nucleus was calculated using the formula: Plasmid copies per nucleus = 2 × (pEGFP N1 plasmid copies/genomic GAPDH gene copies), as each nucleus contains two copies of GAPDH.
For qPCR analysis of plasmid DNA abundance per whole cell, total cellular DNA (10 ng) was probed with pEGFP N1 primers using Brilliant SYBRR Green QPCR Master Mix. pEGFP N1 plasmid DNA abundance per rNLSd-, NLSc-, and LP2K-transfected cells was calculated relative to PLL-transfected cells, using the MxPro – QPCR software from Agilent Technologies.
In vivo gene delivery and protein expression
Animal care and procedures were performed in accordance with the University of Texas Medical Branch (UTMB) institutional review board guidelines. rNLSd and NLSc were added to low retention Eppendorf tubes, dissolved in nuclease-free water, and sterilized by filtration. The polymer stock solution was diluted to enable complexation with Luc reporter plasmid DNA (pEF1a-Luc, Addgene) at N/P 8. DNA (12.5 or 45 μg) was added to nuclease-free water, mixed 1:1 with polymer solution, and allowed to equilibrate for a minimum of 35 min under sterile conditions. Following polyplex formation, sterile Micromarker microbubbles (2.2 μL) were added to each polyplex solution. The polyplex solution (50 μL) was then injected intramuscularly to the hind legs of male mice (C57/BL6) at n=4 (rNLSd, NLSc). After applying ultrasound gel, polymer-mediated gene delivery of pLuc plasmid was facilitated by irradiating the muscles using a Sonigene sonoporator (VisualSonics), using settings of 1 MHz, 20% duty cycle, 2 W/cm2, 60 sec. The left hind leg, injected with the same volume and type of polyplex, served as a non-sonodelivery control. In vivo imaging for luciferase expression in muscles was performed 3, 6, 9, 15 and 30 days following polyplex injection and sonoporation, using previously published procedures, by intravenous coelenterazine substrate administration and collection of images within 10 min using a Xenogen IVIS100 CCD apparatus.25,26
3. RESULTS AND DISCUSSION
3.1. Synthesis of NLS-containing homopolymers
Cyclooctene macromonomer 1, bearing the Boc- and Pfp-protected sequence VK(Boc)R(Pbf)K(Boc)K(Boc)K(Boc)P, was prepared by solid-phase peptide synthesis (SPPS), and the structure confirmed by 1H and 13C NMR spectroscopy in DMSO-d6 and fast atom bombardment (FAB) mass spectrometry ([M+H]+ calculated 1672.00, found 1672.01). Macromonomer 1 was homopolymerized by ring-opening metathesis polymerization (ROMP) in a mixture of 2,2,2-trifluoroethanol (TFE) and dichloromethane using the bromopyridine-substituted Grubbs metathesis catalyst24 to afford rNLSa-e (Scheme 1). Polymerization at 40 °C gave 64% monomer conversion (rNLSe, Table S1). NLSa-c polymers were synthesized similarly as rNLSa-e, using a macromonomer having the NLS attached to cyclooctene through a proline residue.11 Monomer conversion was determined by 1H NMR spectroscopy, integrating the relative intensity of the cyclic olefin proton resonance (5.6 ppm) vs. the polymer olefin resonance (5.3 ppm). The molecular weights and polydispersities of the Boc- and Pbf-protected polymers were estimated by gel permeation chromatography (GPC) in N,N-dimethylformamide (DMF) in the presence of 0.01 M LiCl (Figure 2A, S1). Removal of the Boc and Pbf protecting groups gave the corresponding cationic structures (soluble in water at 70 mg/mL) which were purified by dialysis in 30% acetic acid (once) and water (three times). The polymer structures were evaluated by 1H and 13C NMR spectroscopy. Though comb polycations with several counterions were prepared, the counterion exerted no impact on transfection efficiency (Figure S3). The molecular weight and molecular weight distribution of the deprotected cationic polymers, with both acetate and trifluoroacetate (TFAc) counterions, were estimated by GPC eluting with TFE having 0.02 M sodium trifluoroacetate (Figure 2). The narrow molecular weight distribution of the Boc- and Pbf- protected polymers, evaluated by GPC in DMF (PMMA standards), and of deprotected cationic polymers, evaluated by GPC in TFE (PMMA standards), may be attributed to steric bulk of the macromonomer that acts to reduce secondary metathesis events.27
Scheme 1.
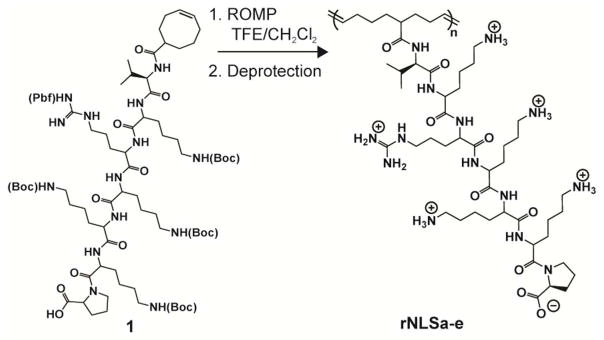
ROMP of rNLS macromonomer 1 to give the corresponding rNLSa-e.
Figure 2.

(A) Molecular weight and molecular weight distribution of protected polymers (estimated by GPC in DMF) and deprotected polymers following dialysis (estimated by GPC in TFE). Molecular weights were estimated relative to PMMA standards; (B) GPC traces of rNLSa, c, and e, eluting with TFE.
3.2. Polyplex formation and characterization
Polyplexes formed upon incubating the plasmid DNA (reporter mRFP-IRES-Puro) with NLSc, rNLS, and PLL were evaluated by access of PicoGreen® to the complexed DNA, measured by PicoGreen® fluorescence relative to experiments using naked DNA. Figure S4 shows that all of the polymers formed tight, dye excluding polyplexes at N/P > 2, and remained intact for over 16 h, as judged by the lack of increase in PicoGreen® fluorescence over that timeframe.
Polyplexes for transfection should optimally be nanoscale structures with a net positive surface charge for complexing DNA and facilitate cellular uptake. Zeta-potential characterization of the polyplexes (N/P = 7) formed from PLL, NLSc, rNLSa-e, and PEI, and diluted with NaCl to a final concentration of 10 mM NaCl solution, showed net charge ranging from +26–41 mV (Table 1). Polyplex size measurements by dynamic light scattering (DLS) in 10 mM NaCl(aq) at 25 °C revealed structures ranging from 50 to 80 nm in diameter (Table 1). Furthermore, AFM images of the PLL, NLSc, rNLS and JetPEI™ polyplexes also revealed approximately spherical, nanoscopic structures (Figure S5).
Table 1.
Polyplex characterization by dynamic light scattering (DLS) and zeta potential in 10 mM NaCl. Error bars indicate ± S.D.
| Polymer | Size (DLS) (nm)† | Zeta potential (mV) |
|---|---|---|
| PLL | 62 ± 2 | 41 ± 4 |
| NLSc | 73 ± 1 | 33 ± 1 |
| rNLSa | 76 ± 1 | 33 ± 1 |
| rNLSb | 78 ± 2 | 28 ± 2 |
| rNLSc | 74 ± 2 | 32 ± 1 |
| rNLSd | 76 ± 1 | 26 ± 2 |
| rNLSe | 76 ± 2 | 28 ± 5 |
| JetPEI | 50 ± 4 | 28 ± 3 |
Average of three sets of 12 measurements, intensity average size
3.3. Serum stability, nuclease protection, and DNA accessibility
Prior to cellular uptake, polyplexes must tolerate physiological conditions (serum and salt) that may affect their colloidal stability. Serum proteins may non-specifically adsorb to polyplexes, while salt may induce polyplex aggregation or even decomplexation to the detriment of transfection efficiency.28 An increase in polyplex diameter in salt and serum, due to the screening of charge repulsion and adsorption of serum proteins, is frequently reported for polyplexes formed from PLL, PEI, and other polycations not modified with a hydrophilic moiety, such as poly(ethylene glycol) (PEG).29–31 While PEG-modified polyplexes groups do not aggregate in salt and serum, they often exhibit decreased cellular uptake and thus lower transfection efficiency.32 Polyplex stability in salt and serum was evaluated by incubation with 10% fetal bovine serum (FBS) and 150 mM NaCl(aq). DLS-derived size (intensity average) of rNLSc and rNLSd polyplexes at 37 °C (Figure 3) changed little over time (from t = 1 to 60 min), demonstrating their resistance to aggregation into very large (micron scale) structures that would be problematic in transfection experiments.33
Figure 3.

DLS measurements of polyplex diameter in 150 mM NaCl(aq) and 10% fetal bovine serum (FBS). The intensity-average diameter is plotted for the polyplexes in water, as well as in 150 mM NaCl and 10% FBS at t = 1 and 60 min after incubation. The data represents the average and standard deviation of three sets of at least 12 measurements.
The ability of rNLS and NLS comb polymers to protect DNA as polyplexes was evaluated by incubation with DNase I, an enzyme that degrades naked DNA, followed by heparin-induced decomplexation. Following decomplexation, the solutions were evaluated on an agarose gel against a DNA ladder. Polymer-complexed DNA showed very little degradation on the gel (Figure S6a), confirming the ability of all the polyplexes to sufficiently protect DNA in the DNase I solution. This suggests their ability to protect DNA against intra- and extracellular nucleases upon incubation with live cells. Next, DNA accessibility in the polyplexes was quantified using the PicoGreen® reagent by incubating polyplexes (N/P 7) with heparin, an anionic polymer that competes with DNA for binding polycations. Relative fluorescence intensities at several time points after heparin incubation demonstrated significant differences in PicoGreen® access to the plasmid DNA (Figure S6b). Tight DNA-polymer interactions for PLL are evident from the relatively low percentage of DNA accessible after 16 h, while NLS-containing structures show higher DNA accessibility, confirming faster decomplexation and weaker plasmid-DNA association (Figure 4). Encouraged by the ability of the comb polymers to both protect and release DNA, we extended this evaluation to transfection efficiency of the NLS-containing polymers in cell culture.
Figure 4.
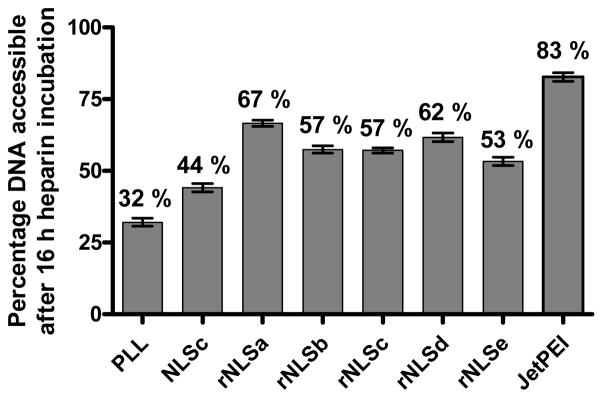
Determination of DNA accessibility using a PicoGreen® plate reader assay. Percentage of DNA accessible to PicoGreen® relative to free DNA (100% accessible) after incubation with heparin for 16 h. Error bars indicate ± S.D.
3.4. Effect of NLS orientation on nuclear translocation
As intranuclear delivery, essential for protein expression, relies on polyplex uptake by the nuclear import machinery, we evaluated the effect of NLS orientation on nuclear translocation by quantitative PCR (qPCR) following the transfection of SKOV3 cells. These experiments (Figure 5a) showed a three-fold greater plasmid copy number per cell nuclei delivered by rNLSd (~8.7 × 106) than by NLSc (~2.3 × 106). These results can be contrasted to those of Branden and coworkers, who reported a complete absence of nuclear translocation of DNA/PNA-NLS complexes when NLS was conjugated to PNA by the proline residue (as in NLSa-c).20 These NLS comb polymers, irrespective of NLS orientation, exhibit evidence of nuclear translocation, but proved more efficient in the ‘rNLS’ orientation. Since nuclear localization of plasmid DNA depends on both cellular uptake and nuclear translocation of polyplexes, we determined the relative intracellular abundance of plasmids per whole SKOV3 cell by qPCR (Figure 5b). Cells transfected with PLL-, NLSc-, and rNLSd-based polyplexes showed a statistically similar intracellular abundance of plasmid DNA, whereas Lipofectamine™ 2000 (LP2K) gave an approximately 50 times higher abundance of plasmid DNA. These results confirm that the higher nuclear abundance of plasmid DNA afforded by rNLSd-based polyplexes, relative to those formed from NLSc (Figure 5a), stems from efficient nuclear translocation rather than differences in cellular entry.
Figure 5.
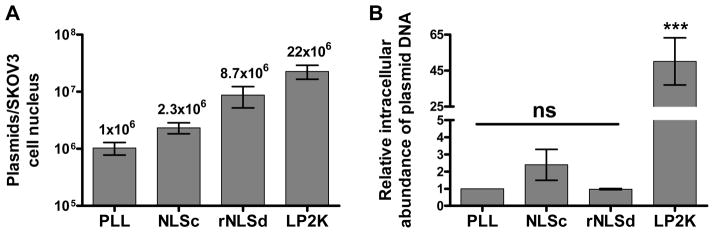
qPCR analysis of plasmid abundance per SKOV3 A) cell nuclei, and B) whole cells, 24 h post transfection with PLL, NLSc, and rNLSd polyplexes and Lipofectamine 2000 (LP2K). Error bars indicate ± S.D. ‘ns’ indicates no statistically significant difference relative to PLL transfected cells, and ‘***’ indicates p<0.001 of LP2K transfected cells compared to PLL, NLSc and rNLSd transfected cells.
3.5. Protein expression and cytotoxicity profiles
Since the polyplexes used in this study are of comparable size, surface charge, and stability, we next examined the combined effects of differences in nuclear translocation and polyplex decomplexation on protein expression. Transfection studies in cell culture showed significantly higher protein expression derived from rNLSc and rNLSd relative to NLSa-c, PLL, JetPEI™, and LP2K in SKOV3 cells (Figure 6). Even the lowest expressing rNLS polymer (rNLSe) exceeded that of the NLS-based structures. The superior protein expression of rNLSa-e relative to NLSa-c suggests that reorienting the PKKKRKV sequence improves transfection efficiency. Moreover, PLL, rNLSa-e, and NLSa-c polyplexes exhibited high cell viability, in strong contrast to JetPEI™ and LP2K. The utility of these polymers for gene therapy was further gauged in human ovarian (SKVLB1) cancer cells, a derivative of SKOV3 cells, having high p-glycoprotein expression and significantly reduced sensitivity to chemotherapeutic drugs.34,35 Transfection experiments in SKVLB1 cells reiterate the combined biocompatibility and high protein expression of rNLS-based polyplexes relative to cationic liposomal formulations (Figure S7). We note that while polymers with different molecular weights afforded differing amounts of protein expression, we found no discernable trend of molecular weight on transfection efficiency.
Figure 6.

Transfection performance of polyplexes in SKOV3 cells: (A) RFP expression; (B) cell viability 48 h post transfection. Cells in each well of a 96-well plate were exposed to NLSa-c or rNLSa-e polyplexes having polymer concentrations ranging from 6.24 ng/μL (for N/P 4) to 10.8 ng/μL (for N/P 7). The concentration of plasmid DNA was 2 ng/μL. Each well contained a total volume of 125 μL. Error bars indicate ± S.E.M.
3.6. In vivo performance
The high transfection efficiency of rNLS-based polyplexes in cell culture encouraged us to evaluate the performance of these polyplexes in vivo. Renilla Luciferase reporter plasmid DNA (pRluc) expression from rNLSd- and NLSc-based polyplexes was calculated from measured intramuscular luminescence intensities. We note that rNLSd- and NLSc-based polyplexes were selected for this study because they provided the highest transfection efficiency in SKOV3 cells. These polymers outperformed even NLS2, a copolymer of 5-PKKKRKV-1-cyclooctene and 5-tetralysine-1-cyclooctene that had previously proven superior to JetPEI in ultrasound mediated intramuscular delivery in mice.26 In the present study, we utilized polyplexes at N/P 8 and measured gene expression following sonoporation (ultrasound stimulus) of firefly luciferase reporter plasmid DNA (pLuc) at 3, 6, 9, 15 and 30 days following intramuscular injection of mice hind legs with, and without, sonoporation.26,27 In accord with cell culture experiments, in vivo results showed the impact of NLS orientation. Polyplexes formed from rNLSd gave 2–10 times greater protein expression than those formed from NLSc using sonoporation (right hind legs). Even in the absence of an ultrasound stimulus, protein expression afforded by rNLSd exceeded that of NLSc (Figure 7a, left hind legs). In all cases, sonoporation increased protein expression 100–500 % relative to non-ultrasound controls (Figure 7c). Moreover, Luciferase expression resulting from rNLSd-based polyplexes increased 200–500 % upon application of ultrasound, whereas that of NLSc-based polyplexes increased only 100 %. Since rNLSd- and NLSc-based polyplexes exhibit similar size, net charge, nuclease protection, DNA accessibility and cellular uptake, and sonoporation permits non-specific uptake of extracellular molecules independent of their composition,36 the superior Luciferase expression by rNLSd-based polyplexes may be attributed to efficient nuclear translocation stemming from this more favorable NLS orientation.
Figure 7.

Intramuscular ultrasound-mediated gene delivery in mice by NLSc- and rNLSd-based polyplexes. (A) Representative bioluminescence images of mice transfected with luciferase reporter plasmid after 5 min acquisition time using a Xenogen IVIS100 CCD camera (9 days after sonoporation). The right hind legs represent protein expression resulting from intramuscular ultrasound-mediated delivery; the left hind legs represent intramuscular delivery in the absence of ultrasound. The color bar indicates the luminescence intensity in photons/sec/cm2/steradian (p/s/cm2/sr). (B) Luciferase expression from right hind legs. (C) Percentage change in luciferase expression upon application of an ultrasound stimulus (the ratio of the expression from the right hind leg to that from the left hind leg). Bars indicate mean ± S.E.M (error bars) with p< 0.05 for rNLSd 12.5 μg dose as compared to all other groups.
4. CONCLUSIONS
In summary, we find the orientation of the PKKKRKV heptapeptide sequence as pendent groups on comb polymers to have a profound impact on transfection performance, both in vitro and in vivo. Given that the higher protein expressing rNLSd delivers more plasmids to the nucleus than NLSc and PLL, these results suggest that the enhanced nuclear localization provided by NLS pendent groups contributes constructively to the transfection performance of these reagents. Though LP2K delivers more plasmid DNA to the nucleus than rNLSd, lower protein expression is seen, due in part to the significant cytotoxicity of this reagent. Taken together, we have shown that amphiphilic comb polymers having rNLS pendent groups afford high transfection efficiency in cell culture and in vivo, as well as low cytotoxicity, making them attractive reagents for deeper investigation as gene therapy reagents.
Supplementary Material
Acknowledgments
The authors acknowledge financial support from the National Science Foundation (DMRBMAT-1207775) and facilities support from the UMass Amherst Genomics, Bioinformatics facility, and the Materials Research Science and Engineering Center (MRSEC DMR-0820506) on Polymers at the University of Massachusetts Amherst. The authors acknowledge Dr. Lawrence Sowers and the University of Texas Medical Branch Department of Pharmacology and Toxicology for support of a bioluminescence optical imaging core. ABBREVIATIONS
Footnotes
Notes
The authors declare no competing financial interests.
Supporting Information. Characterization of polymers, polyplex complexation, polyplex characterization by AFM, PicoGreen® assays, DNase I Assays, SKVLB1 transfection efficiency and cytotoxicity experiments are available in the supporting information. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.De Geest BG, De Koker S, Sukhorukov GB, Kreft O, Parak WJ, Skirtach AG, Demeester J, De Smedt SC, Hennink WE. Soft Matter. 2009;5:282–291. [Google Scholar]
- 2.Boudou T, Crouzier T, Ren K, Blin G, Picart C. Adv Mater (Weinheim, Ger) 2010;22:441–467. doi: 10.1002/adma.200901327. [DOI] [PubMed] [Google Scholar]
- 3.Zhang J, Chua LS, Lynn DM. Langmuir. 2004;20:8015–8021. doi: 10.1021/la048888i. [DOI] [PubMed] [Google Scholar]
- 4.Pack DW, Hoffman AS, Pun S, Stayton PS. Nat Rev Drug Discovery. 2005;4:581–593. doi: 10.1038/nrd1775. [DOI] [PubMed] [Google Scholar]
- 5.Hacein-Bey-Abina S, von Kalle C, Schmidt M, Le Deist F, Wulffraat N, McIntyre E, Radford I, Villeval JL, Fraser CC, Cavazzana-Calvo M, Fischer A. N Engl J Med. 2003;348:255–256. doi: 10.1056/NEJM200301163480314. [DOI] [PubMed] [Google Scholar]
- 6.Couzin J, Kaiser J. Science. 2005;307:1028. doi: 10.1126/science.307.5712.1028b. [DOI] [PubMed] [Google Scholar]
- 7.Tang MX, Szoka FC. Gene Ther. 1997;4:823–832. doi: 10.1038/sj.gt.3300454. [DOI] [PubMed] [Google Scholar]
- 8.Yamagata M, Kawano T, Shiba K, Mori T, Katayama Y, Niidome T. Bioorg Med Chem. 2007;15:526–532. doi: 10.1016/j.bmc.2006.09.033. [DOI] [PubMed] [Google Scholar]
- 9.Intra J, Salem AK. J Pharm Sci. 2010;99:368–384. doi: 10.1002/jps.21840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Welsh DJ, Jones SP, Smith DK. Angew Chem, Int Ed Engl. 2009;48:4047–4051. doi: 10.1002/anie.200900401. [DOI] [PubMed] [Google Scholar]
- 11.Parelkar SS, Chan-Seng D, Emrick T. Biomaterials. 2011;32:2432–2444. doi: 10.1016/j.biomaterials.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 12.Chen C, Jones CH, Mistriotis P, Yu Y, Ma X, Ravikrishnan A, Jiang M, Andreadis ST, Pfeifer BA, Cheng C. Biomaterials. 2013;34:9688–9699. doi: 10.1016/j.biomaterials.2013.08.063. [DOI] [PubMed] [Google Scholar]
- 13.Kow SC, McCarroll J, Valade D, Boyer C, Dwarte T, Davis TP, Kavallaris M, Bulmus V. Biomacromolecules. 2011;12:4301–4310. doi: 10.1021/bm201199c. [DOI] [PubMed] [Google Scholar]
- 14.Kim YK, Minai-Tehrani A, Lee JH, Cho CS, Cho MH, Jiang HL. Int J Nanomed. 2013;8:1489–1498. doi: 10.2147/IJN.S42949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tian H, Chen J, Chen X. Small. 2013;9:2034–2044. doi: 10.1002/smll.201202485. [DOI] [PubMed] [Google Scholar]
- 16.Kanazawa T, Takashima Y, Murakoshi M, Nakai Y, Okada H. Int J Pharm. 2009;379:187–195. doi: 10.1016/j.ijpharm.2009.06.015. [DOI] [PubMed] [Google Scholar]
- 17.Hu Q, Wang J, Shen J, Liu M, Jin X, Tang G, Chu PK. Biomaterials. 2012;33:1135–1145. doi: 10.1016/j.biomaterials.2011.10.023. [DOI] [PubMed] [Google Scholar]
- 18.Opanasopit P, Rojanarata T, Apirakaramwong A, Ngawhirunpat T, Ruktanonchai U. Int J Pharm. 2009;382:291–295. doi: 10.1016/j.ijpharm.2009.08.029. [DOI] [PubMed] [Google Scholar]
- 19.Boulikas T. Gene Ther Mol Biol. 1998;1:713–740. [Google Scholar]
- 20.Branden LJ, Mohamed AJ, Smith CE. Nat Biotechnol. 1999;17:784–787. doi: 10.1038/11726. [DOI] [PubMed] [Google Scholar]
- 21.Ashby EC, Coleman D. J Org Chem. 1987;52:4554–4565. [Google Scholar]
- 22.Hillmyer MA, Laredo WR, Grubbs RH. Macromolecules. 1995;28:6311–6316. [Google Scholar]
- 23.Hartley D. J Am Chem Soc. 1962:4722–4723. [Google Scholar]
- 24.Love JA, Morgan JP, Trnka TM, Grubbs RH. Angew Chem, Int Ed Engl. 2002;41:4035–4037. doi: 10.1002/1521-3773(20021104)41:21<4035::AID-ANIE4035>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 25.Zolochevska O, Xia X, Williams BJ, Ramsay A, Li S, Figueiredo ML. Hum Gene Ther. 2011;22:1537–1550. doi: 10.1089/hum.2011.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zolochevska O, Parelkar S, Ellis J, Chan-Seng D, Emrick T, Wei J, Patrikeev I, Motamedi M, Figueiredo ML. Hum Gene Ther. 2013 doi: 10.1089/hum.2013.091. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boydston AJ, Helcombe TW, Unruh DA, Frechet JMJ, Grubbs RH. J Am Chem Soc. 2009;131:5388–5389. doi: 10.1021/ja901658c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Santander-Ortega MJ, Fuent M, Lozano MV, Bekheet ME, Protgatzky F, Elouzi A, Ucheghu IF, Schazlein AG. Soft Matter. 2012;8:12080–12092. [Google Scholar]
- 29.Johnson RN, Burke RS, Convertine AJ, Hoffman AS, Stayton PS, Pun SH. Biomacromolecules. 2010;11:3007–3013. doi: 10.1021/bm100806h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burke RS, Pun SH. Bioconjugate Chem. 2010;21:140–150. doi: 10.1021/bc9003662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anderson K, Sizovs A, Cortez M, Waldron C, Haddleton DM, Reineke TM. Biomacromolecules. 2012;13:2229–2239. doi: 10.1021/bm300471n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lai TC, Kataoka K, Kwon GS. Biomaterials. 2011;32:4594–4603. doi: 10.1016/j.biomaterials.2011.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Y, Reineke TM. Bioconjugate Chem. 2006;17:101–108. doi: 10.1021/bc050275+. [DOI] [PubMed] [Google Scholar]
- 34.Smith CD, Carmeli S, Moore RR, Patterson GML. Cancer Research. 1993;53:1343–1347. [PubMed] [Google Scholar]
- 35.Lan K, Yen S, Liu R, Shih H, Tseng F, Lan K. Clin Exp Metastasis. 2007;24:461–470. doi: 10.1007/s10585-007-9083-9. [DOI] [PubMed] [Google Scholar]
- 36.Zolochevska O, Figueiredo ML. Front Biosci, Scholar Ed. 2012;4:988–1006. doi: 10.2741/s313. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.