

Abstract
Glucokinase is a glucose-phosphorylating enzyme that regulates insulin release and hepatic metabolism, and its loss-of-function is implicated in the pathogenesis of diabetes. Glucokinase activators (GKAs) are attractive therapeutics in diabetes, however, clinical data indicate that their benefits can be offset by hypoglycemia due to marked allosteric enhancement of the enzyme’s glucose affinity. We show that a phospho-mimetic of the BCL-2 homology 3 (BH3) alpha-helix derived from human BAD, a glucokinase binding partner, increases the enzyme catalytic rate without dramatically changing glucose affinity, providing a new mechanism for pharmacologic activation of glucokinase. Remarkably, BAD BH3 phospho-mimetic mediates these effects by engaging a novel region near the enzyme’s active site. This interaction increases insulin secretion in human islets and restores the function of naturally-occurring human glucokinase mutants at the active site. Thus, BAD phospho-mimetics may serve as a novel class of GKAs.
INTRODUCTION
Loss of glycemic control in type 2 diabetes (T2D) is the outcome of combined defects in insulin action and insulin secretion, which have strong genetic and environmental components1,2. A major challenge in T2D therapy is to achieve durable glycemic control while minimizing side effects and the risk of hypoglycemia3. Small molecule activators of glucokinase (GK, hexokinase IV) are among multiple investigational agents currently under development for their glucose-lowering capacity in T2D4,5. GK is a key component of the mammalian glucose sensing machinery with tissue-specific roles in insulin secretion by beta-cells, glucose utilization and storage in hepatocytes, as well as central glucose sensing and counterregulatory responses to hypoglycemia5. Compared to other hexokinase isoforms, unique kinetic properties such as lack of product inhibition, a high S0.5 (Km) value for glucose, and positive substrate cooperativity render GK particularly well-suited for regulation of blood glucose levels. As such, gain-of-function mutations in GK are central to the molecular pathogenesis of persistent hyperinsulinemic hypoglycemia of infancy (PHHI), while loss-of-function mutations in the enzyme are linked to maturity onset diabetes of the young type 2 (MODY2) and permanent neonatal diabetes mellitus (PNDM)5. Beyond the fundamental relevance of GK in beta-cell function, emerging evidence also suggests that increased GK activity and glucose metabolism in these cells can impart mitogenic and pro-survival benefits, leading to beta-cell mass expansion6. Improvement of both beta-cell function and mass through increased GK activity may well expand the potential utility of synthetic GK activators beyond T2D to restoration and maintenance of functional beta-cell mass for the treatment of type 1 diabetes (T1D).
All small molecule GK activators (GKAs) reported to date bind to the allosteric site in the enzyme, which is located 20 Å from its active site, and markedly increase the affinity of the enzyme for glucose, typically from 7–8 mM to approximately 1–2 mM4,5. Structural analysis of several GK conformations in the presence and absence of glucose and GKAs has provided insights into the mechanism of its enzymatic action7–14. While the precise transitions between GK conformations are still under active investigation, several studies indicate that, in the presence of increasing glucose concentrations, GK transitions from an inactive super open conformation with low affinity for glucose to an active, closed conformation that is catalytically competent for binding of substrates and release of products7–11,13–16. Gain-of-function mutations in GK, which typically cluster in the allosteric site, and similarly binding of allosteric GKAs to this region cause this transition to occur at lower glucose concentrations5. While several allosteric activators of GK have shown promising short term effects, a drawback of these compounds is the risk of developing hypoglycemia over time3,17–19, thus warranting an exploration of alternative pharmacologic modalities for GK activation, particularly those with a more tempered effect on glucose affinity5.
The BCL-2 family protein BAD binds GK and regulates glucose metabolism in liver and beta-cells20–23. BAD belongs to the BH3-only subclass of pro-apoptotic BCL-2 family proteins, which share sequence homology only within an alpha-helical BH3 domain. Using mutational studies, mouse models and genetic reconstitution assays, we have shown that BAD phosphorylation on a conserved serine residue within its BH3 domain, Ser155 in mouse BAD corresponding to Ser118 in the human sequence, acts as a molecular switch that enables it to enhance glucose metabolism and support insulin secretion in beta-cells22. Concomitant with these metabolic benefits, phosphorylation of the BH3 domain blocks BAD’s capacity to engage the pro-survival proteins BCL-2, BCL-XL and BCL-W, and thereby neutralizes its pro-apoptotic function20,22. Importantly, BAD phosphorylation is sensitive to glucose, nutritional (fed and fasted) states, and hormones or growth factors known to regulate metabolic adaptations to nutritional states, suggesting that BAD phosphorylation and its functional crosstalk with GK may serve as a homeostatic sensor of the nutrient milieu (reviewed in20).
The BAD BH3 domain is not only required but sufficient to mimic BAD’s ability to influence GK, as evidenced by the capacity of Stabilized Alpha-Helices of BCL-2 domains (SAHBs) modeled after the phospho-BH3 helix of BAD to activate GK and restore insulin secretion in BAD-deficient islets22. However, the region of GK engaged by the BAD BH3 domain and the mechanism of enzyme activation was not known. Hydrocarbon-stapled peptides that recapitulate the natural structure of bioactive helices have proven to be both powerful tools for dissecting signaling pathways and prototype therapeutics for targeting protein interactions22,24–26. Given the functional link between phospho-BAD BH3 and GK22, we set out to investigate the molecular and structural properties of BAD SAHBs as synthetic GK activators. Here, we define the enzymatic and structural characteristics of GK bound to the BAD phospho-BH3 helix and demonstrate fundamental mechanistic distinctions from allosteric GK activators that were not previously anticipated. Thus, phospho-BAD BH3 mimetics may represent a novel class of synthetic GK activators.
RESULTS
The effect of phospho-BAD mimetic compounds on GK kinetics
To determine how the inherent characteristics of GK are altered by phospho-BAD BH3, we synthesized a panel of stapled peptides modeled after the human BAD BH3 domain for use in kinetic assays with recombinant GK. This panel included the phospho-mimic BAD SAHBA (S118D) and the non-phosphorylatable BAD SAHBA (S118L) variants (Supplementary Table 1). We examined GK activity as a function of the Hill equation in order to evaluate the effect of these stapled peptides on the maximal rate of reaction (Vmax), the glucose concentration that allows half maximal activity (S0.5), and substrate cooperativity reflected by the sigmoidal response to increasing glucose concentration (Hill coefficient, nH). We also tested the effect of an archetypal allosteric activator of GK, RO0281675 (ref. 9) for comparison.
The kinetic profile of recombinant GK treated with vehicle (DMSO) or RO0281675 showed good agreement with published data9 (Table 1). Treatment with RO0281675 increased the Vmax by approximately 40%, and the affinity for glucose, as reflected by a change in S0.5 from 7.49 ± 0.18 mM to 1.62 ± 0.13 mM, with no significant effect on the Hill coefficient (Table 1). Similarly, treatment with BAD SAHBA (S118D) did not change the Hill coefficient but increased the Vmax by 44%, an increase comparable to that induced by RO0281675, (Fig. 1a and Table 1). However, treatment with BAD SAHBA (S118D) resulted in only a minimal change in the S0.5 for glucose (6.54 ± 0.15 from 7.49 ± 0.18 mM) despite the substantial increase in Vmax (Table 1). This is markedly different from the observed increase in GK affinity for glucose upon treatment with RO0281675 (Table 1).
Table 1.
Modulation of glucokinase kinetic parameters by BAD BH3 stapled peptides
| Treatment | S0.5 (mM) | Vmax (% veh.) | n H | S0.5 Veh/S0.5 Treat | |
|---|---|---|---|---|---|
| Vehicle | 7.49 ± 0.18 | 100 | 1.61 ± 0.03 | 1.00 ± 0.05 | n = 18 |
| GKA (RO0281675) | 1.62 ± 0.13*** | 138 ± 6.15*** | 1.71 ± 0.06 | 4.62 ± 0.47*** | n = 6 |
| BAD SAHBA (S118D) | 6.54 ± 0.15** | 144 ± 4.64*** | 1.71 ± 0.03 | 1.15 ± 0.05 | n = 18 |
| BAD SAHBA (S118pS) | 6.00 ± 0.68** | 129 ± 6.54** | 1.69 ± 0.18 | 1.25 ± 0.17 | n = 3 |
| BAD SAHBA (S118L) | 8.14 ± 0.41 | 89.6 ± 5.01 | 1.62 ± 0.05 | 0.92 ± 0.07 | n = 7 |
| BAD SAHBA (S118D) + GKA | 1.35 ± 0.07*** | 124 ± 1.54* | 1.84 ± 0.07* | 5.55 ± 0.41*** | n = 4 |
Summary of the effects of BAD BH3 stapled peptides on GK activity. Kinetic parameters were derived using the Hill equation as described in Methods. S0.5 Veh/S0.5 Treat in the fourth column calculates the fold change in glucose affinity resulting from each treatment. Data represent means ± s.e.m.
P < 0.05,
P < 0.01,
P < 0.001, compared with vehicle control (veh.); one-way ANOVA.
Figure 1.
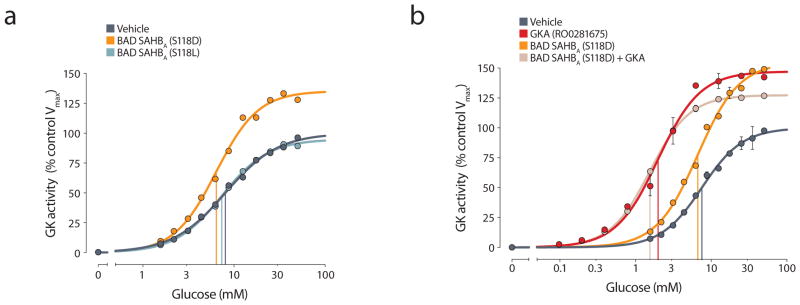
Effects of BAD BH3 stapled peptides on glucokinase (GK) activity. (a) GK activity in the presence of BAD SAHBA (S118D), BAD SAHBA (S118L), or vehicle alone. (b) Activation of GK by BAD SAHBA (S118D) in the presence or absence of the allosteric GK activator (GKA) RO0281675. Vertical lines in (a) and (b) indicate the glucose concentration corresponding to the S0.5 values obtained from the representative experiment shown. Data in (b) show the means and range from a representative experiment, see Table 1 for the summary of all independent experiments performed similarly (n = 3–18 per treatment).
Mutation of Ser118 to leucine [BAD SAHBA (S118L)] blunted the capacity of the BAD BH3 helix to activate GK (Fig. 1a and Table 1), consistent with the notion that phosphorylation of serine at this site is important for GK activation by BAD22. To further examine the effect of Ser118 phosphorylation, we also tested the kinetic profile of GK treated with a phospho-BAD BH3 stapled peptide in which the residue corresponding to Ser118 was replaced with a phosphoserine [BAD SAHBA (S118pS)], and found that it activates GK in a similar manner to BAD SAHBA (S118D) with minimal effect on glucose affinity or substrate cooperativity (Table 1). To our knowledge, this is the first chemical mode of GK activation that has little effect on the S0.5 of the enzyme for glucose. This predicts a mechanism of GK activation that is markedly different from all allosteric GKAs reported to date9,10,12,14,15,17,27–32.
Given the distinct modes of GK activation by RO0281675 and BAD SAHBA (S118D), we next tested their combined effects. Co-treatment with RO0281675 and BAD SAHBA (S118D) produced changes in glucose affinity that were similar to RO0281675 treatment alone, indicating that BAD SAHBA (S118D) does not interfere with RO0281675 binding to the allosteric site (Fig. 1b and Table 1).
Localization of the phospho-BAD BH3 interaction site on GK
The differences between the modes of GK activation by BAD SAHBs and the allosteric activator RO0281675 predicted distinct binding sites for the two compounds. To identify the interaction site for the phospho-BAD BH3 domain on GK, we generated biotinylated and photoreactive BAD SAHB analogs, further derivatized by phosphorylation at Ser118. This allowed for capture of the covalent BAD SAHBA–GK complex and binding site analysis by MS/MS sequencing26,33. We placed the photoreactive benzoylphenylalanine (Bpa) moiety near either the N-terminus [BAD SAHBA (Y110Bpa S118pS)] or C-terminus [BAD SAHBA (S118pS D123R F125Bpa)] of the BAD BH3 domain (Supplementary Table 1). We further installed a C-terminal tryptic site in BAD SAHBA (S118pS D123R F125Bpa) by D123R mutagenesis in order to ensure that the resultant Bpa-containing BAD peptide fragment would be sufficiently small for optimal MS/MS sequencing, as previously reported33. Importantly, these biotinylated and photoreactive stapled peptides retained their functional interaction with GK as demonstrated by their capacity to increase the Vmax while minimally affecting the S0.5 for glucose (Supplementary Table 2).
Exposure of the BAD SAHBA–GK (2:1) mixtures to UV light led to a crosslinked complex as apparent from biotin western analysis (Fig. 2a and Supplementary Fig. 1). Importantly, the presence or absence of glucose did not affect the crosslinking of BAD SAHBA–GK complexes (Fig. 2a and Supplementary Fig. 1). These data predict that the binding site for phospho-BAD BH3 helix on GK is distinct from the allosteric site, which becomes available only in the presence of glucose5,10,12,32,34.
Figure 2.

Phospho-BAD BH3 peptides crosslink near the active site of GK. (a) Phospho-BAD BH3 peptides crosslink to GK after UV exposure in the presence or absence of glucose. See supplementary Fig. 1 for uncropped image of the gel. (b,c) Normalized spectral count frequency of BAD BH3 helix-crosslinked sites along the GK protein sequence in the presence of BAD SAHBA (Y110Bpa S118pS) (b) or BAD SAHBA (S118pS D123R F125Bpa). (c) Labeled residues are unique to each peptide and were found to occur ≥ 25% of the time. Protein sequences for the corresponding BAD SAHB peptide are included above each plot. Relevant substitutions to the wild-type sequence are denoted. (d,e) Mapping of BAD SAHBA (Y110Bpa S118pS) (d) or (S118pS D123R F125Bpa) (e) crosslinked residues onto surface view representations of the inactive super open, active intermediate, and active closed conformations of GK (PDB entries 1V4T10, 4DCH12 and 1V4S10, respectively) locates the phospho-BAD BH3 binding interface to the enzyme’s active site. To display an unobstructed view of the enzyme active site in the closed structure, the Active Site Loop (ASL) comprised of residues L164–N180 is presented in cartoon form rather than in surface view to show residues located behind the loop. Glucose is shown in green, allosteric GKAs in red, the ASL in white.
We next determined the explicit sites of crosslinking using mass spectrometry. We affinity purified BAD SAHBA–GK covalent adducts by streptavidin pull down and visualized them as coomassie-stained bands on SDS-PAGE migrating slightly higher than input protein. After excision and in-gel trypsin digestion, we analyzed the crosslinked adducts by nano-LC-MS/MS26,33. We identified Bpa-crosslinked tryptic fragments by searching the MS data for a variable modification corresponding to the mass of the tryptic GK fragment plus the Bpa-containing BAD SAHBA fragment. We identified the crosslinked amino acids within GK by MS/MS sequencing of the crosslinked peptides, and used these MS/MS spectral count frequencies as a measure of the abundance of these modifications (Fig. 2b,c and Supplementary Table 3). Whereas the phospho-BAD peptide with the Bpa moiety closer to the N-terminus [BAD SAHBA (Y110Bpa S118pS)] predominantly crosslinked to GK in the vicinity of M115 (Fig. 2b), the photoreactive SAHB with Bpa installed near the C-terminus [BAD SAHBA (S118pS D123R F125Bpa)] uniquely modified the region of GK containing residues I181 (V181 in human GK), T206, and R333 (Fig. 2c). In addition, both peptides crosslinked to residues centered around L79 and F150, which lie between the unique interaction sites for each photoreactive BAD SAHBA. Strikingly, mapping of the crosslinked residues onto each of the three previously defined conformational states of GK (PDB entries 1V4T10, 4DCH12 and 1V4S10) demonstrates engagement of the phospho-BAD BH3 domain near the active site of the enzyme (Fig. 2d,e). The interaction surface identified by phospho-BAD peptides is distinct from that of any published GKA. Consistent with this conclusion, we observed little effect on the crosslinking pattern upon co-incubating RO0281675 and the photoreactive BAD SAHBA compounds (Supplementary Fig. 2). These data are also consistent with the absence of glucose dependence in crosslinking (Fig. 2a), as well as the distinct GK kinetic profiles of RO0281675 as compared to BAD SAHBs (Table 1).
In the absence of glucose, GK adopts an inactive super open conformation (PDB entry 1V4T10), while in the presence of glucose, two distinct active structures have been reported: an active closed10 and a glucose-bound intermediate structure12 (PDB entries 1V4S10 and 4DCH12, respectively). Based on the crosslinking of photoreactive BAD SAHBA compounds to a host of surface residues that would otherwise be buried in the active closed conformation, the mapped binding site data are more consistent with phospho-BAD BH3 engagement of a structure that resembles the published inactive super open or the active intermediate conformations of GK 10,12 (Fig. 2d,e). Furthermore, mapping of the most frequently crosslinked residues for each BAD SAHBA (≥25% threshold) onto each of the three GK structures suggests a discrete orientation for phospho-BAD BH3 at the binding interface (Fig. 3). In the context of the inactive super open and the active intermediate conformations, phospho-BAD BH3 appears to almost exclusively engage the small domain, with N-terminus oriented toward M115 and the C-terminus toward V181, T206 and R333 (Fig. 3a,b). In contrast, when mapped to the active closed conformer, the N-terminus continues to localize to the M115 region of the small domain, but the C-terminus would only be capable of engaging the exposed R333 residue of the large domain (Fig. 3c). Taken together, these data indicate that the phospho-BAD BH3 helix directly engages GK at a novel interaction site that is distinct from that of small molecule allosteric GK activators, consistent with the unique modulatory effect of BAD observed on GK enzyme kinetics (Table 1).
Figure 3.

Mapping the phospho-BAD BH3 helix at the GK interaction site. (a–c) Phospho-BAD BH3 peptides bind to residues in the small domain of the inactive super open and the active intermediate, with the N-terminus localized around residue M115 and the C-terminus oriented toward residues V181, T206 and R333. The most frequently crosslinked residues for each BAD SAHB (≥25% threshold) are mapped onto the inactive super open (a), active intermediate (b), and active closed (c) conformers of GK. Residues selectively crosslinked by BAD SAHBA (Y110Bpa S118pS) and (S118pS D123R F125Bpa) are colored orange and blue, respectively, whereas residues crosslinked by both photoreactive BAD SAHBs are colored tan. For clarity, all of the associated residues are colored, but not all are labeled. Crosslinked residues common to both peptides are among those listed in Supplementary Table 3. Glucose is depicted in green, the Active Site Loop in white.
Effect of the phospho-BAD BH3 helix on human donor islets
We previously have shown that phospho-BAD BH3 stapled peptides can restore the insulin secretory defect of BAD-deficient murine islets treated with stimulatory glucose concentrations22. However, the capacity of these compounds to further enhance insulin release in the presence of endogenous wild-type BAD has not previously been tested. Importantly, we sought to expand our efficacy testing of phospho-BAD SAHBs to human islets. We treated human islets overnight with 3 μM BAD SAHBA (S118D) or SAHBA (S118L) and assessed insulin release after incubation with basal (1.67 mM) or stimulatory (20 mM) glucose concentration. At 20 mM glucose, islets pre-treated with BAD SAHBA (S118D) showed a marked increase in insulin release, while islets pre-treated with BAD SAHBA (S118L) secreted comparable levels of insulin to vehicle-treated control islets (Fig. 4a). The distinct capacities of BAD SAHBA (S118D) and SAHBA (S118L) to stimulate insulin secretion paralleled their effects on GK activation (Table 1). Importantly, pre-treatment with BAD SAHBA did not augment insulin secretion at 1.67 mM glucose (Fig. 4a), a concentration that is insufficient to trigger GK activation. Collectively, these data indicate that direct modulation of GK activity by BAD SAHBA (S118D) augments glucose-stimulated insulin secretion, a specific functional outcome in human islets.
Figure 4.

Effects of BAD BH3 stapled peptides on human islet function and human GK mutations. (a) Stimulation of insulin release by glucose in donor islets treated with BAD BH3 stapled peptides. Error bars represent means ± s.e.m. * P < 0.05, *** P < 0.001, n.s., non-significant; two-way ANOVA. (b,c) Effects of BAD SAHBA (S118D) on the activity of GK active site mutations. Kinetic analyses of two human GK mutants, M298K (b) and E300K (c), in the absence or presence of BAD SAHBA (S118D). Wild-type human GK treated with vehicle was included in each assay as a control. Vertical lines indicate the glucose concentration corresponding to the S0.5 obtained from the representative experiment shown. The data in (b) and (c) show means and range from representative experiments, see Table 2 for the summary of all independent experiments performed similarly (four experiments per GK mutant).
Kinetics of active site GK mutants treated with phospho-BAD mimetic
The location of the BAD SAHBA binding interface near the active site of GK prompted us to examine whether BAD SAHBs could alter the function of active site GK mutants, perhaps stabilizing otherwise impaired structures, or whether these mutations might prevent BAD SAHBA binding altogether. GK activators can be expected to promote the protein stability of GK mutants in addition to increasing their activity34. We selected two naturally-occurring MODY2 mutations located near the active site, M298K and E300K, for analysis35–39. Both mutations render the enzyme highly unstable8,39,40. Previous reports on the M298K mutation have noted substantial deficiencies in multiple enzymatic parameters34,36,39,41. In agreement with these reports, we observed impaired kinetic constants for this mutant, including decreased Vmax, diminished glucose affinity, and a decreased Hill coefficient (Fig. 4b and Table 2). Upon treatment with BAD SAHBA (S118D), these three kinetic parameters showed marked improvement, including restoration of Vmax from 57% to 95% of the wild-type enzyme (Fig. 4b and Table 2). A similar series of studies using the M298K mutant in the presence of an allosteric GKA treatment reported Vmax increase from approximately 60% to 79% of wild-type34. However, the restoration of Vmax in the presence of BAD SAHBA (S118D) occurred with modest effects on S0.5 of this mutant, which changed from approximately 11 mM to near normal values of 8 mM (Table 2), considerably different from drastic lowering of S0.5 to approximately 1 mM when M298K is treated with an allosteric GKA34. These findings reinforce the mechanistic distinction between allosteric GKAs and SAHBA (S118D) (Table 1).
Table 2.
Enhancement of GK activity by phospho-mimic BAD BH3 in select MODY2 mutations
| Treatment | S0.5 (mM) | Vmax (% WT Vmax) | n H | |
|---|---|---|---|---|
| GK (Wild-type) + Vehicle | 5.60 ± 0.06 | 100 | 1.73 ± 0.03 | n = 8 |
| GK (M298K) + Vehicle | 11.3 ± 0.90*** | 57.1 ± 4.02*** | 1.24 ± 0.05*** | n = 4 |
| GK (M298K) + BAD SAHBA (S118D) | 8.08 ± 0.57††† | 95.7 ± 9.47††† | 1.43 ± 0.04† | n = 4 |
| GK (E300K) + Vehicle | 8.39 ± 0.12*** | 84.3 ± 1.89* | 1.83 ± 0.03 | n = 4 |
| GK (E300K) + BAD SAHBA (S118D) | 8.02 ± 0.38 | 103 ± 8.35† | 2.11 ± 0.08†† | n = 4 |
Summary of mutant GK activity in the absence or presence of BAD SAHBA (S118D). Kinetic parameters were derived using the Hill equation. Data represent means ± s.e.m.
P < 0.05,
P < 0.001 compared with vehicle treated recombinant human wild-type (WT) GK;
P < 0.05,
P < 0.01,
P < 0.001 compared with vehicle treated mutant GK; one-way ANOVA.
The active site mutant E300K has previously been reported to display variable kinetics. Some studies indicate near normal kinetics36,37, while others have documented more moderate and even severe8,38 changes in the kinetic profile. We observed near normal nH and S0.5 values but a noticeable reduction in Vmax (84.3% ± 1.89 vs. wild-type GK), comparable to previous reports by Davis et al.8 and Kesavan et al.37. Moreover, this mutant had low expression levels as previously reported8,35,37,40. Addition of BAD SAHBA (S118D) fully restored the Vmax of this mutant to wild-type levels (Fig. 4c and Table 2). These data suggest that binding of BAD SAHBA (S118D) in the vicinity of the active site stabilizes the M298K and E300K mutant forms of GK, markedly improving their enzymatic function.
DISCUSSION
Our biochemical and structural dissection of the interaction between GK and the phospho-BAD BH3 helix demonstrates a novel binding region and distinct mode of enzyme activation compared to allosteric GK activators. Binding of the BAD BH3 helix near the active site of GK augments Vmax while preserving the native nH and S0.5 values. The relevance of this interaction mode is further underscored by the ability of the BAD BH3 helix to restore the function of two independent MODY2 mutations residing near the active site of GK. Direct engagement and activation of GK by the BAD BH3 helix also imparts a functional benefit to human donor islets, warranting exploration of BAD BH3 mimetic compounds as a novel class of GK activators.
The positive cooperativity of monomeric GK for glucose is central to its role as a glucose sensor. This has been explained at the mechanistic level by the ligand-induced slow transition model42 or the mnemonic model10, which has been further refined as the pre-existing equilibrium model12,13,16,32, all involving distinct conformations of GK with different affinities for glucose that interconvert slowly. Structural information for several GK conformers corroborates the mechanistic properties of the enzyme and the glucose dose-dependent conversion between conformations7–11,13–16. GK exists in a super open conformation in low glucose concentrations and transitions to an active closed conformation in the presence of glucose and allosteric activators10. A glucose-bound intermediate conformation between the inactive super open and the active closed conformers of GK was recently described12. It has been suggested that the degree to which allosteric activators enlarge the allosteric site may determine the extent to which the active site is closed and the affinity for glucose is increased12. The transition from this active intermediate to the active closed form is thought to allow higher affinity glucose binding at the active site, enabling catalytic activity12. Given the inherent mobility of GK, it is likely that the spectrum of GK conformations in the presence or absence of glucose and other ligands will continue to expand11,12 and provide new information on the mechanism of enzyme action.
The BAD BH3 domain is the minimal region required for BAD’s capacity to activate GK and stimulate glucose metabolism22. Our integrative dissection of the mode of engagement and activation of GK by stapled peptides modeled after the BAD BH3 domain indicates that they directly bind GK in the immediate vicinity of the active site and activate the enzyme through a mechanism that is markedly distinct from all known allosteric activators, as evidenced by minimal alterations in the enzyme’s affinity for glucose. To our knowledge, BAD BH3 mimetic compounds constitute the first example of synthetic glucokinase activators that exhibit a non-allosteric mode of action.
Crosslinking patterns of the phospho-BAD BH3 helix on GK are largely unaffected by the presence or absence of either glucose or an allosteric GK activator, both of which dramatically shift GK conformational equilibrium away form the inactive super open conformation. MS-based mapping of the amino acids most frequently crosslinked by photoreactive BAD SAHBs onto published GK structures suggests that the conformation of BAD SAHB-bound GK most likely resembles a structure similar to the inactive super open or the active intermediate conformers in which the N-terminus of the BAD BH3 domain solely engages the apex of the small domain of GK and the C-terminus binds near the interface between the small and large domains proximal to the glucose binding site (Fig. 3). Several reports have proposed multiple intermediate GK conformations based on tryptophan fluorescence7,13,34,43,44, X-ray scattering12, and NMR spectroscopy45. Such conformational dynamics were decreased, but not eliminated, in the presence of high glucose concentrations45. Thus, the explicit conformational structure of GK upon BAD BH3 domain engagement may indeed be distinct from the crystal structures published to date. While the spectrum of GK conformers limits mechanistic interpretation of the activating nature of the BAD BH3 helix bound to GK, our data firmly place the BAD BH3 domain near the active site of the enzyme. As a result of this direct interaction, the BAD BH3 domain may stabilize and enable conformational transitions of GK facilitating catalysis or enhancing release of products. This scenario would be consistent with the functional restoration of the M298K and E300K active site mutants in the presence of BAD SAHBA (S118D).
GK activating mutations predominantly cluster in the allosteric site and lower the S0.5 of the enzyme5,39,46,47. The residues identified in our crosslinking studies tend to be closer to a region between the small and large domains of GK than any of the residues associated with GK activating mutations reported to date, which largely exist away from the glucose binding site and the proposed BAD interface. Among residues altered in GK activating mutations, V452 is closest to any of those proposed within the interface between GK and the BAD BH3 helix in the inactive open conformation and active intermediate conformation. However, the kinetic profile of the corresponding activating mutation, V452L, differs greatly from the kinetic profiles we have observed in the presence of BAD SAHBs34. In light of these considerations and the results of our binding site localization studies, it appears that the interface between GK and the BAD BH3 helix is independent from the known GK activating mutations.
To date, all published small molecule GK activators significantly increase the affinity of the enzyme for glucose9,10,14,15,17,19,27,30. This is similar to kinetic alterations reported for GK activating mutations in PHHI5,39,46,47. Beyond a shared capacity to augment the affinity of GK for glucose, GK activators can have diverse effects on substrate cooperativity and Vmax of the enzyme5. Several studies examining the short term effects of allosteric GKAs (less than one week) in human subjects with early T2D have reported promising outcomes, including improved beta-cell function, decreased hepatic glucose output, and a net lowering of glucose levels in glucose tolerance tests that are consistent with known physiologic roles of GK in the beta-cell and in liver17,18. However, a concern with allosteric GKAs is their ability to decrease the glucose threshold for GK activation, resulting in marked glucose lowering capacity and the risk of hypoglycemia, as well as other undesired effects such as increased triglyceride levels19. This latter effect is expected upon chronic activation of hepatic GK and the associated stimulation of lipogenesis and increased hepatic fat content48–50. Lower doses of GKAs or combination therapies with other anti-diabetic agents may circumvent these undesirable outcomes5. Furthermore, studies that tested the long term effects of certain allosteric GKAs (up to four months) in T2D subjects reported initial improvement of glucose homeostasis with loss of efficacy over time that is not fully understood19,51,52. These complications are also relevant if pharmacologic activation of GK is to be used as a potential therapeutic approach for expansion of functional beta-cell mass in T1D6.
The successes and challenges of allosteric GK activators in T2D therapy have spurred active investigation of other chemical and enzymatic modes of GK activation, including development of different classes of GKAs that preserve the native enzymatic properties of the enzyme, especially its affinity for glucose5, or “partial agonists” that more moderately lower the S0.5 for glucose12. To our knowledge, BAD SAHBs are the first example of a class of synthetic GK activators that raise the Vmax with minimal effect on the affinity of the enzyme for glucose. This is mediated by engaging the enzyme near its active site, revealing a novel druggable site on GK. By preserving the natural kinetic properties of GK, compounds modeled after the phospho-BAD BH3 helix may have important therapeutic benefits, in addition to serving as powerful tool compounds for further biophysical, structural, and enzymatic dissection of GK and its pathologic mutants.
METHODS
Recombinant GK expression and purification
Mouse GK isoform 1 was cloned in the pET-43.1 Ek/LIC vector (Novagen) containing an N-terminal hexahistidine tag that was further engineered to express a TEV cleavage site (ENLYFQS) preceding the GK sequence. The resultant recombinant GK was purified as follows and used in experiments documented in Fig. 1, 2, and 3, and Table 1, and Supplementary Tables 2 and 3. Briefly, Escherichia coli BL21(DE3) pLysS transformed with the construct were grown at 37 °C to an optical density of 0.5–0.6 at 595 nm, and the expression of GK fusion protein was induced with 0.6 mM IPTG for 6 h at 30 °C. The cells were harvested, resuspended in 100 mM sodium phosphate, pH 7.4, 500 mM NaCl, 5 mM BME, 1 mM PMSF and EDTA-free complete protease inhibitor cocktail (Roche), and sonicated at 4 °C (Misonix, Inc. S-4000). The lysates were passed over a Ni-NTA agarose matrix (Qiagen) followed by ion-exchange chromatography with a Mono Q 5/50 GL (GE Life Sciences) using a NaCl gradient and gel filtration using a Superdex 75 10/300 GL column (GE Life Sciences) in 50 mM Tris, 150 mM, 5 mM DTT, pH 7.4. Aliquots of purified GK were stored at −80 °C with 20% glycerol (v/v).
N-terminal GST-tagged constructs (pGEX3) expressing the recombinant human wild-type GK isoform 1 and the M298K and E300K GK active site mutations used in Fig. 4 and Table 2 have been previously described37,38. Briefly, BL21(DE3) transformed with the constructs were induced with IPTG overnight at 25 °C, lysed in buffer containing 4 mM KH2PO4, 16 mM K2HPO4, 150 mM KCl and 5 mM DTT as previously described38. Tagged GST-hGK was captured using glutathione agarose beads (Sigma), cleaved on-column by incubation with Factor Xa (GE Life Sciences), and subjected to gel filtration.
SAHB synthesis
Peptide synthesis, olefin metathesis, amino terminal derivatization (e.g. biotin-βAla), reverse phase HPLC purification, and amino acid analysis were performed as previously described22,33.
Steady-state kinetics
Glucokinase activity was measured by monitoring the rate of NADH formation using a G6PDH-coupled reaction as previously described38, with minor modifications. Briefly, assays were performed at 37 °C in 100 μL total volume per well of a Costar 3596 plate using a SpectraMax M5 microplate reader (Molecular Devices). Absorbance at 340 nm was recorded every two min and the change in absorbance per min was calculated using data between 30 and 60 min, where the rate of reaction was linear. Reaction wells were prepared on ice, and contained final concentrations of 7.5 nM GK, 100 mM HEPES pH 7.4, 150 mM KCl, 6 mM MgCl2, 1 mM DTT, 1 mM NAD, 0.05% BSA, 2.5 units G6PDH and 5% DMSO in the presence or absence of 5 μM BAD SAHBs (a concentration deemed near-saturating based on dose-response assays) and/or 3 μM of RO0281675 (http://www.axonmedchem.com). To determine glucose-dependent kinetic parameters, glucose was varied (0–50 mM) while maintaining ATP at 5 mM. Kinetic parameters were derived from the Hill equation using GraphPad Prism 6.0a.
Crosslinking of photoreactive BAD SAHBs and glucokinase
Photoreactive BAD SAHBs were applied in protein target crosslinking as previously reported33. Briefly, SAHBs (10 μM) containing benzophenone moieties were mixed with recombinant mouse glucokinase (5 μM) in buffer A (50 mM Tris, 5mM DTT, 200 mM NaCl, pH 7.5) with or without the addition of 50 mM glucose as indicated, incubated for 15 min, and then irradiated with UV light (365 nm) for 2 h on ice. Unreacted SAHBs were removed by dialysis overnight at 4 °C in buffer B (50mM Tris, 200mM NaCl, pH 7.5) using 6–8 kDa MWCO D-tube dialyzers (EMD Biosciences). Crosslinking in the presence of RO0281675 was performed with 50 mM glucose and 3 μM RO0281675. Biotin affinity capture, elution, and gel electrophoresis were performed as described33.
Western analysis of photoaffinity captured proteins
Following electrophoresis and transfer to PDVF, membranes were blocked in PBS containing 5% milk for 30 min at room temperature. The membranes were rinsed with PBS containing 3% bovine serum albumin (BSA), and subsequently probed with purified goat anti-biotin antibody conjugated to horseradish peroxidase (Cell Signaling Technology, #7075) at a dilution of 1:1000 in PBS containing 3% BSA overnight as previously described33. After washing 3 times for 15 min with PBS containing 0.1% Tween-20, bands were detected by chemiluminescence (PerkinElmer).
Mass spectrometry analysis
Samples were subjected to in-gel digestion and mass spectrometry as described33. Searches were done using the sequest algorithm53, with a sequence-reversed search database comprised of GK, trypsin, and common keratin contaminants. Search results were limited to those peptides containing a BAD SAHB crosslinked adduct. False positive identifications (considered to be any crosslinked peptides derived from trypsin or keratins, from the reversed database, or peptides with masses corresponding to more than one crosslinked adduct) were limited to less than 5% via linear discriminant analysis of multiple variables including mass accuracy, XCorr, tryptic state, and charge state. MS/MS spectral counting was performed as described33. Spectral counts for crosslinking to each residue were normalized in each experiment such that the most frequently detected residue was assigned a value of 100%, while 0% indicated no crosslinking. The normalized values for each experiment were then combined and averaged to generate frequency distribution plots. For Fig. 3, a value of 25% for this normalized average frequency of spectral counts was chosen for depicting the differential crosslinking pattern of N- and C- terminal photoreactive BAD SAHBs. This 25% cutoff was chosen in accordance with observations from both published26,33 and unreported experiments that when crystal structures of an interaction are known, higher relative crosslinking frequency generally correlates with better agreement with these structures.
Human islets culture, SAHB treatment and insulin release assays
Human donor islets were obtained through the Integrated Islet Distributing Program (http://iidp.coh.org/). Islets were cultured according established protocols54, at 37 °C (5% CO2) in CMRL 1066 media (Gibco) containing 5.5 mM glucose and supplemented with human albumin serum (10%, Gemini)55, L-Glutamine (2 mM, Sigma) and HEPES (1 mM, Sigma), and 0.1% insulin-transferrin-selenium, pH 7.4. Islets were cultured at least an additional 24 h prior to treatment with 3 μM BAD SAHBs or vehicle (DMSO 0.3%) in CMRL 1066 medium containing 5.5 mM glucose, supplemented with 10% human serum (pH 6.2), overnight at 37 °C. Islets were then incubated for 90 min in basal conditions (1.67 mM glucose), hand picked and incubated in either basal (1.67 mM glucose) or stimulatory glucose condition (20 mM glucose) for 45 min54, and insulin secretion was measured as previously described56.
Statistical analysis
Statistical significance was calculated in GraphPad Prism 6.0a (www.graphpad.com) using one-way ANOVA followed by an appropriate post-test. When comparing the effects of BAD SAHB compounds on wild-type GK activity (Table 1 and Supplementary Table 2), comparisons were made using one-way ANOVA followed by Dunnett’s test using the vehicle data as the control in each case. When comparing mutant GK activity in the absence or presence of BAD SAHBA (S118D) (Table 2), analyses used one-way ANOVA followed by a Holm-Šídák test involving four comparisons in total. When comparing the effects of BAD SAHB compounds on insulin secretion in human islets, two-way ANOVA was used followed by a Tukey multiple comparisons test. For all analyses, thresholds for significance used multiplicity adjusted P values to account for multiple comparisons.
Supplementary Material
Acknowledgments
We thank K. Robertson and P. Chen for technical assistance, F. Bernal, S. Devarakonda, and C. Buettger for advice on protein purification, E. Gavathiotis, A. West, and R. McNally for advice on structural studies, M. Eck, N. Gray, S. Blacklow, G. Yellen, and members of the Danial and Walensky laboratories for valuable discussions. This work was supported by the U.S. National Institutes of Health grants (R01DK078081 to N.N.D. and R01GM090299 to L.D.W.), Burroughs Wellcome Fund Career Award in Biomedical Sciences (N.N.D.), the Juvenile Diabetes Research Foundation Grant 17-2011-595 (N.N.D.), a Claudia Adams Barr Award in Innovative Basic Cancer Research (N.N.D.), a Stand Up to Cancer Innovative Research Grant (L.D.W.), a National Sciences and Engineering Research Council of Canada postgraduate scholarship (C.R.B.), a Swiss National Science Foundation postdoctoral fellowship (S.L.), and a Juvenile Diabetes Research Foundation postdoctoral fellowship (M.A.O.).
Footnotes
Author contributions
B.S, E.P., M.A.O., and N.N.D. purified recombinant proteins and performed enzyme kinetic analyses. C.R.B., G.H.B., and L.D.W. designed, synthesized, and characterized SAHB compounds. C.R.B. and L.D.W. performed crosslinking, mass spectrometry, and structural analyses. S.L. and N.N.D. performed analyses in human donor islets. B.S., C.R.B., L.D.W., and N.N.D. wrote the manuscript. F.M.M. provided critical advice and reviewed the manuscript.
Competing financial interests
L.D.W. is a consultant and scientific advisory board member for Aileron Therapeutics.
References
- 1.Ashcroft FM, Rorsman P. Diabetes mellitus and the beta cell: the last ten years. Cell. 2012;148:1160–1171. doi: 10.1016/j.cell.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell. 2012;148:852–871. doi: 10.1016/j.cell.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Majumdar SK, Inzucchi SE. Investigational anti-hyperglycemic agents: the future of type 2 diabetes therapy? Endocrine. 2013;44:47–58. doi: 10.1007/s12020-013-9884-3. [DOI] [PubMed] [Google Scholar]
- 4.Grimsby J, Berthel SJ, Sarabu R. Glucokinase activators for the potential treatment of type 2 diabetes. Curr Top Med Chem. 2008;8:1524–1532. doi: 10.2174/156802608786413483. [DOI] [PubMed] [Google Scholar]
- 5.Matschinsky FM. Assessing the potential of glucokinase activators in diabetes therapy. Nat Rev Drug Discov. 2009;8:399–416. doi: 10.1038/nrd2850. [DOI] [PubMed] [Google Scholar]
- 6.Dadon D, et al. Glucose metabolism: key endogenous regulator of beta-cell replication and survival. Diabetes Obes Metab. 2012;14 (Suppl 3):101–108. doi: 10.1111/j.1463-1326.2012.01646.x. [DOI] [PubMed] [Google Scholar]
- 7.Antoine M, Boutin JA, Ferry G. Binding kinetics of glucose and allosteric activators to human glucokinase reveal multiple conformational states. Biochemistry. 2009;48:5466–5482. doi: 10.1021/bi900374c. [DOI] [PubMed] [Google Scholar]
- 8.Davis EA, et al. Mutants of glucokinase cause hypoglycaemia- and hyperglycaemia syndromes and their analysis illuminates fundamental quantitative concepts of glucose homeostasis. Diabetologia. 1999;42:1175–1186. doi: 10.1007/s001250051289. [DOI] [PubMed] [Google Scholar]
- 9.Grimsby J, et al. Allosteric activators of glucokinase: potential role in diabetes therapy. Science. 2003;301:370–373. doi: 10.1126/science.1084073. [DOI] [PubMed] [Google Scholar]
- 10.Kamata K, Mitsuya M, Nishimura T, Eiki J, Nagata Y. Structural basis for allosteric regulation of the monomeric allosteric enzyme human glucokinase. Structure. 2004;12:429–438. doi: 10.1016/j.str.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 11.Larion M, Salinas RK, Bruschweiler-Li L, Bruschweiler R, Miller BG. Direct evidence of conformational heterogeneity in human pancreatic glucokinase from high-resolution nuclear magnetic resonance. Biochemistry. 2010;49:7969–7971. doi: 10.1021/bi101098f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu S, et al. Insights into mechanism of glucokinase activation: observation of multiple distinct protein conformations. J Biol Chem. 2012;287:13598–13610. doi: 10.1074/jbc.M111.274126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petit P, et al. The active conformation of human glucokinase is not altered by allosteric activators. Acta Crystallogr D Biol Crystallogr. 2011;67:929–935. doi: 10.1107/S0907444911036729. [DOI] [PubMed] [Google Scholar]
- 14.Pfefferkorn JA, et al. Discovery of (S)-6-(3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanamido)nicotini c acid as a hepatoselective glucokinase activator clinical candidate for treating type 2 diabetes mellitus. J Med Chem. 2012;55:1318–1333. doi: 10.1021/jm2014887. [DOI] [PubMed] [Google Scholar]
- 15.Futamura M, et al. An allosteric activator of glucokinase impairs the interaction of glucokinase and glucokinase regulatory protein and regulates glucose metabolism. J Biol Chem. 2006;281:37668–37674. doi: 10.1074/jbc.M605186200. [DOI] [PubMed] [Google Scholar]
- 16.Pfefferkorn JA, et al. Pyridones as glucokinase activators: identification of a unique metabolic liability of the 4-sulfonyl-2-pyridone heterocycle. Bioorg Med Chem Lett. 2009;19:3247–3252. doi: 10.1016/j.bmcl.2009.04.107. [DOI] [PubMed] [Google Scholar]
- 17.Bonadonna RC, et al. Piragliatin (RO4389620), a novel glucokinase activator, lowers plasma glucose both in the postabsorptive state and after a glucose challenge in patients with type 2 diabetes mellitus: a mechanistic study. J Clin Endocrinol Metab. 2010;95:5028–5036. doi: 10.1210/jc.2010-1041. [DOI] [PubMed] [Google Scholar]
- 18.Ericsson H, et al. Tolerability, pharmacokinetics, and pharmacodynamics of the glucokinase activator AZD1656, after single ascending doses in healthy subjects during euglycemic clamp. Int J Clin Pharmacol Ther. 2012;50:765–777. doi: 10.5414/CP201747. [DOI] [PubMed] [Google Scholar]
- 19.Meininger GE, et al. Effects of MK-0941, a novel glucokinase activator, on glycemic control in insulin-treated patients with type 2 diabetes. Diabetes Care. 2011;34:2560–2566. doi: 10.2337/dc11-1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Danial NN. BAD: undertaker by night, candyman by day. Oncogene. 2008;27 (Suppl 1):S53–70. doi: 10.1038/onc.2009.44. [DOI] [PubMed] [Google Scholar]
- 21.Danial NN, et al. BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature. 2003;424:952–956. doi: 10.1038/nature01825. [DOI] [PubMed] [Google Scholar]
- 22.Danial NN, et al. Dual role of proapoptotic BAD in insulin secretion and beta cell survival. Nat Med. 2008;14:144–153. doi: 10.1038/nm1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu S, et al. Insulin signaling regulates mitochondrial function in pancreatic beta-cells. PLoS One. 2009;4:e7983. doi: 10.1371/journal.pone.0007983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gavathiotis E, et al. BAX activation is initiated at a novel interaction site. Nature. 2008;455:1076–1081. doi: 10.1038/nature07396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.LaBelle JL, et al. A stapled BIM peptide overcomes apoptotic resistance in hematologic cancers. J Clin Invest. 2012;122:2018–2031. doi: 10.1172/JCI46231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leshchiner ES, Braun CR, Bird GH, Walensky LD. Direct activation of full-length proapoptotic BAK. Proc Natl Acad Sci U S A. 2013;110:E986–995. doi: 10.1073/pnas.1214313110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bebernitz GR, et al. Investigation of functionally liver selective glucokinase activators for the treatment of type 2 diabetes. J Med Chem. 2009;52:6142–6152. doi: 10.1021/jm900839k. [DOI] [PubMed] [Google Scholar]
- 28.Brocklehurst KJ, et al. Stimulation of hepatocyte glucose metabolism by novel small molecule glucokinase activators. Diabetes. 2004;53:535–541. doi: 10.2337/diabetes.53.3.535. [DOI] [PubMed] [Google Scholar]
- 29.Castelhano AL, et al. Glucokinase-activating ureas. Bioorg Med Chem Lett. 2005;15:1501–1504. doi: 10.1016/j.bmcl.2004.12.083. [DOI] [PubMed] [Google Scholar]
- 30.Efanov AM, et al. A novel glucokinase activator modulates pancreatic islet and hepatocyte function. Endocrinology. 2005;146:3696–3701. doi: 10.1210/en.2005-0377. [DOI] [PubMed] [Google Scholar]
- 31.Fyfe MC, et al. Glucokinase activator PSN-GK1 displays enhanced antihyperglycaemic and insulinotropic actions. Diabetologia. 2007;50:1277–1287. doi: 10.1007/s00125-007-0646-8. [DOI] [PubMed] [Google Scholar]
- 32.Ralph EC, Thomson J, Almaden J, Sun S. Glucose modulation of glucokinase activation by small molecules. Biochemistry. 2008;47:5028–5036. doi: 10.1021/bi702516y. [DOI] [PubMed] [Google Scholar]
- 33.Braun CR, et al. Photoreactive stapled BH3 peptides to dissect the BCL-2 family interactome. Chem Biol. 2010;17:1325–1333. doi: 10.1016/j.chembiol.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zelent B, et al. Mutational analysis of allosteric activation and inhibition of glucokinase. Biochem J. 2011;440:203–215. doi: 10.1042/BJ20110440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gidh-Jain M, et al. Glucokinase mutations associated with non-insulin-dependent (type 2) diabetes mellitus have decreased enzymatic activity: implications for structure/function relationships. Proc Natl Acad Sci U S A. 1993;90:1932–1936. doi: 10.1073/pnas.90.5.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gloyn A, et al. Glucokinase and the regulation of blood sugar: a mathematical model predicts the threshold for glucose stimulated insulin release for GCK gene mutations that cause hyper- and hypoglycaemia. In: MM, FM, editors. Glucokinase and glycemic diseases: from the basics to novel therapeutics. Karger; Basel: 2004. pp. 92–109. [Google Scholar]
- 37.Kesavan P, et al. Structural instability of mutant beta-cell glucokinase: implications for the molecular pathogenesis of maturity-onset diabetes of the young (type-2) Biochem J. 1997;322 ( Pt 1):57–63. doi: 10.1042/bj3220057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liang Y, et al. Variable effects of maturity-onset-diabetes-of-youth (MODY)-associated glucokinase mutations on substrate interactions and stability of the enzyme. Biochem J. 1995;309 ( Pt 1):167–173. doi: 10.1042/bj3090167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Osbak KK, et al. Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum Mutat. 2009;30:1512–1526. doi: 10.1002/humu.21110. [DOI] [PubMed] [Google Scholar]
- 40.Cullen KS, Matschinsky FM, Agius L, Arden C. Susceptibility of glucokinase-MODY mutants to inactivation by oxidative stress in pancreatic beta-cells. Diabetes. 2011;60:3175–3185. doi: 10.2337/db11-0423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barrio R, et al. Nine novel mutations in maturity-onset diabetes of the young (MODY) candidate genes in 22 Spanish families. J Clin Endocrinol Metab. 2002;87:2532–2539. doi: 10.1210/jcem.87.6.8530. [DOI] [PubMed] [Google Scholar]
- 42.Larion M, Miller BG. Global fit analysis of glucose binding curves reveals a minimal model for kinetic cooperativity in human glucokinase. Biochemistry. 2010;49:8902–8911. doi: 10.1021/bi1008672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin SX, Neet KE. Demonstration of a slow conformational change in liver glucokinase by fluorescence spectroscopy. J Biol Chem. 1990;265:9670–9675. [PubMed] [Google Scholar]
- 44.Molnes J, Bjorkhaug L, Sovik O, Njolstad PR, Flatmark T. Catalytic activation of human glucokinase by substrate binding: residue contacts involved in the binding of D-glucose to the super-open form and conformational transitions. FEBS J. 2008;275:2467–2481. doi: 10.1111/j.1742-4658.2008.06391.x. [DOI] [PubMed] [Google Scholar]
- 45.Larion M, Miller BG. Homotropic allosteric regulation in monomeric mammalian glucokinase. Arch Biochem Biophys. 2012;519:103–111. doi: 10.1016/j.abb.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barbetti F, et al. Opposite clinical phenotypes of glucokinase disease: Description of a novel activating mutation and contiguous inactivating mutations in human glucokinase (GCK) gene. Mol Endocrinol. 2009;23:1983–1989. doi: 10.1210/me.2009-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kassem S, et al. Large islets, beta-cell proliferation, and a glucokinase mutation. N Engl J Med. 2010;362:1348–1350. doi: 10.1056/NEJMc0909845. [DOI] [PubMed] [Google Scholar]
- 48.Ferre T, Riu E, Franckhauser S, Agudo J, Bosch F. Long-term overexpression of glucokinase in the liver of transgenic mice leads to insulin resistance. Diabetologia. 2003;46:1662–1668. doi: 10.1007/s00125-003-1244-z. [DOI] [PubMed] [Google Scholar]
- 49.O’Doherty RM, Lehman DL, Telemaque-Potts S, Newgard CB. Metabolic impact of glucokinase overexpression in liver: lowering of blood glucose in fed rats is accompanied by hyperlipidemia. Diabetes. 1999;48:2022–2027. doi: 10.2337/diabetes.48.10.2022. [DOI] [PubMed] [Google Scholar]
- 50.Peter A, et al. Hepatic glucokinase expression is associated with lipogenesis and fatty liver in humans. J Clin Endocrinol Metab. 2011;96:E1126–1130. doi: 10.1210/jc.2010-2017. [DOI] [PubMed] [Google Scholar]
- 51.Kiyosue A, Hayashi N, Komori H, Leonsson-Zachrisson M, Johnsson E. Dose-ranging study with the glucokinase activator AZD1656 as monotherapy in Japanese patients with type 2 diabetes mellitus. Diabetes Obes Metab. 2013;15:923–930. doi: 10.1111/dom.12100. [DOI] [PubMed] [Google Scholar]
- 52.Wilding JP, Leonsson-Zachrisson M, Wessman C, Johnsson E. Dose-ranging study with the glucokinase activator AZD1656 in patients with type 2 diabetes mellitus on metformin. Diabetes Obes Metab. 2013;15:750–759. doi: 10.1111/dom.12088. [DOI] [PubMed] [Google Scholar]
- 53.Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 54.Murdoch TB, McGhee-Wilson D, Shapiro AM, Lakey JR. Methods of human islet culture for transplantation. Cell Transplant. 2004;13:605–617. [PubMed] [Google Scholar]
- 55.Barbaro B, et al. Increased albumin concentration reduces apoptosis and improves functionality of human islets. Artif Cells Blood Substit Immobil Biotechnol. 2008;36:74–81. doi: 10.1080/10731190701857819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mahdi T, et al. Secreted frizzled-related protein 4 reduces insulin secretion and is overexpressed in type 2 diabetes. Cell Metab. 2012;16:625–633. doi: 10.1016/j.cmet.2012.10.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.