

Abstract
Immunotherapy has the potential to provide a possible treatment therapy to prevent or delay Alzheimer Disease. In a clinical trial (AN1792) in which patients received this immunotherapy and received active Aβ1–42 peptide immunizations, treatment was stopped when 6% of patients showed signs of meningoencephalitis. Follow up on these patients led to the conclusion that the antibody response was beneficial in removing Aβ1–42 from brain but an accompanying inflammatory Th1 T cell response was harmful. As a safe alternative treatment targeting the same self protein, Aβ1–42, in brain, we and others are working on a DNA Aβ1–42 immunization protocol as the immune response to DNA immunizations differs in many aspects from immunizations with peptide antigens. Because the immune response to DNA vaccination has different kinetics and has a significantly lower antibody production, we evaluated two different prime boost regimens, Aβ1–42 DNA prime/ Aβ1–42 peptide boost and Aβ1–42 peptide prime/ Aβ1–42 DNA boost for their effectiveness in antibody production and possible side effects due to inflammatory T cell responses. While both boost regimes significantly enhanced the specific antibody production with comparable antibody concentrations, the absence of the Aβ1–42 T cell response (no proliferation and no cytokine production) is consistent with our previous findings using this DNA Aβ1–42 trimer immunization and greatly enhances the safety aspect for possible clinical use.
Keywords: Alzheimer Disease, Amyloid beta 1–42, immunotherapy, DNA vaccination, antibody production, regulatory immune response, peptide prime/DNA boost
1. Introduction
Alzheimer Disease (AD) has been strongly associated with the accumulation, aggregation and deposition of amyloid beta (Aβ) peptides in brain and it was postulated 20 years ago that Aβ deposition is an initial event in the pathogenesis of AD (Hardy, 1992, Selkoe et al. 1992, Hardy and Selkoe, 2002). A major component of senile plaques is aggregated extracellular amyloid (Aβ1–42 fibrills) and is thus, a major target for the development of an AD therapy. Mice transgenic for human FAD genes causing overproduction of human Aβ peptides and plaque development in mouse brain were developed as mouse models of AD (Games et al. 1995, Dodart et al. 2002) and in these mice immunization with Aβ1–42 peptide lead to anti-Aβ1–42 antibody responses and reduction of Aβ1–42 from the brain, as well as a reduction in total number of plaques (Schenk et al. 1999). In addition, increased learning and memory have been described for Aβ42 immunized mice compared to the respective control animals (Janus et al. 2000, Morgan et al. 2000) and these findings lead to a first clinical trial, in which AD patients were vaccinated with Aβ1–42 peptide. This trial was stopped when 6% of immunized patients developed meningo-encephalitis (Fox et al. 2005, Gilman et al. 2005). A follow-up showed that Aβ1–42 peptide vaccination did indeed lead to a reduction in plaque load in patients who had been treated with Aβ1–42 peptide compared to the placebo control patients providing proof that it is possible to remove brain amyloid by immunotherapy (Holmes et al, 2008).
In an attempt to develop a safe treatment for Alzheimer Disease we and others are working on a DNA Aβ1–42 immunization protocol (Qu et al, 2004, 2006, 2007. 2010, Kim et al. 2007, Movsesyan et al. 2008, DaSilva 2009, Davtyan et al. 2010). The immune response to DNA immunizations in general is different from protein or peptide based immunizations and we have shown that in our model, a gene gun delivered DNA Aβ1–42 trimer immunization, a predominant Th2 antibody response and the absence of antigen specific T cell responses were the main characteristics. Both of these are indicative of a non-inflammatory immune response (Lambracht-Washington et al. 2009, Qu et al. 2010, Lambracht-Washington et al. 2011).
In regard to immunizations with whole organisms, proteins and peptides, DNA vaccines in general are considered as third generation vaccines consisting of genetically engineered plasmids designed to produce one or two specific proteins against which the immune response is elicited. To overcome a significantly lower antibody production following DNA immunizations, so called prime-boost regimens have been shown to be highly effective. (Kim et al. 2007, Subramanian & Divya Shree, 2008, Davtyan et al. 2010). This greater level of immunity is based on a heterologous prime boost protocol in which the antigen is applied via different routes and immunization sites. The first immunization initiates the priming of the immune response and subsequent immunizations trigger further expansion of antigen specific cells and selection of cells with high antigen avidity to boost the specific responses (Woodland, 2004). We show here the results from two different boost regimens: The peptide boost of a DNA primed immune response and the DNA boost of a peptide primed immune response. Both appear to be effective to increase the antibody responses but they show marked differences in respect to the cellular immune response.
2. Material and Methods
2.1 Mice
All experiments were performed in 4 to 6 month old B6SJLF1/J female mice (n=24) purchased from The Jackson Laboratory (Bar Harbor, Maine). Animal use for this study was approved by the UT Southwestern Medical Center Animal Research Committee. In the first set of experiments we immunized four mice 3x with the DNA Aβ42 trimer followed by 3 immunizations with Aβ42 peptide (DNA prime/peptide boost) and another group of four mice received 3 Aβ42 peptide immunizations first followed by 3 immunizations with DNA Aβ42 trimer (peptide prime/DNA boost). The experiments were repeated with 16 mice in 4 groups: 6x DNA Aβ42 trimer, 3x DNA Aβ42 trimer/ 3x Aβ42 peptide, 6x Aβ42 peptide, 3x Aβ42 peptide/3x DNA Aβ42 trimer. All mice received a total of six immunizations.
2.2 Immunization with Aβ1–42
DNA vaccinations with a double plasmid DNA system (activator plasmid encoding Gal4 transcription factor and responder plasmid encoding three copies of human Aβ1–42) were performed using a Helios gene gun (Bio-Rad, Hercules, CA) with a helium pressure of 300 psi into the mouse ears without the use of aduvant. Human Aβ1–42 peptide was mixed with QuilA as adjuvant which is very similar to Qs21 which had been used in the clinical trial and was given by i.p. injections. The immunizations procedures have been described in detail previously (Qu et al. 2007, Lambracht-Washington et al. 2009, Qu et al. 2010, Lambracht-Washington et al. 2011).
2.3 Plasma and splenocyte collections
The mice were killed ten days following the final immunization. Blood was collected by cardiac puncture; spleens were aseptically removed and processed for tissue culture as previously described (Lambracht-Washington et al. 2009).
2.4 Analysis of cell proliferation by CFSE dilution
This method has been described in detail previously (Lambracht-Washington et al. 2011). Fluorescence of the cells was measured using an Accuri C6 Flow Cytometer (Ann Arbor, MI) and analyzed with CFlow Plus and FCS Express Version 3 software programs.
2.5 ELISA and ELISPOT assays
ELISA assays for antibody levels in mouse plasma and cytokine concentrations from cell culture supernatants and ELISPOT assays to determine the numbers of cytokine producing cells were done as described previously (Lambracht-Washington et al. 2009).
2.6 Statistics
For statistics of proliferation, ELISA and ELISPOT data points, which were all performed in triplicate samples we used GraphPad Prism version 5.02 for Windows (San Diego, CA, www.graphpad.com). Mean values and standard deviations were calculated using an unpaired t test with two tailed p values and column statistics. P-values of ≤ 0.05 were considered significant.
3. Results
3.1 Th1 and Th2 T cell subsets provide help for increased antibody production
The antibody response after DNA Aβ1–42 immunization delivered via gene gun bombardment into the skin is a characteristic Th2 response with antibodies of the IgG1 and IgG2b isotypes only. In the prime boost experiments performed here we found that boosting with Aβ1–42 peptide as well as DNA Aβ1–42 trimer, lead to remarkable increases in specific antibody production (Figure 1). The antibody levels found were similar to levels found in peptide only immunized mice, 341.3 ± 9.25 μg/ml in the peptide boosted mice and 217.9 ± 16.5 μg/ml in the DNA boosted mice, but the signature Th2 antibody response characteristic for the DNA regimen disappeared following the boost immunizations in both groups.
Figure 1. Boost immunizations strongly increase the Aβ1–42 specific antibody response.

Bars show the amount of antigen specific IgG levels in mouse plasma combined from the individual median antibody levels for each mouse. The number of mouse samples analyzed is indicated as n. Both of the tested boost immunizations (DNA prime/Peptide boost and Peptide prime/DNA boost) significantly increased the levels of Aβ1–42 specific antibodies in mouse plasma compared to the DNA Aβ1–42 trimer
After 3 immunizations with DNA Aβ1–42 trimer or Aβ1–42 peptide, respectively, we found good antibody production in both groups: 191.6 μg/ml ± 14.52 in the peptide immunized mice (n=8) and 92.05 μg/ml ± 65.05 in the DNA immunized mice (n=8), P = 0.0009 for the comparison of the two groups. The antibody isotypes after 3 immunizations showed a mixed antibody response in the peptide immunized mice with IgG1/IgG2a ratios of 1.0005 (Table 1) and a predominant IgG1 antibody response in the DNA immunized mice (IgG1/IgG2a ratio of 7.677, Table 1) consistent with our published results (Lambracht-Washington et al. 2009). Following the switch in the immunization procedure (peptide boost and DNA boost) the isotype profile changed completely. Now three of the groups, peptide only, peptide primed/DNA boost, DNA primed/peptide boost, showed a mixed anti-Aβ42 antibody isotype profile. Only the mice which had received 4 DNA Aβ42 peptide immunizations (n=4) kept the predominant IgG1 antibody profile. Due to the mixed background in the mice we were using (B6SJLF1) and the known allelic differences for the IgG2a locus in the B6 and SJL mouse strains, we included the IgG2c (IgG2ab) isotype in our final analyses of the Th1/Th2 antibody ratios (Table 1, IgG1/IgG2a-IgG2c). We had seen in our isotype analyses a substantial amount of the IgG2c antibody from B6SJLF1 mouse plasma (data not shown).
Table 1.
Analysis of changes in the IgG1/IgG2a ratios due to different boost regimens
| IgG1/IgG2a-2c ratio
| ||||
|---|---|---|---|---|
| Immunization times | DNA Aβ42 trimer | DNA prime/peptide boost | Aβ42 peptide | Peptide prime/DNA boost |
| 3 × | 7.677 | 1.005 | ||
| 4 × | 10.5 | 4.438 | 1.026 | 1.033 |
| 5 × | 9.717 | 1.198 | 1.113 | 1.023 |
| 6 × | 9.293 | 1.385 | 1.113 | 1.008 |
3.2 Epitope analyses from plasma of prime-boost mice revealed a previously undetected B cell epitope difference and showed the influence of T cells during the priming immunizations
The antibody epitope for Aβ1–42 specific antibodies lays with in the N-terminal peptide sequence, Aβ1–15. To search for different antibody binding features in the mouse groups we tested antibody binding to a number of overlapping peptides within the known B cell epitope, Aβ1–17, Aβ2–17, Aβ3–17, Aβ4–17, Aβ5–17, and Aβ6–17. Furthermore, we dissected the antibody binding in regard to IgG isotypes, IgG1 and IgG2a/IgG2c. While IgG1 and IgG2a/c antibodies from Aβ1–42 peptide immunized mice detected Aβ1–17, Aβ2–17, and Aβ3–17, some of the IgG1 antibodies from DNA Aβ1–42 trimer immunized mice detected also Aβ4–17 and Aβ5–17. Due to the high antibody levels this new epitope becomes very obvious in the prime-boost mouse groups: In the DNA prime/peptide boost mice IgG1 antibodies from five of eight plasma samples reacted with Aβ4–17 and Aβ5–17 and for the IgG2a/c isotypes four of eight plasma samples detected Aβ4–17 and Aβ5–17 (Figure 2). In the peptide prime/DNA boost mice only one from seven plasma samples recognized Aβ4–17 and Aβ5–17. Thus, it appears as if the DNA priming guided the antibody response towards this new B cell epitope. Antibodies reacting with Aβ4–17 can be found in 62.5% of the plasma samples from DNA primed mice and only 14.2% of the plasma samples from peptide primed mice.
Figure 2. Detection of the B cell epitope Aβ4–17 in DNA immunized mice.

Plasma samples from the 6x immunization time point of all the mice used in this study were analyzed by ELISA in parallel. DNA Aβ1–42 immunized mice had no anti-Aβ1–42 IgG2a antibodies and thus no anti-Aβ4–17 or anti-Aβ5–17 IgG2a antibodies were detected (B and D) while some (2/4 and 1/4) mouse samples showed specific IgG1 antibody binding to this epitope (A and C). Despite high antibody levels against Aβ1–42 in the Aβ42 peptide immunized mice which detected the B cell epitope Aβ1–17, no antibody binding was found for Aβ4–17 and Aβ5–17 (A, B, C, and D). IgG1 antibodies reacting with Aβ4–17 can be found in 62.5% (5/8) of the plasma samples from DNA primed mice and only 14.2% (1/7) of the plasma samples from peptide primed mice (A). IgG2a antibodies reacting with Aβ4–17 can be found in 50% (4/8) of the plasma samples from DNA primed mice and again 14.2% (1/7) of the plasma samples from peptide primed mice (B). Identical reactivities were found for the Aβ5–17 epitope (C and D)
In addition, we never found Aβ4–17 antibody binding in other Aβ1–42 peptide immunized mice but in some of the DNA Aβ1–42 trimer immunized mice from mouse strains B6SJLF1/J and B6C3F1/J carrying the mixed H2 haplotypes b/s and b/k (data not shown).
3.3 DNA boosted T cells down-regulate T cell proliferation in peptide primed mice
T cell proliferation was analyzed with a CFSE dilution assay and staining for CD4 and CD8 T cells as described previously (Lambracht-Washington et al. 2011). In the first set of experiments we found no T cell proliferation in the mice which had received the peptide prime/DNA boost regimen (CD4 T cell proliferation index of 1, n=3), while for the mice which had received the DNA prime/peptide boost regimen elevated CD4 T cell proliferation was observed (CD4 T cell proliferation index of 1.66 ± 0.14, and Aβ42 specific CD4 T cell precursor frequencies of 7.41% ± 1.6, n=4) (Figure 3). Thus, even though both groups had received three DNA and three peptide immunizations, the latter immunization, DNA or peptide, dominated in the T cell response, which meant that DNA boosted T cells prevented the T cell proliferation in the peptide primed mice, while the peptide boosted T cells proliferated despite the presumed presence of DNA primed Aβ1–42 specific T cells. In these experiments we had also included wells in which we used plate-bound CD28 antibody to provide a secondary signal in addition to TCR signaling to improve T cell proliferation. No differences to the above described results were found (data not shown).
Figure 3. No CD4 T cell proliferation in mice which received the peptide prime/DNA boost Aβ42 immunizations.
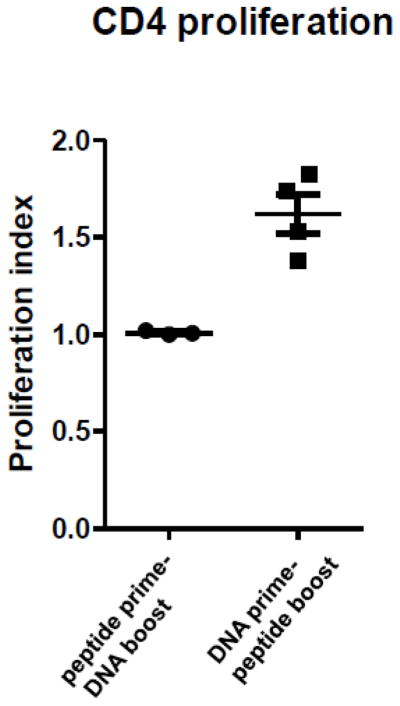
Pooled splenocytes from mice which had received the respective 6 immunizations were re-stimulated in-vitro with Aβ1–42 peptide and CFSE dilution was analyzed at day 6. No proliferation was found in mice which had received the DNA boost (CD4 T cell proliferation index of 1), while the DNA boosted mice which had received 3 peptide boost immunizations showed elevated CD4 T cell proliferation (CD4 T cell proliferation index of 1.6).
These results were confirmed in the second set of experiments in which we tested in parallel mice which had received six DNA Aβ1–42 trimer immunizations and six Aβ1–42 peptide immunizations respectively.
3.4 Cytokine secretion in the prime-boost models
Splenocytes from six times Aβ1–42 peptide immunized mice secreted IFNγ (shown by ELISPOT, Figure 4), IL-17, and IL-5, whereas the splenocytes from six times DNA Aβ1–42 trimer immunized mice secreted none of these three cytokines. The ELISPOT analysis showed that indeed the DNA boost regimen apparently suppressed the T cell response in peptide primed mice. While the peptide prime/DNA boost mice showed only a slight increase in IFNγ secreting cells compared to the respective medium controls and the levels found in the DNA only immunized mice (26.3 ± 6 spots per 106 cells after Aβ1–42 re-stimulation and 56 ± 14.7 spots after Aβ10–26 re-stimulation), the DNA prime/peptide boost mice showed 105 ± 27.7 IFNγ ELISPOTs after Aβ1–42 re-stimulation and 85.6 ± 0.5 IFNγ ELISPOTs after re-stimulation with Aβ10–26 peptide. This difference in IFNγ producing cells in these two groups of mice, peptide prime/DNA boost and DNA prime/peptide boost, is highly significant (P = 0.0067). Furthermore, while the peptide prime/DNA boost mice showed no IL-17 secreting cells in the ELISPOT analysis, the DNA prime/peptide boost mice showed an increase in IL-17 spot count (36.33 ± 18.18) compared to the spot count in the peptide prime/DNA boost mouse group. However, due to the high standard deviation this difference is not significant (P = 0.06). For the Th2 cytokine IL-5 the DNA prime/peptide boost mouse group has the highest number of cytokine secreting cells with 37.67 ± 17.01 spots per 106 cells, but again due to the high standard deviation the difference to the peptide prime/DNA boost group as well as the peptide only immunized mouse group were not significant (P values of 0.037 and 0.252, respectively).
Figure 4. ELISPOT analysis of prime boost groups indicates down-regulation of cytokine secretion due to the DNA boost immunizations.
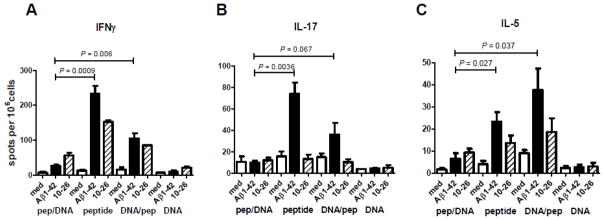
Pooled splenocytes from the four immunization groups were analyzed for IFNγ (A), IL-17 (B) and IL-5 (C) secreting cells after Aβ42 peptide re-stimulation in-vitro in ELISPOT assays. The four immunization groups are indicated below the graphs (peptide prime/DNA boost, peptide, DNA prime/peptide boost, DNA). White bars show the respective number of cytokine secreting cells in the medium controls, black bars show cytokine secretion after re-stimulation with full-length Aβ1–42 peptide, and the striped bars show cytokine secreting cells after re-stimulation with the T cell epitope peptide, Aβ10–26. The differences for IFNγ secreting cells in the two prime boost groups (peptide prime/DNA boost and DNA prime/peptide boost) is significant (P value 0.006) and it appears that the DNA boost regimen down regulates the inflammatory cytokine response in these mice. The comparison of IFNγ and IL-17 secreting cells in the peptide prime/DNA boost group and the peptide group shows for both of these cytokines significantly less cytokine secreting cells in the peptide prime/DNA boost group (P values of 0.0009 and 0.0036 respectively) which is indicative for the down regulated immune response due to the DNA boost immunizations.
4. Discussion
We present here data for two different immune boost regimens to enhance the antibody production in a wild-type mouse model for DNA Aβ1–42 immunization as possible therapy for Alzheimer Disease. The immune response following DNA immunizations in general is much lower compared to peptide immunizations and to enhance the antigen specific response boost immunizations are performed to enhance this response. Most commonly used is a protein/peptide or viral boost immunization to enhance the antibody response in DNA immunizations (Kim et al. 2007, Subramanian & Divya Shree, 2008, Davtyan et al. 2010). Our results show that it is also very efficient to augment a peptide immunization with a DNA boost. To our knowledge this is also the first time, DNA immunizations were used to boost an existing immune response. The benefits from this vaccination approach (peptide prime/DNA boost) may lay in the down regulation of a T cell response which harbors complications of an inflammatory immune response. Therefore, the protocol described here with a peptide prime/DNA boost approach provides the basis for future vaccination protocols.
Mice were primed for three immunization time points to establish a well directed immune response which was tested with the antibody isotype analysis after the initial immunizations. In line with our previous data, we found a mixed immune response (IgG1/IgG2a antibodies) in the peptide primed mice and a predominant Th2 immune response with almost exclusively IgG1 antibodies in the DNA primed mice as shown with the high IgG1/IgG1a ratio (Table 1).
This changed immediately with the boost immunizations: The DNA prime/peptide boost mice switched from a predominant IgG1 response (IgG1/IgG2a ratio of 7.7) to the mixed isotype profile with an IgG1/IgG2a ratio of 4.4 after four immunizations and ratios of 1.1 and 1.3 after five and six immunizations which is similar to the peptide immunizations with ratios of 1.0 and 1.1 indicative of equal IgG1 and IgG2a Aβ1–42 antibody isotype levels.
Antibodies from the DNA primed group maintained the small overlapping epitope specificity and bound strongly to Aβ4–17 and Aβ5–17, whereas this epitope is undetected in the peptide primed/DNA boost group or the Aβ1–42 peptide only group. Thus, a large portion of the high level antibodies and the respective antibody secreting B cells is derived directly from the three DNA or peptide priming vaccinations and enhanced with the latter peptide boost immunizations. After six immunizations both groups which had received the boost immunization, peptide or DNA respectively, had similar high antibody levels with a mixed antibody isotype pattern. As both showed similar levels of IgG1 and IgG2a antibodies indicative for the type of ongoing immune response, Th1 or Th2, we expected to find a similar cellular immune response as well. While we do find strong T cell reactivity in the DNA boost/peptide primed mice, we do not find T cell responses as tested by proliferation assays or the analysis of cytokine secretion in the peptide primed/DNA boost mice. T cells from DNA prime/peptide boost mice showed good proliferation and secretion of IFNγ and IL-17 while T cells from peptide prime/DNA boost mice did not proliferate and made no IFNγ or IL-17. The DNA boost dominated in the T cell immune response as it down regulated antigen specific T cells consistent with what we have shown before (Lambracht-Washington et al. 2011) and a peptide prime/DNA boost immunization regimen is thus beneficial as it minimizes the risk of an inflammatory immune response.
A number of positive functions of Aβ immunotherapy were directly associated with the anti-Aβ42 antibodies. Several studies have shown that the removal of the toxic Aβ42 depositions in brain with antibody therapy preserves synaptic structures and neuron morphology (Buttini et al. 2005, Serrano-Pozo et al. 2010) and again the positive effects were attributed to the antibodies only. Recently it has been shown that antibodies binding to the N-terminal portion of Aβ1–42, which is the known B cell epitope, bind and neutralize soluble Aβ species and prevent synaptotoxicity (Zago et al. 2012). In regard to the mixed antibody response (IgG1 and IgG2a) in our peptide prime/DNA boost model, the beneficial antibodies used in the Zago study were all of the IgG2a (Th1) isotype. Thus, the antibody itself even with the IgG2a (Th1) isotype does not appear to be harmful per se. Furthermore, as stated in this paper the mouse antibody 3D6, which has the IgG2a isotype is the parent of the humanized anti-Aβ42 antibody, bapineuzumab, which is FDA approved and in phase III clinical trials (Zago et al. 2012). Earlier it has been shown that antibodies of the IgG2a isotype are more potent in reducing amyloid burden from APP transgenic mouse brains compared to antibodies of the IgG1 or IgG2b isotypes (Bard et al. 2003).
The published data about the interrupted clinical trial with active Aβ1–42 immunization in AD patients (AN1792) has lead to the conclusion that the antibody response was beneficial but the accompanying inflammatory Th1 T cell response was harmful and was responsible for the meningoencephalitis in 6% of the patients (Gilman et al. 2005, Holmes et al, 2008). Thus, even though we have a mixed antibody response, we assume that the peptide prime/DNA boost regimen is safe and has the advantage that it increases strongly the antibody response and at the same time the DNA boost down regulates the antigen specific T cell immune response which minimizes the risk of an inflammatory autoimmune response.
Acknowledgments
This study was funded by grants from NIH/NIA Alzheimer’s Disease Center (P30AG12300-17), Rudman Partnership; and McCune Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bard F, Barbour R, Cannon C, Carretto R, Fox M, Games D, Guido T, Hoenow K, Hu K, Johnson-Wood K, Khan K, Kholodenko D, Lee C, Lee M, Motter R, Nguyen M, Reed A, Schenk D, Tang P, Vasquez N, Seubert P, Yednock T. Epitope and isotype specificities of antibodies to beta-amyloid peptide for protection against Alzheimer’s disease-like neuropathology. Proc Natl Acad Sci U S A. 2003;100(4):2023–2028. doi: 10.1073/pnas.0436286100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttini M, Masliah E, Barbour R, Grajeda H, Motter R, Johnson-Wood K, Khan K, Seubert P, Freedman S, Schenk D, Games D. Beta-amyloid immunotherapy prevents synaptic degeneration in a mouse model of Alzheimer’s disease. J Neurosci. 2005;5;25(40):9096–9101. doi: 10.1523/JNEUROSCI.1697-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DaSilva KA, Brown ME, McLaurin J. Reduced oligomeric and vascular amyloid-beta following immunization of TgCRND8 mice with an Alzheimer’s DNA vaccine. Vaccine. 2009;27(9):1365–1376. doi: 10.1016/j.vaccine.2008.12.044. [DOI] [PubMed] [Google Scholar]
- Davtyan H, Mkrtichyan M, Movsesyan N, Petrushina I, Mamikonyan G, Cribbs DH, Agadjanyan MG, Ghochikyan A. DNA prime-protein boost increased the titer, avidity and persistence of anti-Abeta antibodies in wild-type mice. Gene Ther. 2010;17(2):261–271. doi: 10.1038/gt.2009.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, DeLong CA, Wu S, Wu X, Holtzman DM, Paul SM. Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer’s disease model. Nat Neurosci. 2002;5(5):452–427. doi: 10.1038/nn842. [DOI] [PubMed] [Google Scholar]
- Fox NC, Black RS, Gilman S, Rossor MN, Griffith SG, Jenkins L, Koller M. Effects of Abeta immunization (AN1792) on MRI measures of cerebral volume in Alzheimer disease. Neurol. 2005;64:1563–1572. doi: 10.1212/01.WNL.0000159743.08996.99. [DOI] [PubMed] [Google Scholar]
- Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature. 1995;373(6514):523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, Eisner L, Kirby L, Rovira MB, Forette F, Orgogozo JM. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurol. 2005;64:1553–1562. doi: 10.1212/01.WNL.0000159740.16984.3C. [DOI] [PubMed] [Google Scholar]
- Hardy J. New insights into the genetics of Alzheimer’s disease. Ann Med. 1996;28:255–258. doi: 10.3109/07853899609033127. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, Jones RW, Bullock R, Love S, Neal JW, Zotova E, Nicoll JA. Long-term effects of Abeta42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372(9634):216–223. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, Chishti MA, Horne P, Heslin D, French J, Mount HT, Nixon RA, Mercken M, Bergeron C, Fraser PE, St George-Hyslop P, Westaway D. A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease. Nature. 2000;408:979–982. doi: 10.1038/35050110. [DOI] [PubMed] [Google Scholar]
- Kim HD, Jin JJ, Maxwell JA, Fukuchi K. Enhancing Th2 immune responses against amyloid protein by a DNA prime-adenovirus boost regimen for Alzheimer’s disease. Immunol Lett. 2007;112(1):30–38. doi: 10.1016/j.imlet.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambracht-Washington D, Qu BX, Fu M, Eagar TN, Stüve O, Rosenberg RN. DNA beta-amyloid (1–42) trimer immunization for Alzheimer disease in a wild-type mouse model. JAMA. 2009;302(16):1796–1802. doi: 10.1001/jama.2009.1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambracht-Washington D, Qu BX, Fu M, Anderson LD, jr, Stüve O, Eagar TN, Rosenberg RN. DNA Immunization against Amyloid beta 42 has high potential as safe therapy for Alzheimer’s Disease as it diminishes antigen specific Th1 and Th17 cell proliferation. Cell Mol Neurobiol. 2011;31:867–874. doi: 10.1007/s10571-011-9680-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemere CA, Masliah E. Can Alzheimer disease be prevented by amyloid-beta immunotherapy? Nat Rev Neurol. 2010;6(2):108–119. doi: 10.1038/nrneurol.2009.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature. 2000;408:982–985. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- Movsesyan N, Ghochikyan A, Mkrtichyan M, Petrushina I, Davtyan H, Olkhanud PB, Head E, Biragyn A, Cribbs DH, Agadjanyan MG. Reducing AD-like pathology in 3xTg-AD mouse model by DNA epitope vaccine - a novel immunotherapeutic strategy. PLoS ONE. 2008;3:2124. doi: 10.1371/journal.pone.0002124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu B, Rosenberg RN, Li L, Boyer PJ, Johnston SA. Gene vaccination to bias the immune response to amyloid-beta peptide as therapy for Alzheimer disease. Arch Neurol. 2004;61:1859–1864. doi: 10.1001/archneur.61.12.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu B, Boyer PJ, Johnston SA, Hynan LS, Rosenberg RN. Aβ42 gene vaccination reduces brain amyloid plaque burden in transgenic mice. J Neurol Sci. 2006;244:151–158. doi: 10.1016/j.jns.2006.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu B-X, Xiang Q, Li L, Johnston SA, Hynan LS, Rosenberg RN. Aβ42 gene vaccine prevents Aβ42 deposition in brain of double transgenic mice. J Neurol Sci. 2007;260:204–213. doi: 10.1016/j.jns.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu BX, Lambracht-Washington D, Fu M, Eagar TN, Stüve O, Rosenberg RN. Analysis of three plasmid systems for use in DNA A beta 42 immunization as therapy for Alzheimer’s disease. Vaccine. 2010;28(32):5280–5287. doi: 10.1016/j.vaccine.2010.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Amyloid beta-protein and the genetics of Alzheimer’s disease. J Biol Chem. 1996;271(31):18295–18298. doi: 10.1074/jbc.271.31.18295. [DOI] [PubMed] [Google Scholar]
- Serrano-Pozo A, William CM, Ferrer I, Uro-Coste E, Delisle MB, Maurage CA, Hock C, Nitsch RM, Masliah E, Growdon JH, Frosch MP, Hyman BT. Beneficial effect of human anti-amyloid-beta active immunization on neurite morphology and tau pathology. Brain. 2010;133(Pt 5):1312–1327. doi: 10.1093/brain/awq056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KM, Pottage L, Thomas ER, Leishman AJ, Doig TN, Xu D, Liew FY, Garside P. Th1 and Th2 CD4+ T cells provide help for B cell clonal expansion and antibody synthesis in a similar manner in vivo. J Immunol. 2000;165(6):3136–3144. doi: 10.4049/jimmunol.165.6.3136. [DOI] [PubMed] [Google Scholar]
- Subramanian S, Divya Shree AN. Enhanced Th2 immunity after DNA prime-protein boost immunization with amyloid beta (1–42) plus CpG oligodeoxynucleotides in aged rats. Neurosci Lett. 2008;436(2):219–222. doi: 10.1016/j.neulet.2008.03.024. [DOI] [PubMed] [Google Scholar]
- Woodland DL. Jump-starting the immune system: prime-boosting comes of age. Trends Immunol. 2004;25(2):98–104. doi: 10.1016/j.it.2003.11.009. [DOI] [PubMed] [Google Scholar]
- Zago W, Buttini M, Comery TA, Nishioka C, Gardai SJ, Seubert P, Games D, Bard F, Schenk D, Kinney GG. Neutralization of soluble, synaptotoxic amyloid β species by antibodies is epitope specific. J Neurosci. 2012;32(8):2696–2702. doi: 10.1523/JNEUROSCI.1676-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]