

Abstract
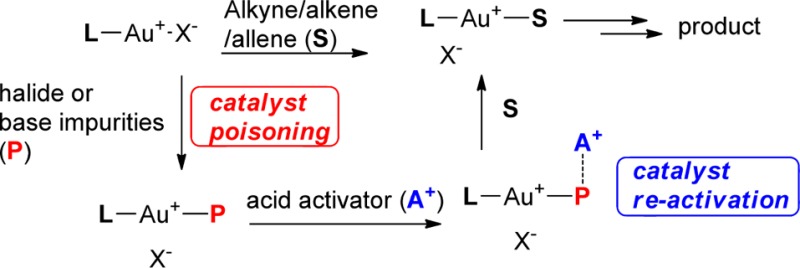
High gold affinity impurities (halides, bases) in solvents, starting materials, filtration, or drying agents could affect the reactivity of gold catalyst adversely, which may significantly reduce the TON of cationic gold-catalyzed reactions. Use of a suitable acid activator (e.g., HOTf, In(OTf)3) reactivates the gold catalyst and makes the reaction proceed smoothly at low gold catalyst loading.
In kinetic studies of catalyzed reactions, it is customary to study the relationship between the rate of reaction and the concentration of catalyst to learn more about the mechanism of the reaction. Although a linear correlation between the concentration of the catalyst and the initial rate data often exists when rate is plotted against concentration, numerous studies have shown that the regression line does not intersect with the origin (eq 1, A ≠ 0; also see Figure 1). If there is no background reaction, the rate should be 0 when the catalyst concentration is 0, so A (intercept) should also be 0, but this is not the case in many reactions (A is usually less than 0),1 indicating that a threshold catalyst concentration is required. Beyond vaguely implying some sort of catalyst poisoning, literature reports have not addressed the causes of this type of threshold. Although this phenomenon is ubiquitous in catalysis, relatively little effort has been spent on the investigation of this anomaly and the possible implications of this threshold phenomenon in catalysis.1
| 1 |
Figure 1.
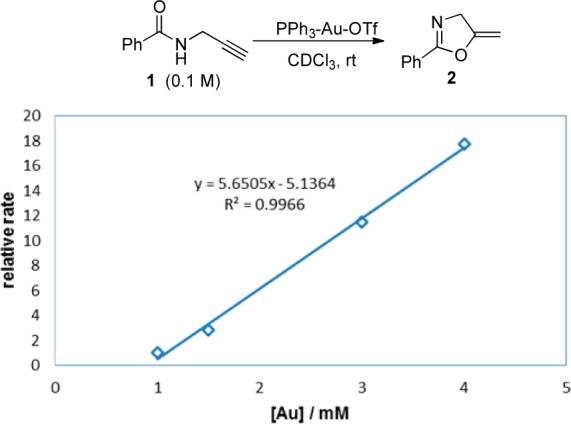
Correlation between rate and catalyst concentration.
During our research to improve the efficiency of gold catalysis,2 we found that this threshold phenomenon is common in gold-catalyzed reactions. We studied the correlation between rate and gold catalyst concentration in the cyclization of propargyl amide 1 (Figure 1).3 We found that there was a linear relationship between the rate of reaction and the concentration of gold catalyst with a negative intercept A (eq 1). If we extrapolate the data, the minimum gold concentration to start the reaction will be 0.9 mM, which represents a 0.9% gold loading. Indeed, the reaction did not start at all when the catalyst loading was low (e.g., 0.3% loading). Other kinetic studies on gold-catalyzed reactions showed a similar trend. For example, Toste and co-workers have reported a linear relationship between rate and gold catalyst loading in the intermolecular hydroamination of allene (rate = 0.0423[Au] – 0.0355).1c Their data suggested that the minimum catalyst concentration to start the reaction was 1.2 mM (0.81% loading).
We have found that the above-mentioned threshold phenomena is not limited to those examples. When we lowered the loading of gold catalyst below a certain level, the reaction simply would not start (rate was 0 for a prolonged time). This occurrence lowers the turnover number (TON) in gold catalysis and could be one of the main reasons why the TON in gold-catalyzed reactions is generally low (1% or higher loading is usually required).
A simple explanation for this threshold phenomenon is the poisoning of the active catalyst. Impurities such as halides and basic alkali components are well-known to contribute to catalyst poisoning in heterogeneous catalysis,4 but there are comparatively few studies on the deactivation of homogeneous catalysts.5 Cationic gold catalyst is sought after because of its high tolerance toward moisture and oxygen (which means we can run reaction without special protection in most cases), but cationic gold has shown a very high affinity toward halides and basic components (e.g., OH–).6 A recent study by Maier and co-workers indicated that OH– or Cl– has approximately 106 times higher affinity toward cationic gold compared to an alkyne.6 This means that just trace amounts of these catalyst poisons, such as halides and basic components present in the solvent or starting material, are enough to block the active site of a gold catalyst (Scheme 1) and may completely inhibit the reaction when a relatively low gold catalyst loading is used.
Scheme 1. Poisoning and Re-activation of Cationic Gold Catalyst.
Of course, use of highly purified starting materials and solvents may solve this problem, but time-consuming purification processes are needed, and often, it is not possible to remove all relevant impurities even after stringent purification protocols. Use of highly purified starting material and solvents is especially impractical in larger scale synthesis. We propose here that it is not necessary to eradicate all traces of possible catalyst poisons; instead, the problem can be solved by directly adding a suitable acid activator to the reaction (A+ in Scheme 1). Acid activators that have high affinity toward P (catalyst poison) may reactivate the gold catalyst (Scheme 1). In other words, an acid activator (A+) acts as a sacrificial reagent to bind to possible catalyst poisons, so that the cationic gold is free to catalyze the reaction. Indeed, Nolan and co-worker had used Brønsted acid acids like HOTf to activate NHC–Au–OH complexes.7
We began our studies with the intramolecular hydroarylation of an alkyne8 (Figure 2). We observed that when this reaction was carried at a higher gold catalyst loading (2% vs starting material), the reaction took place smoothly in CDCl3 (CDCl3 was used as received). But as we reduced the catalyst loading to 0.2%, surprisingly, the reaction did not proceed at all (Figure 2). On the other hand, the reaction proceeded well at 0.2% loading using purified CDCl3 as solvent (K2CO3-treated and freshly distilled). This result suggests that nondistilled CDCl3 has certain impurities (most likely chlorine/chloride containing compounds)9 that inhibit gold catalyst activity.
Figure 2.
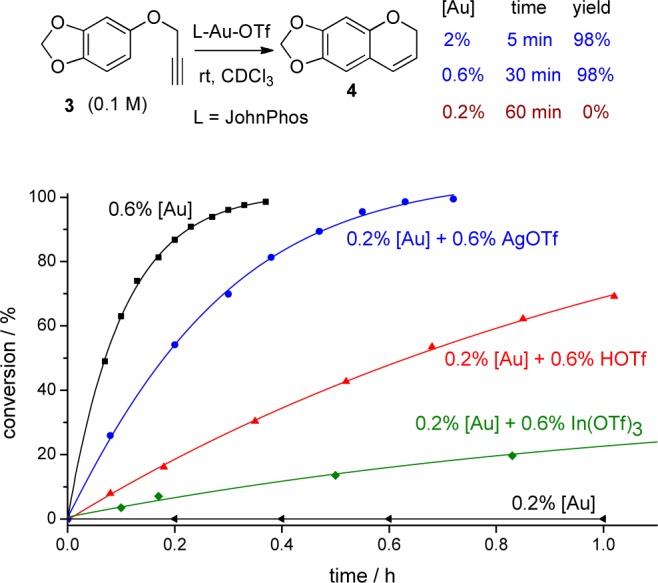
Influence of acid activators in alkyne hydroarylation reaction.
To make the influence of impurity in solvents more significantly, we repeated the same reaction at a more diluted condition (changing [3] from 0.1 to 0.02 M, keeping gold loading at 0.2%); the reaction still did not progress, even in freshly distilled CDCl3. The reaction performed better in CD2Cl2 (probably because there were less halide impurities) at 0.1 M, but when the concentration was decreased to 0.02 M, the reaction became sluggish. This change of behavior may have been caused by catalyst poisons that either could not be completely removed by simple distillation, or were present in the starting material 3 itself.
Next, we inspected the effects of common Brønsted acids or Lewis acids in the reactivation of the gold catalyst (Figure 2). We found that AgOTf and TfOH worked very well. Other Lewis acids like Ga(OTf)3, Eu(OTf)3, Y(OTf)3, Sn(OTf)2, Zn(OTf)2, and (CuOTf)2·C6H6 did not show any activity in the reactivation of the gold catalyst. Moreover, we observed that the reaction did not occur in the presence of a Lewis acid or Brønsted acid alone, without gold catalyst, under, otherwise, identical conditions.
We next examined an ester-assisted hydration reaction (Figure 3).10 Again, the reaction proceeded well at 1% gold loading, but when the gold loading was reduced to 0.5%, the reaction did not proceed at all. Use of freshly distilled solvent (acetone) gave better results, but the reaction still did not ensue when the reaction was conducted at more diluted conditions. However, various acid activators restarted the reaction, among them, Ga(OTf)3 provided best result (Figure 3).
Figure 3.
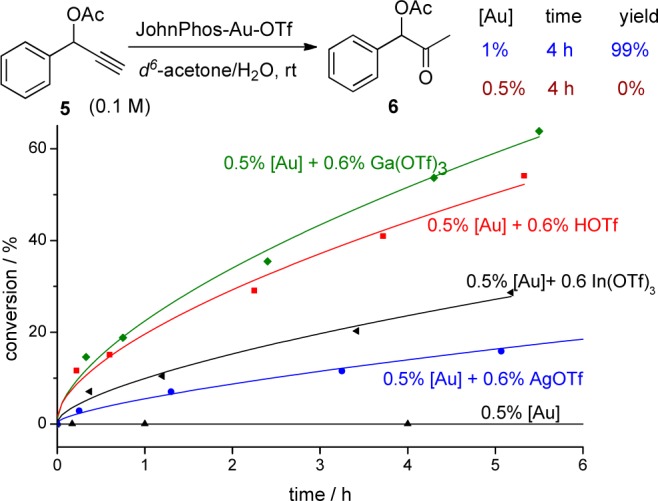
Influence of acid activators in hydration reaction of 5.
In the cycloisomerization of enyne (Figure 4),11 we observed a similar effect as in previous reactions. Cycloisomerization of 7 took place very smoothly at 0.6% catalyst loading, but it became much slower at 0.2% gold catalyst loading, and its rate decreased to zero when the catalyst loading was reduced to 0.02%. A Lewis acid activator, In(OTf)3 caused the reaction to be completed in less than 1 h at very low gold catalyst loading (0.02%). In(OTf)3 was very effective, but a Brønsted acid like HOTf was only marginally effective.
Figure 4.
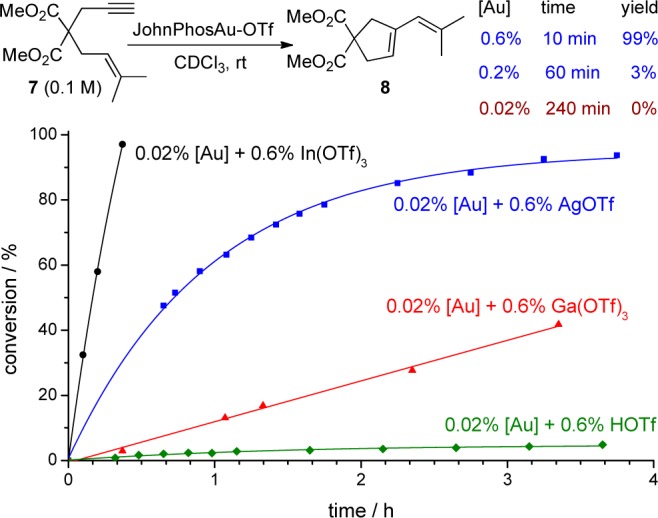
Influence of acid activators in cycloisomerization of 7.
We then focused our attention to the cyclization of hexynoic acid 9 (Figure 5).12 Again, we observed that the cyclization took place quickly at 1% gold catalyst loading, but the reaction rate dropped down to zero at 0.1% gold catalyst loading. Once again, acid activators (AgOTf, In(OTf)3, TfOH) could reactivate the gold catalyst (Figure 5).
Figure 5.
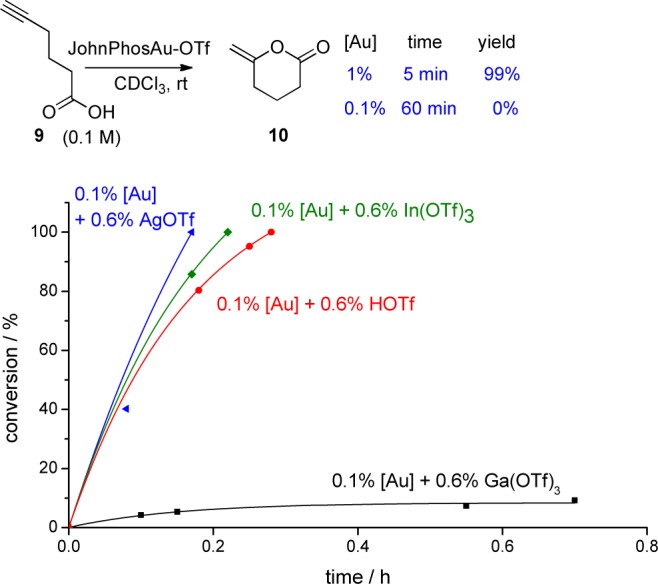
Influence of acid activators in cyclization of 9.
In the cycloisomerization of allenone 11 (Figure 6),13 the reaction with 0.2% gold catalyst loading was very fast. However, the same reaction with 0.04% catalyst loading was very sluggish. Activators like AgOTf and In(OTf)3 sped up the rate of reaction significantly; in contrast, TfOH and Ga(OTf)3 did not have much effect on the reaction rate (Figure 6).
Figure 6.

Influence of acid activators in cycloisomeriation of 11.
Contamination in solvents or starting materials is not the only source of gold catalyst poisons. External reagents, such as drying agents (e.g., molecular sieves) and filtration aids (e.g., Celite) are also commonly used in synthesis. They may also contain possible gold catalyst poisons (P) like halides and alkali bases. For example, a commonly used filtration aid, Celite 545, is prepared from diatomaceous earth treated with a base–sodium carbonate (flux calcined). Indeed, the hydration of 5 proceeded well with untreated gold catalyst (2% loading) (Table 1, entry 1), but when the gold catalyst (a solution in acetone) was pretreated with molecular sieves 4 Å, or filtered through Celite 545, the reaction did not proceed with the same catalyst loading (Table 1, entries 2 and 3). The inactive gold catalyst from entries 2 or 3 could be reactivated by the addition of an acid activator Ga(OTf)3 (Table 1, entries 4 and 5). These results are consistent with literature reports: Yu and co-workers reported that treatment of Ph3PAu-OTf with molecular sieves led to the formation of much less reactive trigold oxonium complex [(Ph3P)Au3O]+OTf – due to the presence of mild bases in molecular sieves.14 The loss of reactivity of gold catalyst by Celite filtration is also consistent with a previous report by Shi and co-workers.10b
Table 1. Influences of Molecular Sieves and Filtration Agent.
| entry | [Au] (%) | cat. pretreatment | time (h) | yield (%) |
|---|---|---|---|---|
| 1 | 2 | none | 3 | 99 |
| 2 | 2 | filtration through Celite 545 | 3 | 0 |
| 3 | 2 | dried over MS 4A | 3 | 0 |
| 4a | 0.2 | Ga(OTf)3 0.6% added | 3 | 25 |
| 5a | 0.2 | Ga(OTf)3 0.6% added | 12 | 89 |
Catalyst from entry 3 was used.
We propose that the possible gold catalyst poisons are the halides, bases, or other high gold affinity impurities. A direct proof would be isolation and identification of these impurities and then investigation of these impurities on the reactivity. It is difficult to do this at this time because routine analytic tools commonly used in organic synthesis (NMR, GC–MS, HPLC–MS) are not effective. In order to give an indirect proof, we investigated the effect of halide and bases on the reactivity and ability of acidic promoters to restore the reactivity (Table 2). We still used hydration of 5 as our model reaction. Indeed, bases and halides like Bu4N+Cl– and Bu4N+OH– effectively inhibited the reaction (Table 2, entries 2 and 3). Similarly, as in Table 1, Ga(OTf)3 effectively restored the reactivity (Table 2, entries 4 and 5).
Table 2. Influence of Halide and Base on Hydration of 5.
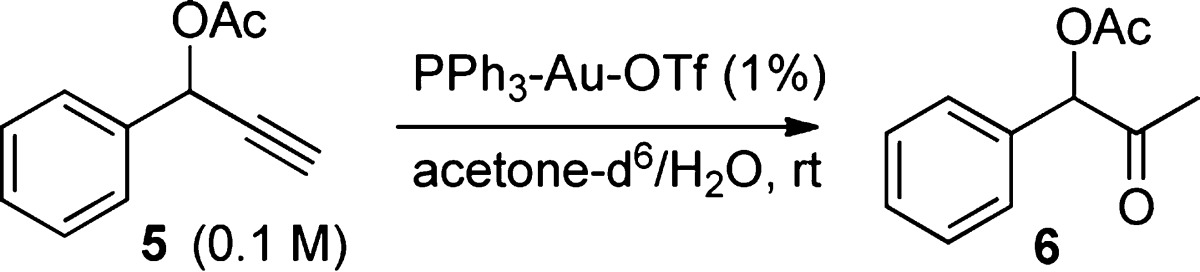
| entry | additive | time (h) | yield (%) |
|---|---|---|---|
| 1 | none | 4 | 99 |
| 2 | Bu4N+OH– (1%) | 24 | 2 |
| 3 | Bu4N+Cl– (1%) | 24 | 8 |
| 4 | Ga(OTf)3 (2%) added to entry 2 | 2 | 99 |
| 5 | Ga(OTf)3 (2%) added to entry 3 | 2 | 99 |
It should be noted that the acid activators not only can reactivate the poisoned gold catalyst but also can positively influence the later stage in the gold catalytic cycle (e.g., protodeauration), acting as co-catalysts to speed-up the reactions.15
In conclusion, high gold affinity impurities (halides, bases) in solvents, starting materials, filtration, or drying agents could affect the reactivity of gold catalysts adversely, which may significantly reduce the TON of cationic gold-catalyzed reactions. The use of a suitable acid activator (e.g., HOTf, In(OTf)3) reactivates the gold catalyst and contributes to making the reaction proceed smoothly at low gold catalyst loading. A similar protocol could benefit other cationic metal catalysis.
Acknowledgments
We are grateful to the National Science Foundation for financial support (CHE-1111316).
Supporting Information Available
Experimental procedure. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Hardacre C.; Katdare S. P.; Milroy D.; Nancarrow P.; Rooney D. W.; Thompson J. M. J. Catal. 2004, 227, 44–52. [Google Scholar]; b Mukherjee A.; Sen T. K.; Ghorai P. K.; Mandal S. K. Sci. Rep. 2013, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Wang Z. J.; Benitez D.; Tkatchouk E.; Goddard Iii W. A.; Toste F. D. J. Am. Chem. Soc. 2010, 132, 13064–13071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Wang W.; Kumar M.; Hammond G. B.; Xu B. Org. Lett. 2014, 16, 636–639. [DOI] [PubMed] [Google Scholar]; b Malhotra D.; Mashuta M. S.; Hammond G. B.; Xu B. Angew. Chem., Int. Ed. 2014, 53, 4456–4459. [DOI] [PubMed] [Google Scholar]; c Kumar M.; Jasinski J.; Hammond G. B.; Xu B. Chem.—Eur. J. 2014, 20, 3113–3119. [DOI] [PubMed] [Google Scholar]; d Kumar M.; Scobie M.; Mashuta M. S.; Hammond G. B.; Xu B. Org. Lett. 2013, 15, 724–727. [DOI] [PubMed] [Google Scholar]; e Wang W.; Hammond G. B.; Xu B. J. Am. Chem. Soc. 2012, 134, 5697–5705. [DOI] [PubMed] [Google Scholar]
- a Hashmi A. S. K.; Schuster A. M.; Rominger F. Angew. Chem., Int. Ed. 2009, 48, 8247–8249. [DOI] [PubMed] [Google Scholar]; b Hashmi A. S. K.; Schuster A. M.; Gaillard S.; Cavallo L.; Poater A.; Nolan S. P. Organometallics 2011, 30, 6328–6337. [Google Scholar]
- a Oxford S. M.; Henao J. D.; Yang J. H.; Kung M. C.; Kung H. H. Appl. Catal., A 2008, 339, 180–186. [Google Scholar]; b Chandler B. D.; Kendell S.; Doan H.; Korkosz R.; Grabow L. C.; Pursell C. J. ACS Catal. 2012, 2, 684–694. [Google Scholar]; c Bartholomew C. H. Appl. Catal., A 2001, 212, 17–60. [Google Scholar]
- Simone D. O.; Kennelly T.; Brungard N. L.; Farrauto R. J. Appl. Catal. 1991, 70, 87–100. [Google Scholar]
- Zhdanko A.; Ströbele M.; Maier M. E. Chem.—Eur. J. 2012, 18, 14732–14744. [DOI] [PubMed] [Google Scholar]
- a Gaillard S.; Bosson J.; Ramón R. S.; Nun P.; Slawin A. M. Z.; Nolan S. P. Chem.—Eur. J. 2010, 16, 13729–13740. [DOI] [PubMed] [Google Scholar]; b Gómez-Suárez A.; Oonishi Y.; Meiries S.; Nolan S. P. Organometallics 2013, 32, 1106–1111. [Google Scholar]
- Nevado C.; Echavarren A. M. Chem.—Eur. J. 2005, 11, 3155–3164. [DOI] [PubMed] [Google Scholar]
- Burfield D. R.; Goh E. H.; Ong E. H.; Smithers R. H. Gazz. Chim. Ital. 1983, 113, 841–3. [Google Scholar]
- a Ghosh N.; Nayak S.; Sahoo A. K. J. Org. Chem. 2010, 76, 500–511. [DOI] [PubMed] [Google Scholar]; b Wang D.; Cai R.; Sharma S.; Jirak J.; Thummanapelli S. K.; Akhmedov N. G.; Zhang H.; Liu X.; Petersen J. L.; Shi X. J. Am. Chem. Soc. 2012, 134, 9012–9019. [DOI] [PubMed] [Google Scholar]
- a Bartolomé C.; Ramiro Z.; Pérez-Galán P.; Bour C.; Raducan M.; Echavarren A. M.; Espinet P. Inorg. Chem. 2008, 47, 11391–11397. [DOI] [PubMed] [Google Scholar]; b Bartolomé C.; Ramiro Z.; García-Cuadrado D.; Pérez-Galán P.; Raducan M.; Bour C.; Echavarren A. M.; Espinet P. Organometallics 2010, 29, 951–956. [Google Scholar]; c Nieto-Oberhuber C.; Muñoz M. P.; Buñuel E.; Nevado C.; Cárdenas D. J.; Echavarren A. M. Angew. Chem., Int. Ed. 2004, 43, 2402–2406. [DOI] [PubMed] [Google Scholar]
- a Tomás-Mendivil E.; Toullec P. Y.; Díez J.; Conejero S.; Michelet V.; Cadierno V. Org. Lett. 2012, 14, 2520–2523. [DOI] [PubMed] [Google Scholar]; b Toullec P. Y.; Genin E.; Antoniotti S.; Genêt J.-P.; Michelet V. Synlett 2008, 2008, 707–711. [Google Scholar]
- a Hashmi A. S. K.; Schwarz L.; Choi J.-H.; Frost T. M. Angew. Chem., Int. Ed. 2000, 39, 2285–2288. [DOI] [PubMed] [Google Scholar]; b Zhou C.-Y.; Chan P. W. H.; Che C.-M. Org. Lett. 2005, 8, 325–328. [DOI] [PubMed] [Google Scholar]
- Tang Y.; Yu B. RSC Adv. 2012, 2, 12686–12689. [Google Scholar]
- Xi Y.; Wang D.; Ye X.; Akhmedov N. G.; Petersen J. L.; Shi X. Org. Lett. 2013, 16, 306–309. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.