

Abstract
The synthesis of stable and isolable iridium nanoparticles with an average core size of ∼1.2 ± 0.3 nm was achieved by employing sodium S-dodecylthiosulfate as a ligand precursor during the modified Brust–Schiffrin reaction. Transmission electron microscopy (TEM) of the isolated Ir nanoparticles revealed a high degree of monodispersity. Further characterizations with 1H NMR, FT-IR, UV–vis spectroscopy, thermogravimetric analysis (TGA), and X-ray photoelectron spectroscopy (XPS) confirmed that the synthesized Ir nanoparticles are stabilized by dodecanethiolate ligands produced upon the adsorption/cleavage of S-dodecylthiosulfate on the growing Ir nanoparticle surface. By comparison, synthetic attempts employing dodecanethiol as a stabilizing ligand led to the formation of Ir-thiolate species (Ir(SR)3) as an intermediate and Ir-hydroxide species at the completion of reaction. Mechanistic investigations of these two reactions using S-dodecylthiosulfate and dodecanethiol provided deeper understandings on the novelty of thiosulfate ligands, which allow the successful formation of stable thiolate-capped Ir nanoparticles. Moreover, these Ir nanoparticles were shown to have strong magnetic properties.
Introduction
Research into the synthesis of noble metal nanoparticles is motivated by the opportunity to harness advantages not present in their bulk equivalents, such as high surface atom to volume ratios,1,2 semiconductor properties,3 and, in some cases, magnetic properties.4 However, nanosized Ir particles have received limited attention from the scientific community compared to that of other noble metal counterparts. Synthetic methods involving highly specified equipment,5,6 extreme reaction conditions,7 and/or specialty chemicals8−10 have been the primary culprits of this conundrum.
In our recent studies, we reported the successful synthesis of dodecanethiolate-capped Pd nanoparticles generated from sodium S-dodecylthiosulfate after the cleavage of the sulfite group.11 The overall lower reactivity of this ligand precursor compared to alkanethiol allowed the successful synthesis of Pd nanoparticles without the formation of oxidized Pd(II) species during and after the reaction.11−13 Further studies from our group using the thiosulfate synthetic method led to the discovery of a way to control ligand surface coverage that has a direct correlation with catalytic activity and selectivity of alkanethiolate-capped Pd nanoparticles.14,15
Many studies have previously revealed that the indistinguishable metal–sulfur bonds result from the adsorption of thiosulfate precursors and from the direct adsorption of thiols on metal nanoparticles and flat metal surfaces.16−21 For Ir nanoparticles, however, such stable alkanethiolate-capped nanoparticles were unattainable by employing alkanethiols directly as a ligand and NaBH4 as a reducing agent thus far.22 In this article, we relate a facile synthesis of ∼1.2 ± 0.3 nm Ir nanoparticles by employing the modified Brust–Schiffrin system along with the aforementioned thiosulfate ligand precursor strategy. New insights into the mechanistic disparities between sodium S-alkanethiosulfate and alkanethiol are obtained by investigating the formation chemistry of alkanethiolate-stabilized Ir nanoparticles using these two organic ligand precursors.
Experimental Methods
Materials
The following reagents were obtained from the indicated suppliers and used as received: tetra-n-octylammonium bromide (98%), 1-dodecanethiol (98%), and sodium borohydride (98%) were obtained from ACROS. 1-Bromododecane (97%) was obtained from Sigma-Aldrich. Potassium hexachloroiridate(IV) (39.8% Ir) was obtained from Alfa Aesar. Sodium thiosulfate pentahydrate (Na2S2O3·5H2O) along with solvents such as toluene, acetone, ethanol, and methanol was purchased from Fisher Scientific. Chloroform-d was obtained from Cambridge Isotope Laboratories. Sodium S-dodecylthiosulfate was synthesized using the published procedure.11,18 Water was purified by using a Barnstead Nanopure Diamond ion exchange resins purification unit.
Synthesis of Ir Nanoparticles
Tetra-n-octylammonium bromide (2.0 mmol) was dissolved in 25 mL of toluene. Precisely measured K2IrCl6 (0.40 mmol) dissolved in 25 mL of water was added to the organic solution. The mixture was continuously stirred until the aqueous layer was cleared signifying the completion of the phase transfer. The organic layer was separated and placed in a 250 mL round-bottomed flask. Sodium S-dodecylthiosulfate (0.80 mmol) along with another portion of tetra-n-octylammonium bromide (2.0 mmol) was added to the round-bottomed flask with the aid of 10 mL of aq methanol (40% v/v). The reaction mixture was placed in 60 °C water bath and stirred for 15 min. Subsequently, 8.0 mL of a freshly prepared solution of sodium borohydride (1 M) was rapidly poured into the reaction mixture. Within 4 min, the color of the reaction changed from red to yellow to brown to finally black indicating the formation of nanoparticles. After stirring for 1 h, the nanoparticle mixture was removed from the water bath. Upon cooling to room temperature, the organic layer was washed with several aliquots of nanopure water and isolated using a separatory funnel. The organic solvent was removed by rotary evaporation. Lastly, the nanoparticle crude was extensively washed with ethanol, methanol, and acetone on a course frit funnel (F). Iridium nanoparticles were ultimately recollected by reconstitution with chloroform, in which the solution was vacuum-dried to obtain solid powders.
Characterization
1H NMR spectra were recorded using Bruker AC400 FT-NMR operating at 400 MHz. UV–vis spectra were obtained by using a Shimadzu UV-2450 UV-spectrometer. Infrared spectra were acquired by attenuated total reflectance (ATR) using a PerkinElmer Spectrum 100 FT-IR spectrometer. Transmission electron microscope (TEM) images were obtained with a JEOL 1200 EX II electron microscope operating at 90 keV. Samples were prepared by placing 25 μL of an Ir nanoparticle toluene solution (∼1 mg/mL) on a 200 mesh copper grid with Formvar film. Size distribution analysis of Ir nanoparticle core microscope images was executed with Scion Image Beta Release 2TM. Background subtraction was done by Rolling Ball at a set radius of 25. Measurement options were done by Ellipse Major Axis. Thermogravimetric analysis (TGA) was conducted using TA Instruments SDT Q600 with a flow rate of 100 mL/min of N2 with heating from room temperature to 600 °C. XPS measurements were performed using a Mg Kα source (XR50, Specs GmbH) and a hemispherical electron energy analyzer (PHOIBOS 100, Specs GmbH). Spectra were acquired with 10 eV pass energy and normalized to the O1s binding energy at 528.5 eV.
Results and Discussion
Similar to the studies by Lennox et al.,23 the mechanistic aspects of the nanoparticle synthesis were evaluated by comparing 1H NMR and UV–vis spectra of the reaction precursors, intermediates, and final products formed by the direct application of either 1-dodecanethiol or sodium S-dodecylthiosulfate as a ligand precursor (Scheme 1). Since the size, composition, and physical properties of the nanoparticles synthesized by the modified Brust–Schiffrin reactions are highly dependent upon the specific reaction conditions, additional mechanistic insights into this procedure regarding different types of capping ligands24−28 and atypical metals29−32 may allow further expansion of available nanomaterials toward nanoparticles with exotic characteristics.
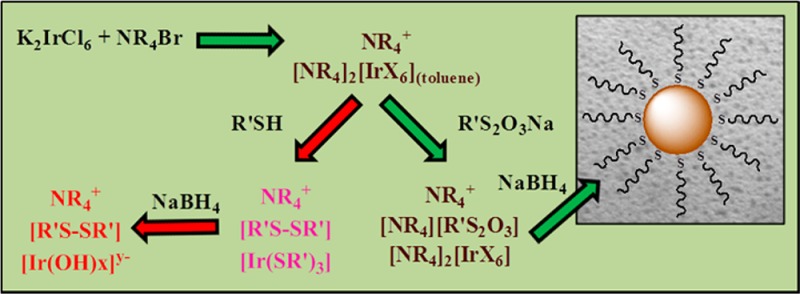
Scheme 1. Proposed Mechanisms for Iridium Nanoparticle Formation during the Modified Brust–Schiffrin Reaction Using Dodecanethiol and S-Dodecylthiosulfate.
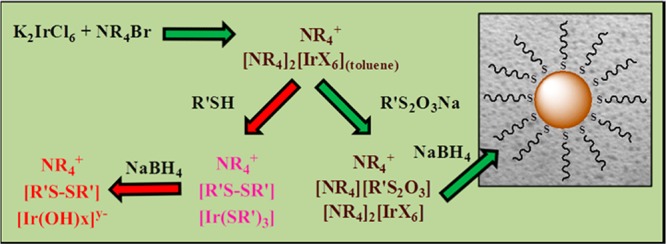
At the onset of this synthesis, aqueous IrCl62– was transferred to toluene-d8 by employing a large excess of tetra-n-octylammonium bromide as a phase transfer agent. This transfer took place just as observed for gold nanoparticles accompanying the colorimetric change of both aqueous and organic phases (Figure 1: from a to b).23 The [R4N+]2[IrX62–] complex in organic phase, however, failed to form a homogeneous solution at room temperature and could only be fully mixed after continuous stirring at a higher temperature of 60 °C. UV–vis spectra of IrX62– in the organic phase showed two strong absorption bands at 440 and 500 nm, which correspond to the presence of Ir4+-X (either Cl or Br) bonds, with a weak absorption band at 595 nm (Figure S1, Supporting Information).33 These absorption bands for Ir4+-X appeared at a slightly higher wavelength, compared to that of the same bands observed for IrCl62– in water before the phase transfer, but were without any significant change in their intensity. The 1H NMR spectra of [R4N+]2[IrX62–] in toluene-d8 revealed the αCH2-N peaks at δ 2.65 ppm, which have been dramatically shifted to more upfield from their original chemical shift of δ 3.40 ppm (Figure 2a). Tong et al. have reported similar dramatic shifts of αCH2-N peaks of the tetra-n-octylammonium complex during the Brust–Schiffrin reaction using HAuCl4 and proposed that this phenomena was due to the formation of inverse micelle structures with AuCl4– complexation in their pore.34 Later, however, Kumar et al. have shown that the same shifts resulted from the ion-pair complex formation without the presence of an inverse micelle of tetraoctylammonium.35,36 On the basis of the latter hypothesis, our NMR data support the formation of an ion-pair complex of tetra-n-octylammonium with IrX62–. The interaction between the positively charged quaternary ammonium nitrogen and IrX62– was the primary reason for the upfield shifts of both αCH2-N peaks and βCH2-CH2-N peaks of tetra-n-octylammonium salts.
Figure 1.

Picture of solutions containing (a) K2IrCl6(aq) (0.4 mmol), (b) [R4N+]2[IrCl62–](toluene), (c) 1-dodecanthiol (0.8 mmol) added to b, (d) NaBH4 (4.0 mmol) added to c, and (e) S-dodecylthiosulfate (0.8 mmol) and NaBH4 (4.0 mmol) added to b.
Figure 2.

1H NMR spectra of (a) [R4N+]2[IrX62–] in toluene-d8 obtained after mixing the organic (toluene-d8) layer of TOAB (2.0 mmol) and the aqueous (D2O) layer of K2IrCl6 (0.4 mmol), (b) 1-dodecanthiol (0.8 mmol) added to a, (c) 1-dodecanthiol (1.6 mmol) added to a, (d) NaBH4 (4.0 mmol) added to c, (e) S-dodecylthiosulfate (0.8 mmol) added to a, and (f) Ir nanoparticle in CDCl3 after purification. The multiplet resonance at δ 2.09 ppm is due to the presence of toluene solvent impurity.
The addition of 2 equiv of 1-dodecanthiol under the reaction temperature of 60 °C resulted in color change from dark brown to bright yellow as demonstrated in Figure 1c and the reaction progression in Figure S2 (Supporting Information). The color change indicates the probable reduction of Ir4+ to Ir3+, which has also been observed from the reduction of IrCl62– (dark brown) to IrCl63– (brownish yellow).33 Previously, Zhu et al. were able to confirm the complete reduction of Au3+ to Au1+ by the addition of 2 equiv of alkanethiols, which were oxidized to disulfides, during the Brust–Schiffrin gold nanoparticle synthesis.37 Similar studies using 1H NMR were attempted for the modified Brust–Schiffrin synthesis of Ir nanoparticles using 1-dodecanethiol, which subsequently showed small peaks representative of dodecyldisulfide (δ 2.54 ppm) and 1-dodecanethiolate (δ 2.18 ppm), along with the αCH2-N peaks of tetra-n-octylammonium downshifted back to their original chemical shift of δ 3.40 ppm. This change in NMR spectra implies that the reduced Ir complex is no longer directly associated with tetra-n-octylammonium salts, which now exclusively form the ion-pair complex with halide ions (Figure 2b).
Clear evidence of the reaction between IrX62– with 1-dodecanethiol was shown in Figure S3 (Supporting Information), which illustrates the UV–vis spectra of the reaction mixture after the addition of 4 equivs of 1-dodecanethiol. The spectra showed complete disappearances of the strong absorption bands at 440 and 500 nm, indicating the cleavage of Ir4+-X (either Cl or Br) bonds.33 The appearance of absorption bands at ∼310 nm is most likely due to the formation of Ir3+-SR bonds.38 The 1H NMR spectra of the reaction mixture after the addition of 4 equivs of dodecanethiol shown in Figure 2c also support the formation of dodecyldisulfide from 1-dodecanethiol after the reaction with IrX62–. The relative intensity of these peaks at δ 2.54 ppm and δ 2.18 ppm was 1 to 3 in favor of dodecanethiolate, confirming the formation of the Ir(SR)3 complex. One equiv of 1-dodecanthiol was, therefore, used for the reduction of Ir4+ to Ir3+, and the other 3 equivs of thiol participated in the complexation with Ir3+. The direct evidence of Ir3+-SR bond formation could be obtained from a closer analysis of 1H NMR spectra. The peaks at δ 2.18 ppm exhibited a multiplicity (triplet of triplet) rather than a triplet typically observed from CH2–αCH2-S– or a quartet observed from CH2–αCH2-SH.37 The unusual splitting pattern of this peak indicates the occurrence of the spin–spin coupling of Ir–H (3J) and H–H (3J) for the Ir(SR)3 complex. To our knowledge, however, such an interaction of three bond (Ir–S-C-H) Ir–H coupling has never been reported. Interestingly, the 1H NMR studies of Ir–H complexes have only shown the tendency for Ir to self-decouple for 1J coupling.39 Considering the spin number (I = 3/2) of Ir, the coupling of Ir–H (3J) should produce a quartet splitting instead of a triplet splitting as shown in Figure 2c, inset. Despite this inconclusive NMR splitting, the presence of this abnormal multiplicity strongly supports the presence of an Ir–H (3J) interaction and thus the formation of the Ir–S bond.
The addition of NaBH4, a highly basic reducing agent, was enacted in order to produce Ir nanoparticles from an Ir(SR)3 complex. Upon the addition of NaBH4, the color of the solution turned into strong orange as shown in Figure 1d. This color change was followed by the formation of insoluble gel-like materials during further stirring of the reaction mixture (Figure S4, Supporting Information). Moreover, UV–vis spectra of the reaction mixture did not resemble that of a typical nanoparticle solution but instead showed strong absorption bands centered at ∼330 nm. This result indicated the probable formation of the hydrolysis product rather than the desired reduction of Ir3+ to Ir0 after the addition of basic NaBH4 solution. The stable product of this hydrolysis reaction would be Ir(OH)62– or Ir(OH)63–.33 Insoluble Ir(OH)6y-(Na+)y would precipitate out in toluene as shown in Figure S4 (Supporting Information), and more soluble Ir(OH)6y-(R4N+)y would result in the appearance of strong absorption bands at ∼330 nm. The UV data also suggested the reaction did not involve the reformation of IrX62– or the formation of IrOx·nH2O, which have strong absorption bands at ∼440/500 nm and ∼580 nm, respectively.33 The NMR spectra of the reaction mixture after the addition of NaBH4 shown in Figure 2d indicated the full conversion of the dodecanethiolate ligand of the Ir(SR)3 complex to its disulfide form in the presence of basic sodium borohydride solution.
In contrast to the alkanethiol, the addition of S-dodecylthiosulfate to the reaction mixture containing the [R4N+]2[IrX62–] adduct did not trigger any change in the solution color and absorption bands of Ir4+-X at 440 and 500 nm in UV–vis spectra. The 1H NMR spectra of [R4N+]2[IrX62–] in toluene-d8 were also nearly unaffected by the addition of S-dodecylthiosulfate, which showed αCH2-S peaks at δ 3.28 ppm (Figure 2e). A slight upfield shift from δ 2.65 ppm to δ 2.48 ppm was observed for the αCH2-N peaks. Since the structure of S-dodecylthiosulfate is highly reminiscent of hydrophobic ionic salts, the formation of an ion-pair complex of [R4N+][R′S2O3–] and the rapid equilibrium with [R4N+]2[IrX62–] would cause such a change in the chemical shifts. The overall spectroscopic results clearly demonstrated the low reactivity of the thiosulfate ligand precursors compared to thiols against the iridium complex.
The addition of NaBH4 to the reaction mixture containing [R4N+]2[IrX62–] and S-dodecylthiosulfate initially turned the solution color from dark brown to faint yellow implying the cleavage of the Ir-X bond, but the solution turned into dark black/blown within 5 min of NaBH4 addition (Figure S5, Supporting Information). This result suggested that the formation of Ir nanoparticles is slower than that of other nanoparticles including Au, Ag, and Pd, which undergo color changes into black/blown immediately upon the addition of NaBH4. UV–vis spectra of the isolated Ir nanoparticles showed an exponential decay in absorbance with a decrease in energy, which is a typical characteristic of small nanosized colloids lacking surface plasmon resonance as is the case with Ir (Figure S6, Supporting Information).11 The 1H NMR of the Ir nanoparticles isolated and redissolved in CDCl3 revealed three broad resonances at δ 1.80–1.60, δ 1.50–1.20, and δ 0.90–0.80 ppm for the β-CH2-CH2-S, -CH2-, and -CH3, respectively (Figure 2f). These peaks resulted from the attached dodecanethiolate stabilizers on the nanoparticle surface, as peak broadening has been indicative of ligand immobilization for other ligand-capped metal nanoparticles.14,17 Interestingly enough, the small core size of these nanoparticles allow the β-protons of attached organic ligand stabilizers to be visible; a rare event only observed for the small and monodisperse metal nanoparticles.40 Moreover, the high purity of the isolated Ir nanoparticles was proven to be attributed to the absence of resonances at δ 3.40 (tetra-n-octylammonium: αCH2-N), δ 3.28 ppm (S-dodecylthiosulfate: αCH2-N), δ 2.54 (didodecyl disulfide: αCH2-S), and δ 2.18 (dodecanethiolate: αCH2-S).
FT-IR spectra of Ir nanoparticles were also identical to those of metal nanoparticles previously generated from both dodecanethiol and S-dodecylthiosulfate (Figure S7, Supporting Information).11,14 Only the νCH2 and νCH3 stretches (3000–2800 cm–1) and bendings (∼1450 cm–1) of dodecanethiolate monolayers were observed in the spectra. Any other significant peaks including sulfonate (−SO3–) stretches at ∼1350 (νasS=O; strong) and ∼1175 cm–1 (νsS=O; strong) were not observed implying the cleavage of the thiosulfate S–S bond after the adsorption of thiosulfate on the Ir nanoparticle surface. The TEM image and the core size histogram of the final purified product are presented in Figure 3. The average core size of the Ir nanoparticles was determined to be 1.2 ± 0.3 nm by counting a total of ∼2300 particles from multiple images. The small size and high monodispersity of these nanoparticles are quite novel for Ir nanostructures because such cluster-like, stable, and isolable Ir nanoparticles have never been reported previously. The overall organic and metallic weight fractions could be determined by thermogravimetric analysis (Figure S8, Supporting Information). The initial major volatilization of these iridium nanoparticles took place close to ∼150 °C, which is quite similar to that in our prior studies involving palladium nanoparticles.11 After the final temperature of 600 °C was reached, 61.5% of the residual sample was still present signifying an organic fraction of 38.5% for these Ir nanoparticles.
Figure 3.

TEM image and the core size distribution histogram of Ir nanoparticles. The scale bar is 20 nm.
XPS spectra of the Ir4f7 (60.8 eV) and Ir4f5 (63.8 eV) region for Ir nanoparticles are shown in Figure 4a.6 The broad XPS S2p signal at 162.2 eV (Figure 4b) revealed the presence of an Ir–S bond on the Ir nanoparticle surface.41 Small traces of oxidized S, which are clearly differentiated from the high intense peaks observed for oxidized S species of the thiosulfate ligand precursor (Figure 4c), were observed at ∼166–8 eV.41 Since the absence of unbound thiosulfate was confirmed by NMR and IR, the small amount of oxidized S species observed in XPS spectra was most likely due to the small presence of chemisorbed oxidized sulfur species such as sulfur trioxide or sulfite that were cleaved off from thiosulfate precursors.18,19 This result demonstrated that the removal of surface adsorbed oxidized sulfur species is somewhat incomplete for Ir nanoparticles.
Figure 4.

(a) Ir4f XPS spectra and (b) S2p XPS spectra of dodecanethiolate-capped iridium nanoparticles (c = 2 mg mL–1). (c) S2p XPS spectra of S-dodecylthiosulfate. The binding energies were referenced by setting the O1s binding energy to 528.5 eV.
Ultimately, one of the main reasons this reaction produces stable and isolable Ir nanoparticles was that all necessary reagents (the Ir complex, ligands, and reducing agent) for nucleation–growth–passivation of Ir nanoparticles form ionic pair complexes with tetraoctylammonium salts and, therefore, are present at close proximity to each other. The activation energy of the reaction is likely decreased thereby aiding the completion of the nanoparticle stabilization stage. In comparison, the thiol protocol involves the nonionic Ir(SR)3 complex as a key intermediate, and the hydrophobic alkyl tail parts of tetraoctylammonium might act as a kinetic barrier for the further reduction of Ir3+ to Ir0 by ion-paired BH4–.
Small Ir clusters have been predicted to have magnetic moments larger than 0.5 μB/atom.42 However, based on our knowledge, there has never been any published report on experimental work related to stable and unsupported Ir nanoparticles with a detectible magnetic moment. The successful synthesis of small Ir nanoparticles capped with dodecanethiolate ligands allowed us, for the first time, to examine the magnetic properties of the cluster-like Ir particles. Macroscopic magnetization measurements have been performed at temperatures ranging from 5 to 300 K using Quantum Design Physical Property Measurement System. The Ir nanoparticles were found to have a relatively large magnetic moment which increases with stronger external fields as shown in Figure 5a. The saturation magnetization, determined using TGA analysis of the nanoparticle sample, was found to be 12.2 emu/g of Ir at 300 K and 13.0 emu/g of Ir at 5 K. This value is higher than the saturation magnetization of dendrimer-encapsulated Ni nanoparticles43 and other alkaenthiolate-capped metal nanoparticles.44,45 The preservation of the magnetic signal from 5 to 300 K (only a decrease of ∼6%) is observed. The hysteresis loop at 300 and 5 K shows the nanoparticles to have a coercitivity of 54 and 105 Oe, respectively. In addition, both the zero-field-cooled (ZFC) and field-cooled (FC) temperature dependence of the magnetization were measured in a 100 Oe applied field (Figure 5b). The maximum in the ZFC curve and divergence in the ZFC and FC curves indicate a blocking temperature, TB, of more than 300 K. The absence of any significant amount of magnetic impurity such as Fe was confirmed from XPS results. The overall characteristics of Ir nanoparticles suggest that they are “soft” magnetic materials with ferromagnetic properties. More in-depth studies on the magnetic behavior of Ir nanoparticles with different core sizes and Ir/thiolate compositions will be done and reported in the near future.
Figure 5.
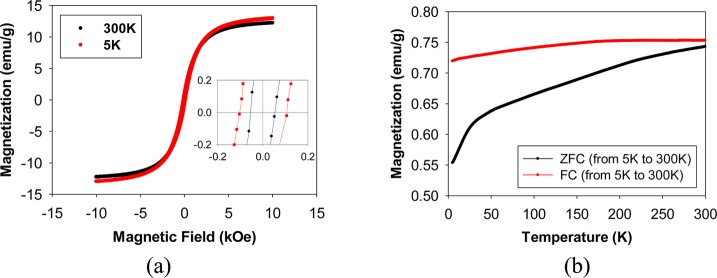
(a) Magnetization curves of dodecanethiolate-capped iridium nanoparticles obtained at 300 and 5 K; the inset shows the similar coercive fields at 300 and 5 K. (b) Temperature dependence of the magnetization of iridium nanoparticles measured in a 100 Oe applied magnetic field.
Conclusions
The synthesis of stable and isolable iridium nanoparticles with an average core size of ∼1.2 nm was achieved by employing sodium S-dodecylthiosulfate as a ligand precursor. It was also demonstrated that employing dodecanethiol directly under identical conditions fails to yield iridium nanoparticles. To our knowledge, our article stands as the lone method for producing stable alkanethiolate-capped Ir nanoparticles using sodium borohydride reduction. Furthermore, our study provided insights into the Brust–Schiffrin mechanism and the reason thiosulfate ligands are preferred over thiol ligands for the synthesis of Ir nanoparticles. Finally, the produced Ir nanoparticles exhibited strong magnetic moments demonstrating the potential of these Ir nanoparticles for various technological applications.
Acknowledgments
We gratefully acknowledge the UCLA Materials Lab of the Molecular Instrumentation Center and Ignacio Martini, Ph.D., the Molecular Instrumentation Center (MIC) Associate Director, for X-ray photoelectric spectroscopy analysis. This research was supported in part by a grant from the National Institute of General Medical Science (#SC3GM089562) and CSULB (RSCA and MGSS awards).
Supporting Information Available
Photographic images of reaction progressions, UV–vis spectra of Ir-precursor species and Ir nanoparticles, and thermogravimetric analysis and FT-IR spectra of Ir nanoparticles. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Philippot K.; Serp P.. Concepts in Nanocatalysis. In Nanomaterials in Catalysis; Serp P., Philippot K., Eds.; Wiley-VCH: Weinheim, Germany, 2013; pp 1–54. [Google Scholar]
- Astruc D.Transition-Metal Nanoparticles in Catalysis: From Historical Background to the State-of-the Art, in Nanoparticles and Catalysis; Astruc D., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008. [Google Scholar]
- Nozik A. J. Nanoscience and Nanostructures for Photovoltaics and Solar Fuels. Nano Lett. 2010, 10, 2735–2741. [DOI] [PubMed] [Google Scholar]
- Laurent S.; Forge D.; Port M.; Roch A.; Robic C.; Elst L. V.; Muller R. N. Magnetic Iron Oxide Nanoparticles: Synthesis, Stabilization, Vectorization, Physicochemical Characterization, and Biological Applications. Chem. Rev. 2008, 108, 2064–2110. [DOI] [PubMed] [Google Scholar]
- Bayram E.; Zahmakiran M.; Özkar S.; Finke R. G. In Situ Formed “Weakly Ligated/Labile Ligand” Iridium(0) Nanoparticles and Aggregates as Catalysts for the Complete Hydrogenation of Neat Benzene at Room Temperature and Mild Pressures. Langmuir 2010, 26, 12455–12464. [DOI] [PubMed] [Google Scholar]
- Zhu Y.; Jang S. H. A.; Tham Y. H.; Algin O. B.; Maguire J. A.; Hosmane N. S. An Efficient and Recyclable Catalytic System Comprising Nano-Iridium(0) and a Pyridinium Salt of nido-Carboranyldiphosphine for the Synthesis of One-Dimensional Boronate Esters via Hydroboration Reaction. Organometallics 2012, 31, 2589–2596. [Google Scholar]
- Rueping M.; Koenigs R. M.; Borrmann R.; Zoller J.; Weirich T. E.; Mayer J. Size-Selective, Stabilizer-Free, Hydrogenolytic Synthesis of Iridium Nanoparticles Supported on Carbon Nanotubes. Chem. Mater. 2011, 23, 2008–2010. [Google Scholar]
- Vollmer C.; Redel E.; Abu-Shandi K.; Thomann R.; Manyar H.; Hardacre C.; Janiak C. Microwave Irradiation for the Facile Synthesis of Transition-Metal Nanoparticles (NPs) in Ionic Liquids (ILs) from Metal-Carbonyl Precursors and Ru-, Rh-, and Ir-NP/IL Dispersions as Biphasic Liquid-Liquid Hydrogenation Nanocatalysts for Cyclohexene. Chem.—Eur. J. 2010, 16, 3849–3858. [DOI] [PubMed] [Google Scholar]
- Yinghual Z.; Chenyan K.; Peng A. T.; Emi A.; Monalisa W.; Louis L. K.-J.; Hosmane N. S.; Maguire J. A. Catalytic Phenylborylation Reaction by Iridium(0) Nanoparticles Produced from Hydridoiridium Carborane. Inorg. Chem. 2008, 47, 5756–5761. [DOI] [PubMed] [Google Scholar]
- Migowski P.; Zanchet D.; Machado G.; Gelesky M. A.; Teixeira S. R.; Dupont J. Nanostructures in Ionic Liquids: Correlation of Iridium Nanoparticles’ Size and Shape with Imidazolium Salts’ Structural Organization and Catalytic Properties. Phys. Chem. Chem. Phys. 2010, 12, 6826–6833. [DOI] [PubMed] [Google Scholar]
- Sadeghmoghaddam E.; Lam C.; Choi D.; Shon Y.-S. Synthesis and Catalytic Property of Alkanethiolate-Protected Pd Nanoparticles Generated from Sodium S-Dodecylthiosulfate. J. Mater. Chem. 2011, 21, 307–312. [Google Scholar]
- Sadeghmoghaddam E.; Gaïeb K.; Shon Y.-S. Catalytic Isomerization of Allyl Alcohols to Carbonyl Compounds using Poisoned Pd Nanoparticles. Appl. Catal. A, General 2011, 405, 137–141. [Google Scholar]
- Sadeghmoghaddam E.; Gu H.; Shon Y.-S. Pd Nanoparticle-Catalyzed Isomerization vs. Hydrogenation of Allyl Alcohol: Solvent-Dependent Regioselectivity. ACS Catal. 2012, 2, 1838–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavia D. J.; Shon Y.-S. Controlling Surface Ligand Density and Core Size of Alkanethiolate-Capped Pd Nanoparticle and Their Effects on Catalysis. Langmuir 2012, 28, 14502–14508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavia D. J.; Maung M. S.; Shon Y.-S. Water-Soluble Pd Nanoparticles Synthesized from ω-Carboxyl-S-alkanethiosulfate Ligand Precursors as Unimolecular Micelle Catalysts. ACS Appl. Mater. Interfaces 2013, 5, 12432–12440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mari A.; Imperatori P.; Marchegiani G.; Pilloni L.; Mezzi A.; Kaciulis S.; Cannas C.; Meneghini C.; Mobilio S.; Suber L. High Yield Synthesis of Pure Alkanethiolate-Capped Silver Nanoparticles. Langmuir 2010, 26, 15561–15566. [DOI] [PubMed] [Google Scholar]
- Shon Y.-S.; Cutler E. Aqueous Synthesis of Alkanethiolate-Protected Ag Metal Nanoparticles using Bunte Salts. Langmuir 2004, 20, 6626–6630. [DOI] [PubMed] [Google Scholar]
- Shon Y.-S.; Gross S. M.; Dawson B.; Porter M.; Murray R. W. Alkanethiolate Protected Gold Clusters Generated from Sodium S-dodecylthiosulfate (Bunte Salts). Langmuir 2000, 16, 6555–6561. [Google Scholar]
- Lohse S. E.; Dahl J. A.; Hutchison J. E. Direct Synthesis of Large Water-Soluble Functionalized Gold Nanoparticles Using Bunte Salts as Ligand Precursors. Langmuir 2010, 26, 7504–7511. [DOI] [PubMed] [Google Scholar]
- Lukkari J.; Meretoja M.; Kartio I.; Laajalehto K.; Rajamäki M.; Lindström M.; Kankare J. Organic Thiosulfates (Bunte Salts): Novel Surface-Active Sulfur Compounds for the Preparation of Self-Assembled Monolayers on Gold. Langmuir 1999, 15, 3529–3537. [Google Scholar]
- Fealy R. J.; Ackerman S. R.; Ferguson G. S. Mechanism of Spontaneous Formation of Monolayers on Gold from Alkyl Thiosulfates. Langmuir 2011, 27, 5371–5376. [DOI] [PubMed] [Google Scholar]
- Yee K. C.; Jordan R.; Ulman A.; White H.; King A.; Rafailovich M.; Sokolov J. Novel One-Phase Synthesis of Thiol-Functionalized Gold, Palladium, and Iridium Nanoparticles Using Superhydride. Langmuir 1999, 15, 3486–3491. [Google Scholar]
- Goulet P. J.; Lennox R. B. New Insights into Brust-Schiffrin Metal Nanoparticle Synthesis. J. Am. Chem. Soc. 2010, 132, 9582–9584. [DOI] [PubMed] [Google Scholar]
- Zhang S.; Leem G.; Lee T. R. Monolayer-Protected Gold Nanoparticles Prepared Using Long-Chain Alkanethioacetates. Langmuir 2009, 25, 13855–13860. [DOI] [PubMed] [Google Scholar]
- Vivek J. P.; Burgess I. J. Insight into Chloride Induced Aggregation of DMAP-Monolayer Protected Gold Nanoparticles Using the Thermodynamics of Ideally Polarized Electrodes. J. Phys. Chem. C 2008, 112, 2872–2880. [Google Scholar]
- Volkert A. A.; Subramaniam V.; Ivanov M. R.; Goodman A. M.; Haes A. J. Salt-Mediated Self-Assembly of Thioctic Acid on Gold Nanoparticles. ACS Nano 2011, 5, 4570–4580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurashige W.; Yamaguchi M.; Nobusada K.; Negishi Y. Ligand-Induced Stability of Gold Nanoclusters: Thiolate versus Selenolate. J. Phys. Chem. Lett. 2012, 3, 2649–2652. [DOI] [PubMed] [Google Scholar]
- Li Y.; Silverton L. C.; Haasch R.; Tong Y. J. Alkanetelluroxide-Protected Gold Nanoparticles. Langmuir 2008, 24, 7048–7053. [DOI] [PubMed] [Google Scholar]
- Huang W.; Kuhn J. N.; Tsung C.-K.; Zhang Y.; Habas S. E.; Yang P.; Somorjai G. A. Dendrimer Templated Synthesis of One Nanometer Rh and Pt Particles Supported on Mesoporous Silica: Catalytic Activity for Ethylene and Pyrrole Hydrogenation. Nano Lett. 2008, 8, 2027–2034. [DOI] [PubMed] [Google Scholar]
- Metin Ö.; Özkar S. Hydrogen Generation from the Hydrolysis of Ammonia-borane and Sodium Borohydride Using Water-soluble Polymer-stabilized Cobalt(0) Nanoclusters Catalyst. Energy Fuels 2009, 23, 3517–3526. [Google Scholar]
- Sharghi H.; Khalifeh R.; Doroodmand M. M. Copper Nanoparticles on Charcoal for Multicomponent Catalytic Synthesis of 1,2,3-Triazole Derivatives from Benzyl Halides or Alkyl Halides, Terminal Alkynes and Sodium Azide in Water as a “Green” Solvent. Adv. Synth. Catal. 2009, 351, 207–218. [Google Scholar]
- Dablemont C.; Lang P.; Mangeney C.; Piquemal J.-Y.; Petkov V.; Herbst F.; Viau G. FTIR and XPS Study of Pt Nanoparticle Functionalization and Interaction with Alumina. Langmuir 2008, 24, 5832–5841. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Hernandez-Pagan E. A.; Vargas-Barbosa N. M.; Dysart J. L.; Mallouk T. E. A High Yield Synthesis of Ligand-Free Iridium Oxide Nanoparticles with High Electrocatalytic Activity. J. Phys. Chem. Lett. 2011, 2, 402–406. [Google Scholar]
- Li Y.; Zaluzhna O.; Xu B.; Gao Y.; Modest J. M.; Tong Y. J. Mechanistic Insights into the Brust-Schiffrin Two-Phase Synthesis of Organo-chalcogenate-Protected Metal Nanoparticles. J. Am. Chem. Soc. 2011, 133, 2092–2095. [DOI] [PubMed] [Google Scholar]
- Perala S. R. K.; Kumar S. On the Mechanism of Phase Transfer Catalysis in Brust-Schiffrin Synthesis of Metal Nanoparticles. Langmuir 2013, 29, 14756–14762. [DOI] [PubMed] [Google Scholar]
- Perala S. R. K.; Kumar S. On the Mechanism of Metal Nanoparticle Synthesis in the Brust-Schiffrin Method. Langmuir 2013, 29, 9863–9873. [DOI] [PubMed] [Google Scholar]
- Zhu L.; Zhang C.; Guo C.; Wang X.; Sun P.; Zhou D.; Chen W.; Xue G. New Insight into Intermediate Precursors of Brust-Schiffrin Gold Nanoparticles Synthesis. J. Phys. Chem. C 2013, 117, 11399–11404. [Google Scholar]
- Sekioka Y.; Kaizaki S.; Mayer J. M.; Suzuki T. Structure and Reactivity of a Pyridine-1-imido-2-thiolato Complex of Iridium(III), Cp*Ir(1-N-2-Spy), Generated by Photolysis of the (Azido)(pyridine-2-thiolato) Complex, Cp*Ir(2-Spy)(N3). Inorg. Chem. 2005, 44, 8173–8175. [DOI] [PubMed] [Google Scholar]
- McLaughlin M. P.; Adduci L. L.; Becker J. J.; Gagnè M. R. Iridium-Catalyzed Hydrosilylative Reduction of Glucose to Hexane(s). J. Am. Chem. Soc. 2013, 135, 1225–1227. [DOI] [PubMed] [Google Scholar]
- Qian H.; Zhu M.; Andersen U. N.; Jin R. Facile, Large-Scale Synthesis of Dodecanethiol-Stabilized Au38 Clusters. J. Phys. Chem. A 2009, 113, 4281–4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacs S. R.; Cutler E. C.; Park J.-S.; Lee T. R.; Shon Y.-S. Synthesis of Tetraoctylammonium-Protected Gold Nanoparticles with Improved Stability. Langmuir 2005, 21, 5689–5692. [DOI] [PubMed] [Google Scholar]
- Pawluk T.; Hirata Y.; Wang L. Studies of Iridium Nanoparticles Using Density Functional Theory Calculations. J. Phys. Chem. B 2005, 109, 20817–20823. [DOI] [PubMed] [Google Scholar]
- Knecht M. R.; Garcia-Martinez J. C.; Crooks R. M. Synthesis, Characterization, and Magnetic Properties of Dendrimer-Encapsulated Nickel Nanoparticles Containing <150 Atoms. Chem. Mater. 2006, 18, 5039–5044. [Google Scholar]
- Garitaonandia J. S.; Insausti M.; Goikolea E.; Suzuki M.; Cashion J. D.; Kawamura N.; Ohsawa H.; Gil de Muro I.; Suzuki K.; Plazaola F.; Rojo T. Chemically Induced Permanent Magnetism in Au, Ag, and Cu Nanoparticles: Localization of the Magnetism by Element Selective Techniques. Nano Lett. 2008, 8, 661–667. [DOI] [PubMed] [Google Scholar]
- Crespo P.; Litrán R.; Rojas T. C.; Multigner M.; de la Fuente J. M.; Sánchez-López J. C.; García M. A.; Hernando A.; Penadés S.; Fernández A. Permanent Magnetism, Magnetic Anisotropy, and Hysteresis of Thiol-Capped Gold Nanoparticles. Phys. Rev. Lett. 2004, 93, 087204. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.