

Abstract
Previously, we developed a panel of nonpeptidic compounds specifically preventing fusion of the measles virus (MV) with target cells at IC50 values of 0.6–3 μM. Mutations in the MV fusion protein (MV F) that render resistance to these blockers were described. The structural basis for both inhibition and resistance was unclear in the earlier work because of the availability of a structural model for only the post-fusion conformation of MV F. We have now developed structural models for both pre- and post-fusion conformers of the latter protein trimer. The models allow investigation of the large-scale conformational changes occurring in the MV fusion machinery and, in conjunction with antisera binding studies, provide a rationale for how inhibitors may arrest a conformational intermediate by interfering with the formation of interactions between the heptad repeat B (HR-B) linker and DIII domains. The models also show that resistance to inhibition can be explained by a predicted destabilizing effect of the mutations on the HR-B domain within the trimeric pre-fusion structure. This viewpoint is supported by the temperature dependent differential fusion activities of MV F variants harboring these mutations.
The measles virus, a member of the paraymyxovirus family, is an important therapeutic target because it remains one of the top ten infectious disease killers in the world, mainly due to low vaccination rates in developing countries (1). We have approached the problem of identifying an inhibitor against measles virus by targeting a major characteristic of the virus: its capacity for fusion of the viral envelope with the target cell plasma membrane and thus entry into target cells at neutral pH. Measles accomplishes fusion by the cooperative action of two transmembrane envelope glycoproteins, the hemagluttinin and fusion proteins (2–6). Hemagluttinin recognizes the cell surface receptor, CD46 or SLAM/CD150W (7–9) depending on virus strain, and triggers the fusion protein in its metastable, pre-fusion state to undergo large-scale conformational changes, ultimately concluding in a stable post-fusion conformation and membrane merger (2–6).
The first atomic level structure of a paramyxovirus fusion protein was solved in 2003 by manipulating the Newcastle Disease virus (NDV) (10). Thereafter, we employed homology modeling to predict the structure of the measles fusion protein (MV F) (11). At the time, it was not clear whether the NDV fusion protein (NDV F) was crystallized in its pre-fusion or post-fusion state, although some evidence pointed toward the post-fusion form. The NDV F structure was later revealed as post-fusion by comparison to the x-ray crystal structure for the uncleaved ectodomain of human parainfluenza virus 3 fusion protein (hPIV3 F), which had crystallized with a six-helix bundle (12).
Comparative characterization of MV strains with different fusogenicity suggested that mutation of MV F residue Val94 modulates F fusogenicity (11, 13). The MV F homology model located this residue at the bottom of a hydrophobic pocket near the C-terminal end of HR-C. Mutatgenesis of Val94 and the surrounding residues supported the idea that this microdomain exerts a critical influence on fusion (11). Structural features of the pocket suggested complementary characteristics of potential small molecule ligands. Examples were designed and tested, triggering identification of the 50 μM lead fusion inhibitor OX-1 (14). Subsequently, a series of compounds based on the latter paved the way to AS-48, a compound with 600–3000 nM activity depending on the viral strain (15). The scaffold of AS-48 was modified extensively in efforts to achieve a more potent compound, yet only marginal gains in activity were achieved (16).
During the course of this work, we demonstrated that viral resistance to AS-48 and analogs can be achieved through mutations at position 462 (N → S, D, or K) in the six-helix bundle (6HB) of the then only available post-fusion conformation and at position 367 (A → T) in the DI domain forming the head of MV F (17). Peptide inhibition studies complemented by molecular dynamics calculations revealed that mutations at 462 most likely confer resistance by lowering the barrier to fusion activation, rather than by explicit binding or by increasing the stability of the post-fusion 6-helical bundle (17). Immunoprecipitation studies of F with soluble HR-B-derived peptides confirmed that the presence of AS-48 increases the precipitation efficiency by the peptides, indicating that the compound arrests F in a refolding-intermediate conformation sensitive to peptide binding (17, 18).
In the present study, we further examine the nature of the AS-48 docking site through competitive binding studies with defined antisera. In parallel, we have developed homology models for MV F based on the X-ray structures of the pre-fusion form of the parainfluenza virus 5 fusion protein (PIV5 F) more recently solved by Yin et al. (6) and the post-fusion form of the human parainfluenza virus 3 fusion protein (hPIV3 F) (12). Using these new models of pre- and post-fusion conformations, we have examined the structural changes that occur in MV F during fusion. In combination with the AS-48/antibody competitive binding study, we have used morphing simulations of these models to understand the development of the binding site for the AS-48 class of inhibitors during fusion and to rationalize how the compounds might arrest an intermediate structure of MV F. A structural explanation for the resistance-conferring mutations occurring at positions 462 and 367 in MV F is proposed and supported by the experimental fusion activities of MV F 462 variants at 37 and 30°C. Finally, we discuss these results in the context of our recent generation of an MV F mutant with reversible fusion activity through engineered disulfide bonds (19).
EXPERIMENTAL DETAILS
Cell culture, transfection and production of virus stocks
Vero cells (African green monkey kidney epithelial) (ATCC CCL-81) were maintained at 37°C and 5% CO2 in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum. 293-3-46 MV rescue cells (20) were incubated at every third passage in the additional presence of G-418 (Geneticin) at a concentration of 100 mg/ml. Lipofectamine 2000 (Invitrogen) was used for cell transfections. To prepare MV stocks, Vero cells were infected at a multiplicity of infection (MOI) of 0.001 plaque-forming units (pfu)/cell and incubated at 37ºC. Cells were scraped in OPTIMEM (Invitrogen), virus released by two freeze-thaw cycles, and titers determined by 50% tissue culture infective dose (TCID50) titration according to the Spearman-Karber method (21) as described (22). To prepare stocks of modified vaccinia virus Ankara expressing T7 polymerase (MVA-T7) (23) DF-1 cells (ATCC CRL-12203) were infected at an MOI of 1.0 pfu/cell and cell-associated viral particles harvested 40 hours post-infection.
Immunoprecipitation
Vero cells were transfected with 4 μg of plasmid DNA encoding MV F (pCG-F) (24). At 30 hours post-transfection, cells were washed with cold PBS and lysed with immunoprecipitation buffer (10mM Tris, pH 7.4, 150 mM NaCl, 1% deoxycholate, 1% Triton X-100, 0.1% sodium dodecyl sulfate (SDS), protease inhibitors (Roche) and 1 mM phenylmethylsulfonylfluoride (PMSF)) in the presence of 300 μM AS-48 (AS-48->Ab) or equal volumes of solvent (DMSO) as indicated. Cleared lysates (20,000 X g; 60 min; 4°C) were incubated with specific antisera directed against epitopes 88–100 or 240–259 in the F head domain at 4°C, followed by precipitation with immobilized protein G (Pierce) at 4°C. Where indicated (Ab->AS-48), AS-48 was added prior to precipitation at a final concentration of 300 μM. Precipitates were washed three times each in buffer A (100 mM Tris, pH 7.6, 500 mM lithium chloride, 0.1% Triton X-100), then buffer B (20 mM HEPES, pH 7.2, 2 mM EGTA, 10 mM magnesium chloride, 0.1% Triton X-100), followed by resuspension in urea buffer (200 mM Tris, pH 6.8, 8 M urea, 5% SDS, 0.1 mM EDTA, 0.03% bromphenolblue blue, 1.5% dithiothreitol). Denatured samples were fractionated on 10% SDS-polyacrylamide gels, blotted onto polyvinylidene difluoride membranes (Millipore), and subjected to chemiluminescence detection (Amersham Pharmacia Biotech) using antisera directed against an epitope in the cytosolic F tail. For comparison, the amount of F1 material present in cell lysates prior to precipitation was determined upon incubation in the presence of AS-48 or equal volumes of solvent (DMSO).
Site-directed mutagenesis
Mutant variants of MV F were generated in the pCG-F plasmid background through site-directed mutagenesis using the QuickChange mutagenesis system (Stratagene) and appropriate primers (sequences available upon request). Prior to further experimentation, all variants were confirmed by DNA sequencing and immunoblotting.
Recovery of recombinant virions
For generation of recombinant MV, NarI/PacI fragments of pCG-F variants harboring mutated F open reading frames were transfered into full-length cDNA copies of the MV genome. Virions were then recovered essentially as previously described (13, 20). Briefly, the helper cell line 293-3-46 stably expressing MV N, MV P, and T7 polymerase was transfected by calcium phosphate precipitation using the ProFection kit (Promega) with plasmids encoding a cDNA copy of the relevant MV genome and MV L polymerase. Helper cells were overlaid on Vero cells 48 hours post-transfection and resulting infectious centers were subsequently passaged on the same cell type. The integrity of recombinant virions was confirmed by RT-PCR and DNA sequencing of the modified area.
Quantitative cell-to-cell fusion assay
To quantify fusion activity, an effector Vero cell population (2 × 105 cells/well) was co-transfected with 2 μg each of H (MV hemagglutinin) and F expression plasmid and target Vero cells (2 × 105 cells/well) were transfected with 2 μg of the reporter plasmid encoding β-galactosidase under the control of the T7 promoter. Single-transfections of plasmid DNA encoding F served as controls. Two hours post-transfection, the effector cells were infected with MVA-T7 at an MOI of 1.0 pfu/cell and incubated in the presence of 400 μM fusion inhibitory peptide (FIP, Bachem) to prevent premature fusion of the effector cell monolayer. Following incubation for 16 hours at 30°C or 37°C as indicated, target cells were detached, washed, overlaid on the effector cells at a (1:1)-ratio and incubated at 30°C or 37°C as before. Seven hours post-overlay, cells were lysed and the β-Galactosidase activity assessed using the β-Galactosidase assay kit [Invitrogen] according to the manufacturer’s instructions. Relative background values provided by the controls were subtracted from all sample values, and the resulting measurements normalized against reference constructs upon incubation at 30°C or 37°C as specified. Averages of four experiments are shown.
Plaque assays
Vero cell monolayers (approximately 90% confluent) in 35 mm wells were infected with recombinant MV-Edm F-462K or unmodified recombinant MV-Edm for 60 minutes at 37°C, followed by removal of inoculum and incubation in the growth medium. Wells were subjected to fixation and crystalviolet-staining (0.1% crystalviolet in 20% ethanol, incubation for 2 hours at room temperature) 30 hours post-infection, followed by photodocumentation of dried plates.
Homology modeling
Sequence alignment of MV F to hPIV3 F and PIV5 F was accomplished with the ClustalW algorithm (25), using a gap open penalty of 10.0, a gap extension penalty of 0.1, and the Gonnet matrix. Sections of MV F corresponding to missing segments of the crystal structures were not modeled. This included the amino- and carboxy-termini from both hPIV3 F and PIV5 F (residues 1–26, 515–550 for the PIV5 F based structure and residues 1–26 and 488–550 for the hPIV3 F based structure). The hPIV3 F based structure also lacks a section from residues 100–144 corresponding to the fusion peptide and a portion of the HR-A domain. The homology models were constructed with the PRIME software (Schrödinger) (26). PRIME does not allow subunits to be built simultaneously, so each subunit was assembled and then merged into a complete trimer. The models were refined using PRIME’s side chain prediction protocol on all residues to generate the lowest energy rotamer for each sidechain. The quality of the structures was evaluated by means of WHAT IF structure validation checks (see below) (27).
Molecular dynamics
Solvated structures were generated using molecular dynamics (MD) with the GROMACS package (28, 29) and simulated using the OPLS-AA force field (30) and the TIP4P water model (31). Hydrated rectangular boxes with periodic boundaries were built around the two models with dimensions of 105nm by 105nm by 170nm and 90nm by 90nm by 180nm for the pre-fusion and post-fusion structures, respectively. A total of 63,000 and 49,000 explicit water molecules were simulated for the pre- and post-fusion structures, respectively. Chloride anions were randomly placed throughout the water to bring the total charge in each system to zero. Both hydrated complexes were subjected to 500 steps of steepest descent minimization using the above parameters to remove any high energy contacts between water and protein, and then simulated for 50 picoseconds of position restrained MD at 300K and 1.0 atm of pressure to allow the waters to soak into the structure. The resulting structures were manually analyzed to determine the number of waters in the cavities and channels.
Lipophilic Connolly surfaces
Lipophilicity-mapped Connolly surfaces were generated using Sybyl’s MOLCAD module (32). The area of hydrophobic surfaces was calculated using only the portion of the surface that showed hydrophobic properties. For these structures, a lower bound of −0.04715 for the lipophilic property (as suggested in the Tripos Bookshelf (33)) was used to exclude surface with more hydrophilic character, leaving surface contributions from only the hydrophobic atoms.
Morphing simulation
A morphing simulation of the DIII domain from the pre-fusion to the post-fusion conformation was accomplished by applying the multi-chain mode of the Yale Morph Server (http://molmovdb.org/morph). The morph was performed in 30 steps using CNS adiabatic mapping (34). The starting and ending structures were aligned by superimposing the backbone atoms of the DI and DII domains, then truncated to residues 47–99 and 194–288 of the DIII domain. Fusion peptide and HR-A residues encompassing residues 113–193 move dramatically from pre-fusion to post-fusion structure, and thus were not included to simplify analysis of the conformational changes in the Val94 microdomain. Using MAESTRO (Schrödinger) (35), AS-48 was manually docked into the Val94 microdomain at each of the 30 timesteps to estimate the point of fusion blockade; namely at which step the pocket opens up sufficiently to permit the ligand to bind without steric congestion.
RESULTS
Modeling the MV F protein trimer in the pre-fusion and post-fusion conformations
To investigate the structural changes that occur during fusion, a homology model of the pre-fusion form of MV F protein has been constructed using the X-ray coordinates reported for PIV5 F (Brookhaven Protein Data Bank access code 2B9B) (6), a paramyxovirus related to measles. The model is based on sequence alignment of F proteins derived from MV-Edmonston and PIV5-W3A strains using ClustalW (25) with a minor adjustment in the HR-C domain to prevent a gap in the helix. The proteins share 29% identity and 48% similarity, with all of the cysteine residues conserved. PRIME (26) was used to build the initial model, which was further refined with PRIME’s side chain prediction module. The same methods were employed to construct a new post-fusion model of MV-F based on the X-ray crystal structure coordinates of hPIV3 F, strain 47885 (Brookhaven Protein Data Bank access code 1ZTM) (12). The latter shares 26% identity and 51% similarity with all the cysteines conserved. The resulting models (Figure 1) score similar to the template structures in WHAT IF structure validation checks (27). The pre-fusion model has a Ramachandran z-score of −4.4 and a local amino acid environment z-score of −1.0, while the 2B9B structure scores −4.0 and −0.5, respectively. For the post-fusion model, the Ramachandran and local amino acid environment z-scores are −4.5 and −1.1, respectively. This compares with −4.6 and −1.0 for 1ZTM.
FIGURE 1.
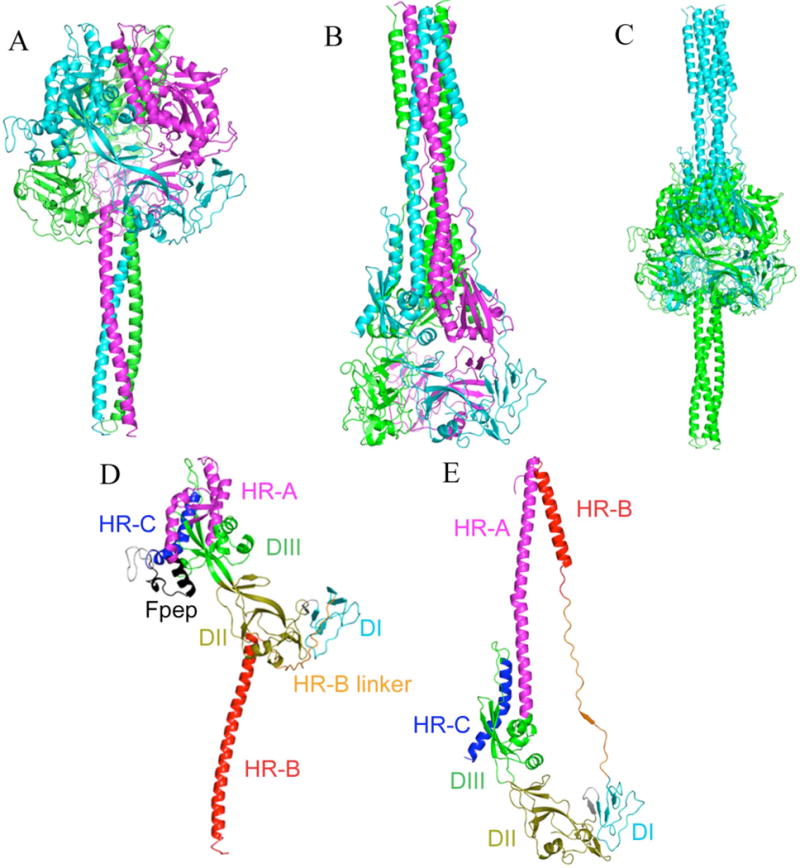
Secondary structure diagrams of MV F protein homology models highlight the domain movements occurring during fusion. Pre-fusion (A) and post-fusion (B) conformations are colored by subunit. Note the 3-helical bundle on the left and the 6-HB on the right. (C) Pre-fusion (green) and post-fusion (cyan) models superimposed by DI and DII domains. Overall backbone RMSD is 50Å. (D) Single subunit of pre-fusion model colored by domains. (E) Post-fusion model of the same subunit as (D), colored by domains, and similarly oriented by DI and DII domains.
Pre- and post-fusion MV F models imply large domain movements during fusion
As seen by comparison of the template structures of Yin et al. (6), the pre- and post- fusion models are trimeric structures in which most of the residues have moved large distances in the transition between the two conformations (Figure 1). The most highly conserved regions in the pre- and post- fusion models are the DI and DII domains (residues 27–46, 289–378; and 385–431, respectively). When DI and DII of the pre-and post-structures are superimposed, the resulting RMSD of the backbone atoms for these regions is 6.2 Å (Figure 1C), similar to the measurement by Yin et al. (6) comparing the DI and DII domains of PIV5 F and hPIV3 F. The RMSD for the entire structure (common residues 27–99 and 145–487), however, is 50 Å. The greatest movement occurs in the HR-A, HR-B, and HR-B linker domains (common residues 145–214 and 432–487) with a combined RMSD of 91 Å (Figure 1D, 1E). Moderate movements in the DIII domain with an RMSD of 13 Å are also evident even without including HR-A (common residues 47–99 and 215–288). These large residue displacements underscore that paramyxovirus fusion involves extraordinarily large and deep-seated rearrangements of protein structure (4–6).
The hydrophobic character of the HR-A and HR-B domains is consistent with the proposed model of fusion
In the pre-fusion structure, HR-B is predicted to form a 3-helical bundle by self-association instead of serving as the outer coil of the 6HB as in the post-fusion structure (Figure 1). Other class I viral fusion proteins such as HIV’s gp41 and influenza’s hemagglutinin also form 6HB structures post-fusion through a similar mechanism, but only in paramyxoviruses are the HR-B domains considered to form self-associated, extracellular 3HBs (4, 36). (Depending on the pathogen under investigation, the N-terminal heptad repeat is referred to as HR-A, HR-N, or HR-1. The C-terminal heptad repeat is referred to as HR-B, HR-C, or HR-2.) The HR-B 3-helical bundle in the pre-fusion model places the hydrophobic residues inward for inter-subunit contacts, while the hydrophilic residues face outward to solvent. However, in the post-fusion model, the hydrophobic residues of HR-B face a hydrophobic groove formed by two subunits of HR-A (Figure 2A–D). HR-B loses a turn in its helix in the transition from pre-fusion to post-fusion and is less hydrophobic overall than HR-A. HR-B (residues 456–487) possesses 534 Å2 and 477 Å2 of hydrophobic surface area in the pre- and post-fusion conformations respectively, while HR-A (residues 146–183) is measured at 1174 and 1271 Å2 of hydrophobic surface, respectively (Figure 2A–D). HR-A presents roughly twice the hydrophobic surface per residue than HR-B, and, in fact, increases its hydrophobic surface upon fusion to obtain favorable hydrophobic contacts with HR-B. HR-B’s lesser hydrophobic surface area relative to HR-A is advantageous for the fusion protein because the HR-B 3-helix bundle is considered to dissociate in order for the protein to achieve the post-fusion conformation.
FIGURE 2.
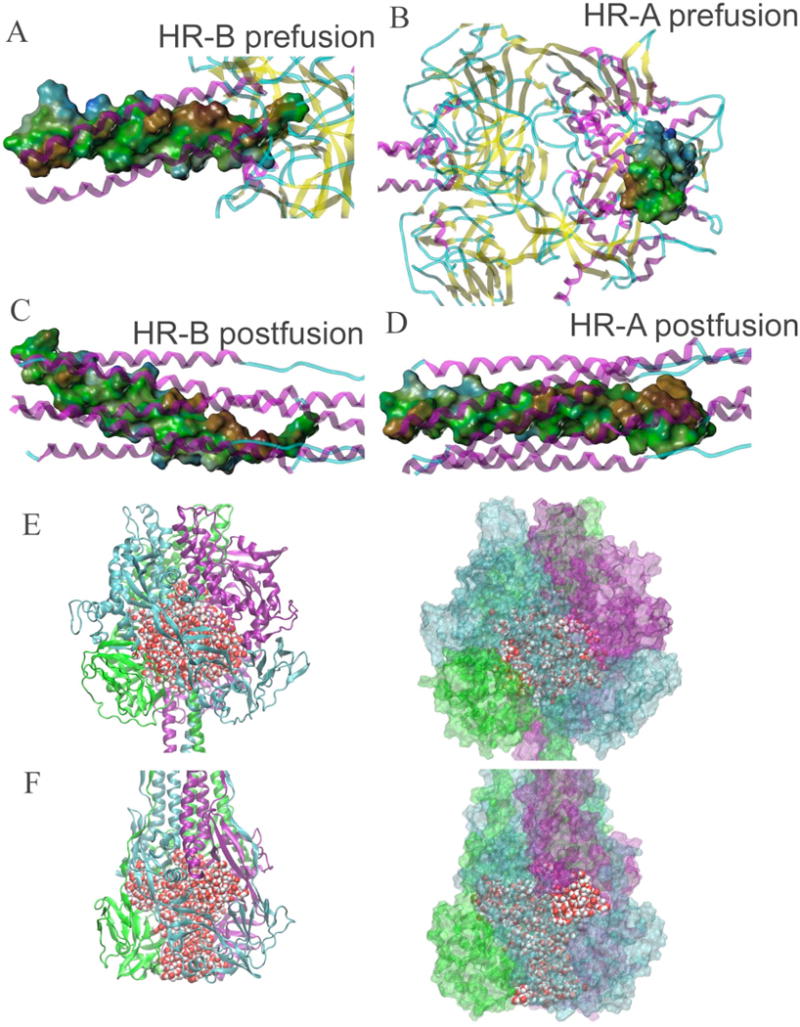
Potential energy sources in fusion refolding. (A–D) Hydrophobic surfaces of one HR-A and HR-B subunit in pre-fusion and post-fusion models (brown = hydrophobic, green = neutral, blue = hydrophilic). (E–F) Water-filled cavity and channels in pre-fusion and post-fusion models. Protein is shown as cartoon or surface, colored by subunit; water is shown as vdW spheres. (E) Pre-fusion model sustains a cavity with 550 water molecules held inside. Surface representation on the right shows that the cavity is almost completely sealed. (F) Post-fusion model has an axial channel and three radial channels filled with 400 water molecules. Surface representation shows the openings of the channels at the top and the sides.
The large, water-filled cavity in the pre-fusion protein is a metastable feature that provides the MV F with a source of potential energy
In the pre-fusion form, the MV F homology model incorporates a feature similar to that present in the X-ray structure of PIV5 F, namely a 35 Å diameter spherical cavity in the head of the protein (Figure 2E). Solvating this space using the GROMACS program placed approximately 550 water molecules in the cavity. In the post-fusion form, the latter is replaced by a quartet of channels (one axial channel, three radial channels), but only 400 water molecules are accommodated by them when solvated in silico (Figure 2F). The entropic cost of transferring a water molecule from liquid to a protein has been estimated to be between 0 and 7 cal mol−1 K−1, corresponding to a free energy cost between 0 and 2 kcal/mol at 300 K (37). Most of the water molecules in the prefusion solvent pool are loosely associated with the protein. It can be expected that individual water molecules will not contribute entropy penalties near the upper bound of 2 kcal/mol, but a sizeable entropic penalty can be associated with the total water pool. On the other hand, water molecules in an open channel (Figure 2F) are expected to experience a reduced entropic penalty relative to water sealed in a cavity. Another useful measure for describing this effect is the additional buried hydrophobic surface area between the pre-fusion and post-fusion forms. Because different fragments of the protein are missing in the two models, a direct comparison cannot be made. However, if the solvent accessible surface area between the common fragments (residues 27–99 and 144–487) are calculated, the post-fusion form buries 4850 Å2 of surface area that was exposed to solvent either in the cavity or on the exterior of the pre-fusion form of the protein. It is generally thought that buried surface area provides 25 cal mol−1 per Å2 of hydrophobic free energy gain at 300 K (38–40). This yields a rough estimate of 121,000 cal/mol or 121 kcal/mol of hydrophobic free energy gain from pre-fusion to post-fusion conformations. Non-hydrophobic energy changes are not addressed here; but the contribution of buried surface area to the relative stability of the post-fusion conformation does serve to illustrate the considerable potential energy stored in the pre-fusion conformation, particularly in the large cavity in the head of the trimer.
Microdomain around Val94 is rearranged and occluded in the pre-fusion model
The microdomain around Val94 has been shown to be important for fusion activity (11, 13). It is formed by contacts between the DIII and HR-C domains from one subunit and the DIII domain of a neighboring subunit. Previously, experiments have demonstrated deduced that mutations disturbing the hydrophobic base and a hydrophilic residue at the rim of the microdomain ablate fusion activity, but mutations to smaller residues in the base (Leu256Ala and Leu257Ala) can be compensated by the mutation to a larger residue at the opposite side of the base (Val94Met). The resulting triple mutant maintains all the fusion activity of wild-type MV F (11). These structural observations are based on the homology model of NDV F. By contrast, this microdomain in the pre-fusion form of MV F adopts an alternative spatial arrangement (Figure 3A–B). Thus, the Cα atoms of leucines 256 and 257 are separated from Val94 by 23 and 25 Å, respectively, while in the new post-fusion model, they are separated by only 7 and 10 Å, respectively. Accordingly, the pre-fusion model does not immediately suggest a structural explanation for how the Val94Met mutation compensates for the other mutations, restores proper folding into a transport-competent conformation, and maintains fusion activity. We cannot rule out the possibility that the model fails to place these residues qualitatively correctly or that long-range effects may play a role.
FIGURE 3.
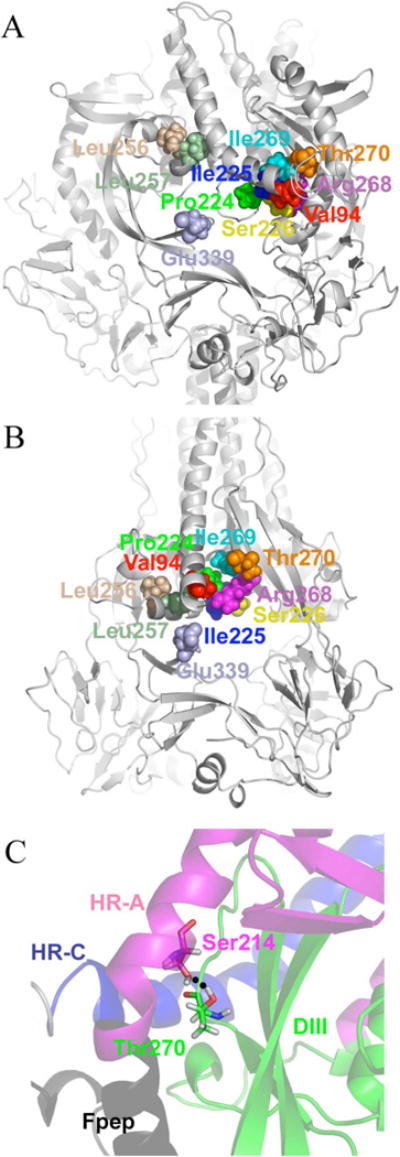
Changes in the Val94 microdomain occurring during fusion. Pre-fusion (A) and post-fusion (B) models are displayed with Val94 microdomain residues as vdW spheres with the protein shown as secondary structure. In the pre-fusion model the microdomain is covered by the fusion peptide and HR-A; structural rearrangements occur in the DIII and HR-C domains to bring the microdomain together. (C) Interaction between Thr270 and Ser144 (shown as sticks) in the pre-fusion model. This interaction appears to anchor HR-A and Fpep to the protein head prior to fusion.
Another possibility is propelled by an independent line of experiments, which show that AS-48 and OX-1 rescue intracellular transport competence and fusion activity in a dose dependent fashion from a Val94Gly/Asn462Ser double mutant that is normally not surface-expressed (17). The Asn462Ser mutation gives resistance to AS-48, but in combination with Val94Gly surface expression is eliminated in the absence of compound. Taking into account that AS-48 recognizes and stabilizes a fusion-intermediate conformation of F (17), this finding suggests that F Val94Gly/Asn462Ser spontaneously assumes an intermediate conformation with a well-formed AS-48 docking site. The latter, if stabilized by AS-48 from premature 6-HB formation, is still remarkably transport competent. Applied to the F 94M/256A/257A situation, by analogy this triple-mutant F variant may spontaneously assume an intermediate conformation that brings residues 94 and 256/257 in close proximity as proposed in our previous studies (11), is still transport-competent and, once matured and surface expressed, mediates fusion in combination with MV H protein. While our experimental data support the latter hypothesis, a definite conclusion is beyond the scope of static models and cannot be reached with the experimental tools currently available.
For mutations that maintain surface expression but lack fusion activity, our models do provide a structural basis. Such is the case for the Thr270Ala mutation, which has 100% surface expression compared to wild-type, but only 55% fusion activity (11). In the pre-fusion model, Thr270 of the DIII domain hydrogen bonds Ser144 of the HR-A domain directly adjacent to the fusion peptide, providing an important point of contact that anchors the fusion peptide to the head of the protein prior to fusion activation (Figure 3C). Mutating Thr270 to Ala removes this interaction and may cause the fusion peptide to dissociate from its interface with the DIII domain without proper activation. Such an event is expected to compel MV F to embark on the conformational pathway to a post-fusion structure without a cellular membrane to target. Non-productive, irreversible conformational changes like this would reduce the ability of the virus to enter cells since more of its fusion proteins are wasted in “misfires.” Mutation of Thr270 to Ser elicits no change in hydrogen bonding causing fusion activity to be maintained at 93% (11). Mutation to His, a larger, polar residue, lowers both surface expression (30%) and eliminates fusion activity. (11) In the pre-fusion model, it would appear that a larger side chain such as His may not be able to preserve the hydrogen bond without other adjustments to the local protein structure.
AS-48 and related entry inhibitors do not bind the Val94 microdomain in the pre-fusion form, but interact with an intermediate structure along the fusion conformational pathway. The structural rearrangements and occlusion of the Val94 microdomain in the pre-fusion form make it clear that AS-48 and related entry inhibitors do not prevent fusion by binding to the pre-fusion structure (Figures 3A–B). To further test whether AS-48 is binding at the Val94 site, we developed two polyclonal antibodies against synthetic peptides including HR-C residues 88–103 and DIII residues 240–259, respectively (Figure 4). Both peptides encompass residues that are near the Val94 microdomain (Figure 4B–C), but only the former peptide covers this residue. Both peptide domains are more exposed and closer to each other in post-fusion than pre-fusion (337 and 79 Å2 increased surface areas for residues 88–104 and 240–259, respectively). In immunoprecipitation experiments with AS-48, the order of addition greatly influences the amount of precipitated protein/antibody complex. Adding AS-48 before the antibody directed against the epitope for 88–103 essentially eliminates subsequent antibody binding. Adding the antiserum before AS-48 does not change the efficiency of precipitation. The antiserum directed against the 240–259 residues confirmed the specificity of these findings: adding AS-48 prior to this antiserum even increases the amount of precipitate, suggesting enhanced accessibility of the epitope. Adding antiserum before AS-48 again does not change the amount of precipitate. These results suggest that AS-48 blocks interaction of the 88–103 antibody to the Val94 microdomain; while with the 240–259 antibody, AS-48 prolongs exposure of this epitope along the fusion conformational pathway. Both results strongly suggest that AS-48 is indeed binding at this location, albeit to an undefined conformational state of the protein.
FIGURE 4.
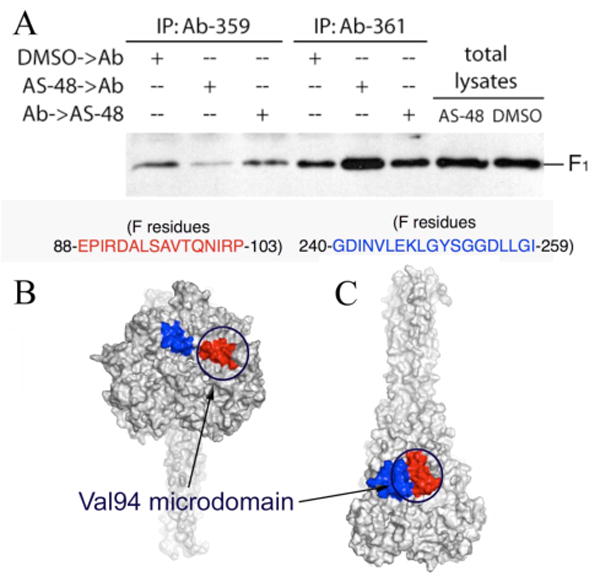
Co-immunoprecipitation experiments with AS-48 and two antibodies directed at epitopes in the Val94 microdomain. (A) Ab-359, directed against residues 88–104, and Ab-361, directed against residues 240–259 have opposing effects on the amount of complex precipitated when added prior to AS-48. When introduced after AS-48 addition, both antibodies precipitated amounts of complex similar to antibody-only conditions. Pre-fusion (B) and post-fusion (C) models are shown as surfaces with residues 88–104 and 240–259 colored red and blue, respectively. The location of the Val94 microdomain in both models is highlighted by circles. The epitopes are closer and considerably more exposed post-fusion (337 Å2 and 79 Å2 increased surface area for residues 88–104 and 240–259, respectively).
Previously, we deployed an epitope-tagged peptide from the HR-B domain of MV F to explore the conformational status of MV F in the presence of AS-48 (17). The compound did not prevent the HR-B peptide from binding to the HR-A coiled-coil, which would be expected if AS-48 acts through physical docking to HR-A. Rather, it increased the co-precipitation efficiency of the epitope-tagged HR-B peptide compared to solvent-only treated controls (17). These results infer that AS-48 arrests MV F in a conformational intermediate that has formed an extended HR-A 3HB, but has not yet formed the 6HB or completed the subsequent refolding process (Figure 5A–C). To visualize the formation of the Val94 microdomain, a morph of the DIII domain from the pre-fusion to the post-fusion structure was carried out (Figure 5D, Movie S1). The binding pocket does not form until late in the simulation. Based on manual docking of AS-48 to the intermediates from the morph, we propose that AS-48 binds an intermediate resembling frame 25 (Figure 5D), and thus prevents fusion by interfering with the final conformational rearrangements necessary to bring the HR-B domains in proximity to the HR-A 3HB. HR-B linker residues 434–441 form beta sheet-like contacts with the defined DIII beta sheet containing residues 48–57. Conceivably, AS-48 could prevent the beta sheet from moving into its final position by disrupting interactions with the HR-B linker and interfering with the placement of HR-B in the 6HB. The 240–259 peptide antibody target exposes 1044 Å2 surface area to solvent in the intermediate predicted to bind AS-48 (Figure 5D, frame 25), as compared with its surface in the post-fusion model (943 Å2). If AS-48 inhibits this intermediate, a prolonged interaction of this epitope with its antibody could occur, resulting in additional protein/antibody precipitate. This is exactly the result of the aforementioned immunoprecipitation experiment when AS-48 is added before the antiserum directed against 240–259 epitope. The Val94 microdomain is expected to be almost fully formed in the intermediate structure, providing the binding site that matches the structure-activity relationships derived for analogs of the compound (16) and the experimental results obtained from F mutants exposed to AS-48.
FIGURE 5.
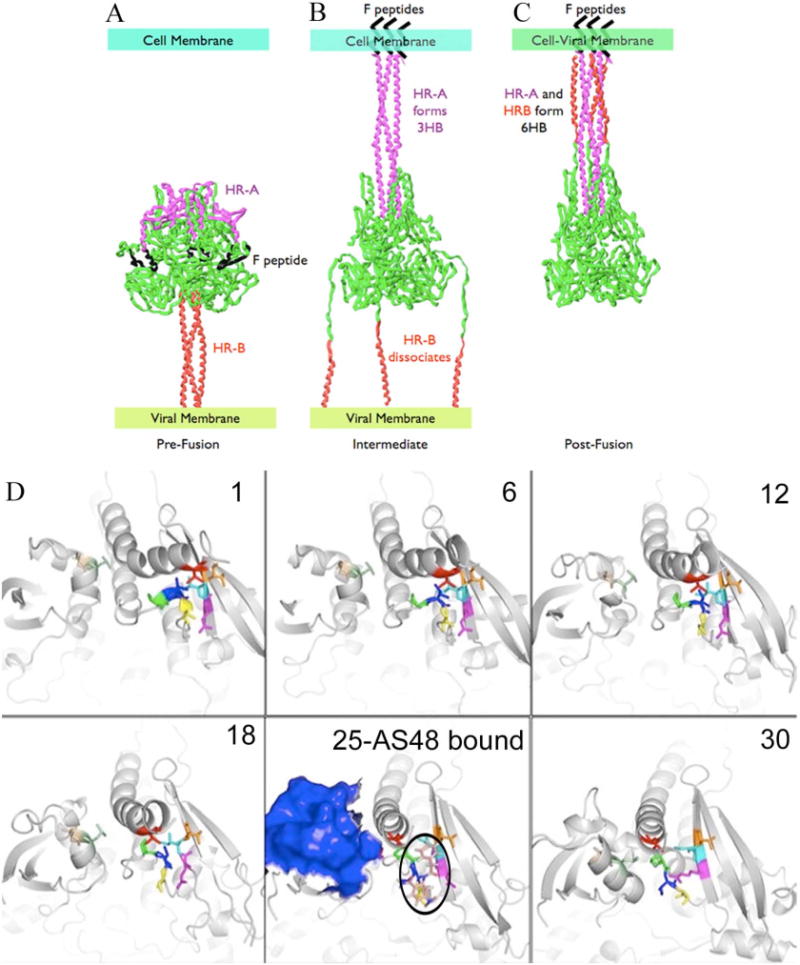
Proposed fusion pathways for the entire MV F (A–C, similar to pathway by Yin et al. (6)) and the DIII domains (D). (A) Prior to fusion, MV F exists in a metastable state with water-filled cavity, HR-B in a 3HB, and the fusion peptide and HR-A wrapped around the head of the protein. (B) Activation causes fusion peptides to bury within the cell membrane, formation of the HR-A 3HB, and dissociation of HR-B. (C) Final post-fusion structure with completely collapsed water-filled cavity and fully formed HR-A and HR-B 6HB. (D) Results of morphing simulation of the DIII domain, shown in 6 of the 30 frames. Residue coloring is the same as Figure 3A–B. AS-48 (shown as sticks, colored with pink carbons and outlined in black) is docked into frame 25 and is proposed to inhibit fusion by disrupting interactions between DIII and the HR-B linker. In this frame, the 240–259 epitope (shown as blue surface) is fully exposed for interaction with the Ab-361 antibody.
MV F residues 462 and 367 are located at the critical interface between the HR-B and DI domains in the pre-fusion conformation of F
To better understand the role of residues 462 and 367 in conferring resistance, we examined the status of these residues in our pre-fusion and post-fusion models. In pre-fusion MV F, Asn462 is found near the beginning of HR-B at the intersection of HR-A and DI domains (Figure 6A). However, in post-fusion MV F, Asn462 is located on HR-B on the outer coil of the six-helix bundle formed by HR-A and HR-B (Figure 6C). In both structures, Asn462 is facing ~90° away from the center of the helical bundle, but in the post-fusion form, it is predicted to hydrogen bond with Glu170 of HR-A found in another subunit. In the pre-fusion form it hydrogen bonds to an adjacent residue (Gly460) on the same subunit. The second mutation, Ala367, is part of the DI domain and is near Asn462 in the pre-fusion form (Cα to Cα distance of 12 Å), but very distant in the post-fusion form (Cα to Cα distance of 114 Å). Ala367 goes from a nonpolar environment in the pre-fusion form to a relatively solvated form in the post-fusion structure. The Asn462 and Ala367 positions in the pre-fusion conformation suggest that mutations here may be conferring resistance by destabilizing a network of non-covalent interactions between the top of the HR-B domains and the base of the pre-fusion F head that must be disengaged for HR-B 3HB to dissociate (vide infra) and form the intermediate structure (Figure 6B). Further experiments described below suggest that destabilization of these interactions could accelerate the fusion kinetics, negating the inhibitory effect of AS-48.
FIGURE 6.
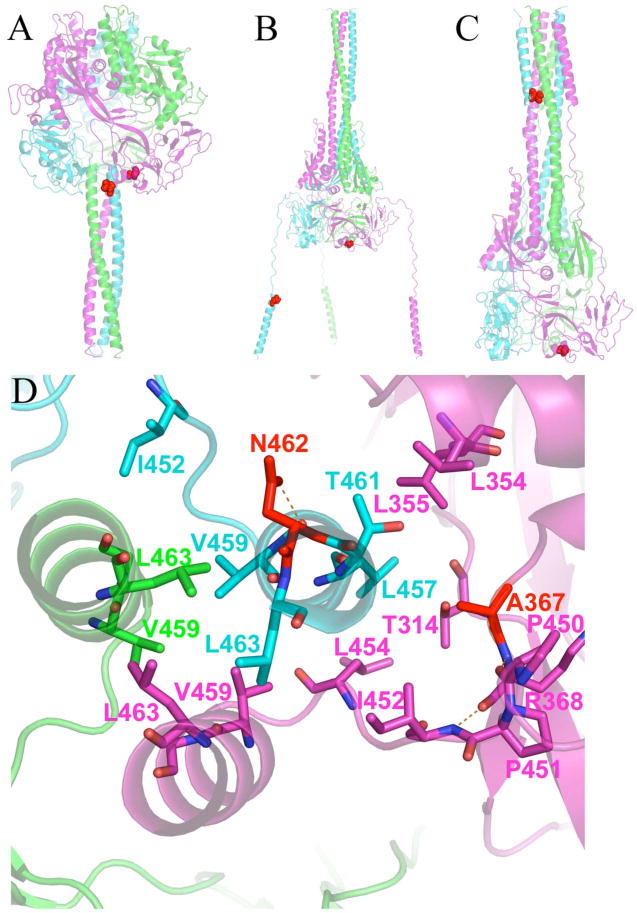
Position of resistance-conferring mutations at Asn462 and Ala367. (A) Pre-fusion model shows that these residues are proximal and part of the key interactions holding HR-B to the head of the protein prior to fusion. (B) Proposed intermediate structure revealing that Asn462 and Ala367 must separate to achieve the conformational intermediate. (C) Post-fusion model showing that Asn462 ends up in the 6HB while Ala367 remains in the DI domain of the MV F head. (D) View of the HR-B/HR-B linker interface with the head of MV F in the pre-fusion model. Residues providing key hydrophobic interactions are shown as sticks. Eight hydrophobic residues provide interaction of the HR-B/HR-B linker to the head. Five of the residues (Leu457, Leu 454, Ile452, Pro451 and Pro450) are from this network. The other three hydrophobic residues (Leu448, Ile446, and Val432, not shown) are located upstream on the HR-B linker.
Mutations at MV F residues 462 and 367 disrupt the hydrophobic interactions holding the HR-B and HR-linker to the DI domain
The models suggest that HR-B and the HR-B linkers are only loosely bound to the DI domain in the head of pre-fusion MV F. Most of the interaction between the HR-B/HR-B linker domains and the rest of the protein occurs at the base of the head where HR-B meets the DI domain. The mutations that confer resistance to AS-48 are located in this critical region (Figure 6D). Ala367 makes up part of the hydrophobic network that holds these three domains (HR-B, HR-B linker, and DI) together. This residue, along with Thr314, Leu354, and Leu355 are part of the DI domain residues in this network. HR-B’s portion of the network is formed by Leu457 and Thr461, while Leu454 and Ile452 form the HR-B linker’s portion of the network. Of the eight hydrophobic residues of HR-B and the HR-B linker forming contacts with the head of the protein, five of these residues (Leu457, Leu454, Ile452, Pro451, and Pro450) are from this network. The remaining three hydrophobic residues (Leu448, Ile446, and Val432; not shown) are dispersed along the length of the HR-B linker. These observations suggest that this concentrated hydrophobic region is critical for holding the HR-B/HR-B linker domains in the pre-fusion conformation. Mutations to more polar or smaller residues in this region will disrupt the hydrophobic network and most likely decrease the barrier for dissociation of HR-B/HR-B linker domains from the head of the fusion protein. Such mutations are seen in AS-48 resistant mutants: Ser, Asp, or Lys replace Asn at position 462, and Thr replaces Ala at position 367, implying that the virus overcomes inhibition by AS-48 through conversion of F to a less stable, constitutively more active variant. To investigate the effect of these changes, we have mutated the corresponding residues in the pre-fusion model and found their lowest energy rotamer using PRIME’s side-chain prediction protocol (Figure 7). In the resulting structures, we find that the Asp and Lys mutations at position 462 introduce a charged species into this hydrophobic network (Figure 7B and 7C), increasing the attraction for water molecules which can break up these interactions. Ser is not more polar than Asn462 (Figure 7A), but when mutated in the pre-fusion model, the smaller residue increases the exposure to solvent (Figure 7D). The Thr mutation at position 367 introduces a polar hydroxyl group into this deeply hydrophobic area (Figure 7E, 7F), likely inducing the same kind of effect as the Asn462 mutations.
FIGURE 7.
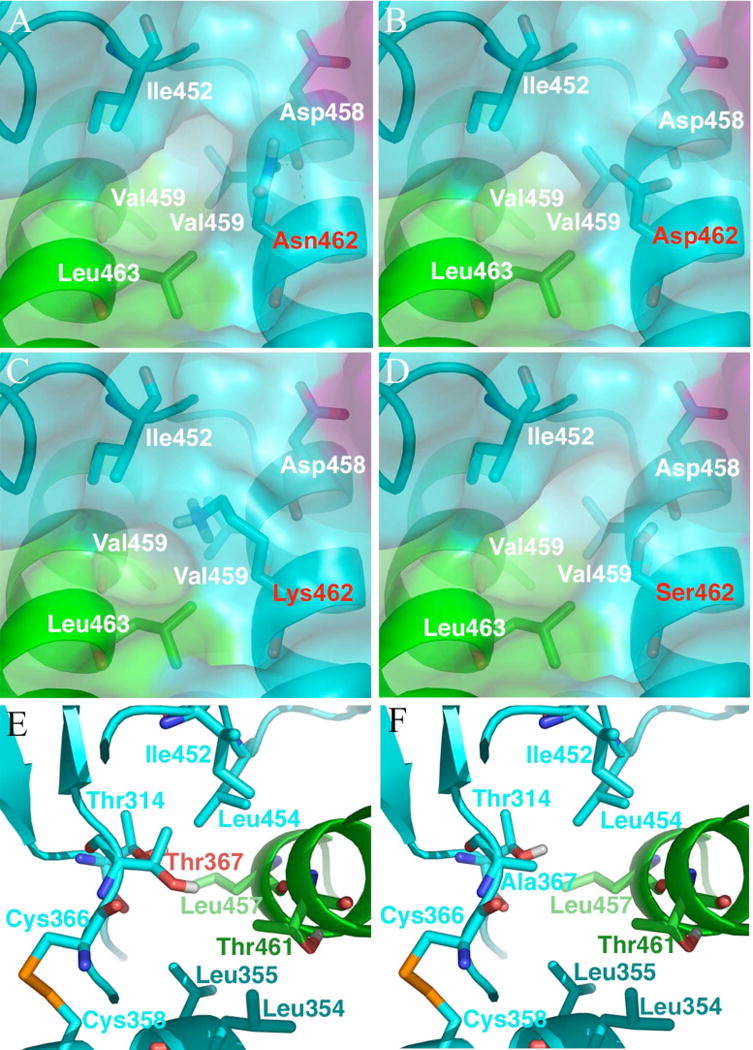
Predicted structures for resistance-conferring mutations. (A–D) Asn462 mutations at the interface of HR-B and DI domains in the pre-fusion model. The protein is shown as secondary structure with its Connolly surface. Selected residues are shown as sticks. In wild-type (A), Asn462 hydrogen bonds the backbone oxygen of Asp458. The Asn462 →Asp mutant (B) introduces a negative charge into this critical hydrophobic environment. The Asn462 → Lys mutant (C) introduces a positive charge. The Asn462 → Ser mutant (D) does not introduce a charge, but as a smaller residue, it increases the solvent exposure of the hydrophobic interactions compared to wild-type. (E–F) Ala367 mutations at the interface of HR-B and DI domains in the pre-fusion model. The protein is shown as secondary structure. Selected residues are shown as sticks. In wild-type (E), Ala367 is part of the network of hydrophobic interactions. The Ala367 → Thr mutant (F) introduces a polar hydroxyl directly into the hydrophobic network.
Mutations at position 462 enhance fusion activity at lower temperatures
Fusion activities of the MV F mutants N462S and N462K upon co-expression with MV H were tested at both 30 and 37°C (Figure 8A). Fusogenicity remains unchanged in unmodified MV F at both temperatures. However, in both mutant constructs, the fusogenicity is markedly increased at 30°C. Even at 37°C, the N462K variant experiences more fusion activity than unmodified MV F at the same temperature. Consistent with this, lateral spread of a recombinant MV harboring F N462K is increased through the cell monolayer (Figure 8B). The N462K variant is increased within the cell monolayer. Such results are consistent with a decrease in the dissociation barrier between the HR-B/HR-B linker domains and the head of the fusion protein, assuming this barrier plays a deciding role in fusion activation. For PIV5 F, Russell et al. have suggested this region to act as a “conformational switch” based on mutations of PIV5 F residues L447 and I449 (MV F equivalent residues L457 and V459, Figure 6D). Thus, aliphatic mutations attenuated fusion activity, but aromatic residues promoted hyperactive fusion (41). A similar effect is implicated in some HIV-1 resistance mechanisms in response to enfuvirtide (42). Accordingly, it appears that mutations promoting hyperactive fusion accelerate fusion kinetics, narrowing the time window for productive AS-48 docking and leading to resistance in MV F.
FIGURE 8.
(A) MV F variants N462S and N462K show enhanced fusion activity at 30°C indicating reduced conformational stability. The 462K variant shows increased fusogenicity also at physiological temperature. Quantification of fusion activity of F variants after co-transfection of Vero cells with equal amounts of plasmid DNA encoding MV H and F, and incubation at 30°C or 37°C as indicated. The values represent means of four experiments and are expressed as the percentage of fusion activity observed for unmodified MV F after incubation at 30°C or 37°C, respectively. (B) In the context of infection with recombinant MV, the F-462K variant confers increased lateral spread through the target cell monolayer. Infected cells were subjected to crystal violet-staining 30 hours post-infection to visualize virus-induced syncytia.
DISCUSSION
Despite extensive structural modification applied to the AS-48 molecular scaffold, the analogous compounds were never able to break the ~1μM inhibition barrier. Prior to the development of the pre-fusion model, it was difficult to justify the lack of increased activity for all of the analogs produced. In light of the pre-fusion structure, it is now possible to understand that the specific conformation of MV F that AS-48 and its related inhibitors is able to target (Figures 5D and 9) is only a brief intermediate within the conformational cascade of the protein. Binding to the post-fusion structure would be non-inhibitory, as the protein has already brought the viral and cell membranes together. Previous work has shown that AS-48 increases the binding of an HR-B based peptide (17). This suggests that the intermediate protein conformation that AS-48 targets is a late stage intermediate in which the HR-A 3HB has formed and the postulated Val94 binding site has been exposed in contrast to being shielded by the HR-A residues. (Figure 3) Based on the morphing simulation of the DIII domain, we suspect that AS-48 disrupts interactions of the HR-B linker necessary to position HR-B near HR-A, interrupting the formation of the 6HB; that is, not by inducing a conformational change in the protein, but by increasing the energy barrier from an intermediate structure to the post-fusion structure. Targeting an intermediate viral protein structure has been successful in some cases, for example the peptide HIV entry inhibitor enfuvirtide (43), which achieves an EC50 in the low nanomolar range, but at the expense of a 36-residue peptide which requires subcutaneous injection. Enfuvirtide also benefits from the relatively long lifetime (several minutes) of the HIV-1 gp41 extended intermediate (44). Other fusion protein intermediates may only exist for a few seconds (36). For structure-based ligand design, lack of detailed structural information for the intermediate poses a significant challenge.
FIGURE 9.
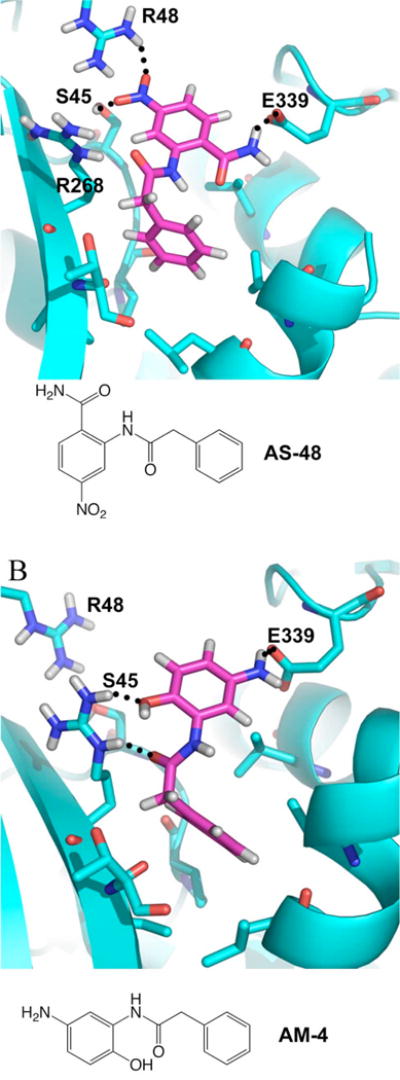
(A) Predicted binding model for AS-48, in the post-fusion model. This binding pocket is not available in the pre-fusion model (vida Figures 3 and 5). (B) Predicted binding model for AM-4, also in the post fusion model.
AM-4 has shown the greatest degree of inhibition of all entry inhibitors related to AS-48, but is unstable at physiological conditions as this compound is essentially a masked quinone which degrades upon facile air oxidation to uncharacterized products (15). Its high level of inhibition can likely be ascribed to the reactivity of its resulting quinone as a Michael acceptor in the binding site, perhaps subject to nucleophilic attack by the hydroxyl of Ser45 (Figure 9), although no attempt was made to determine the labeled residue. Cysteine labeling seems unlikely as all extracellular cysteines are predicted to form disulfide bonds. However, with reactivity comes instability. AM-4 loses all inhibitory activity in 16 hours, corresponding to the loss of NMR spectrum integrity and precipitation in deuterated chloroform (15). Although irreversible binding by AM-4 may result in increased inhibition, such covalent interaction with proteins are undesirable for a compound projected as a possible drug candidate, even if the compound were shelf stable.
Previously, disulfide bonds were modeled into MV F and introduced experimentally by site-directed mutagenesis of residues 452 (on the HR-B linker) and 460 (on HR-B) to cysteines (19). Engineered disulfide bonds have the opposite effect of the acquired mutations occurring at this site: fusion activity is eliminated, but can be restored by reduction of the disulfide bonds to the free cysteines (19). These residues were selected based on the resulting geometry when these residues were mutated to cysteines in the model and subjected to a short molecular dynamics and minimization protocol. (Figure 10) The mutated protein was synthesized and surface-expressed to a degree similar to that for unmodified protein, only as disulfide-linked trimers without fusion activity. Fusion activity can be partially restored by subjecting the protein to reducing conditions, however, indicating that the protein has a native fold that is not interrupted by the introduction of the cysteines.
FIGURE 10.
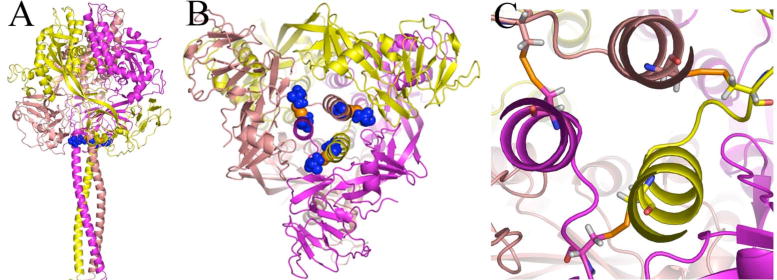
Predicted geometry of the intersubunit disulfide bond between positions 452 and 460 in the pre-fusion MV F model. The protein is shown as secondary structure and colored by subunit. Cysteines at positions 452 and 460 are shown as blue spheres with orange sulfur atoms (A) Side view of the trimer (B) View looking up from the 3HB of HR-B. (C) Close-up view of disulfide bonds with the cysteines shown as sticks
In addition to probing the molecular mechanism of fusion-protein reorganization and its suppression by small molecular weight inhibitors, modeling the MV F structures also provides a predictive tool to guide the planning and interpretation of future experiments. Given the importance of the Asn462 and Ala367 residues and the effect of the engineered disulfides, we propose that a structure-based drug design approach targeting this site may lead to a new class of inhibitors that could stabilize the interface between HR-B/HR-B linker and DI/DII domains. Failing that, small molecule binding to other sites on the pre-fusion structure could interfere with activation by hemagglutinin or other early conformational transitions. Recent work from our group has shown that residues near either side of the fusion cleavage site, as well as a residue in the hairpin turn of HR-A, are functionally involved in activation by hemagglutinin (45). Either site could potentially be exploited by a structure-based design approach using the pre-fusion model, targeting the more abundant form of the protein, instead of a transient intermediate.
Supplementary Material
Acknowledgments
We are grateful to Dennis C. Liotta for helpful discussion and generous support. This work was supported by a research grant from the American Lung Association and Public Health Service grant AI071002 from NIH/NIAID.
ABBREVIATIONS
- MV
measles virus
- F
fusion protein
- 6HB
six helix bundle
- 3HB
three helix bundle
- HR
heptad repeat
- MD
molecular dynamics
- NDV
Newcastle disease virus
- PIV5
parainfluenza virus 5
- hPIV3
human parainfluenza virus 3
- H
hemagglutinin
Footnotes
This work was supported by a research grant from the American Lung Association and Public Health Service grant AI071002 (to RKP) from NIH/NIAID.
Supporting Information Available. MOVIE S1. Movie from results of the DIII domain morphing simulation. Each frame corresponds to a frame of the simulation. The protein is shown as a secondary structure diagram with residues from Figure 5D shown as sticks. Subunits are colored by trimer as in Figure 1. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.World Health Organization. World Health Report, 2002. 2004. Annex 2. [Google Scholar]
- 2.Colman PM, Lawrence MC. The structural biology of type I viral membrane fusion. Nat Rev Mol Cell Biol. 2003;4:309–319. doi: 10.1038/nrm1076. [DOI] [PubMed] [Google Scholar]
- 3.Kielian M, Jungerwirth S. Mechanisms of enveloped virus entry into cells. Mol Biol Med. 1990;7:17–31. [PubMed] [Google Scholar]
- 4.Lamb RA, Jardetzky TS. Structural basis of viral invasion: lessons from paramyxovirus F. Curr Opin Struct Biol. 2007;17:427–436. doi: 10.1016/j.sbi.2007.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lamb RA, Paterson RG, Jardetzky TS. Paramyxovirus membrane fusion: Lessons from the F and HN atomic structures. Virology. 2006;344:30–37. doi: 10.1016/j.virol.2005.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yin HS, Wen X, Paterson RG, Lamb RA, Jardetzky TS. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature. 2006;439:38–44. doi: 10.1038/nature04322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dörig RE, Marcil A, Chopra A, Richardson CD. The human CD46 molecule is a receptor for measles virus (Edmonston strain) Cell. 1993;75:295–305. doi: 10.1016/0092-8674(93)80071-l. [DOI] [PubMed] [Google Scholar]
- 8.Manchester M, Eto DS, Valsamakis A, Liton PB, Fernandez-Muñoz R, Rota PA, Bellini WJ, Forthal DN, Oldstone MB. Clinical isolates of measles virus use CD46 as a cellular receptor. J Virol. 2000;74:3967–3974. doi: 10.1128/jvi.74.9.3967-3974.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Naniche D, Varior-Krishnan G, Cervoni F, Wild TF, Rossi B, Rabourdin-Combe C, Gerlier D. Human membrane cofactor protein (CD46) acts as a cellular receptor for measles virus. J Virol. 1993;67:6025–6032. doi: 10.1128/jvi.67.10.6025-6032.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen L, Gorman JJ, McKimm-Breschkin J, Lawrence LJ, Tulloch PA, Smith BJ, Colman PM, Lawrence MC. The structure of the fusion glycoprotein of Newcastle disease virus suggests a novel paradigm for the molecular mechanism of membrane fusion. Structure. 2001;9:255–266. doi: 10.1016/s0969-2126(01)00581-0. [DOI] [PubMed] [Google Scholar]
- 11.Plemper RK, Lakdawala AS, Gernert KM, Snyder JP, Compans RW. Structural Features of Paramyxovirus F Protein Required for Fusion Initiation. Biochemistry. 2003;42:6645–6655. doi: 10.1021/bi034385k. [DOI] [PubMed] [Google Scholar]
- 12.Yin HS, Paterson RG, Wen XL, Lamb RA, Jardetzky TS. Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc Natl Acad Sci U S A. 2005;102:9288–9293. doi: 10.1073/pnas.0503989102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Plemper RK, Compans RW. Mutations in the putative HR-C region of the measles virus F-2 glycoprotein modulate syncytium formation. J Virol. 2003;77:4181–4190. doi: 10.1128/JVI.77.7.4181-4190.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Plemper RK, Erlandson KJ, Lakdawala AS, Sun A, Prussia A, Boonsombat J, Aki-Sener E, Yalcin I, Yildiz I, Temiz-Arpaci O, Tekiner B, Liotta DC, Snyder JP, Compans RW. A target site for template-based design of measles virus entry inhibitors. Proc Natl Acad Sci U S A. 2004;101:5628–5633. doi: 10.1073/pnas.0308520101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Plemper RK, Doyle J, Sun AM, Prussia A, Cheng LT, Rota PA, Liotta DC, Snyder JP, Compans RW. Design of a small-molecule entry inhibitor with activity against primary measles virus strains. Antimicrob Agents Chemother. 2005;49:3755–3761. doi: 10.1128/AAC.49.9.3755-3761.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun AM, Prussia A, Zhan WQ, Murray EE, Doyle J, Cheng LT, Yoon JJ, Radchenko EV, Palyulin VA, Compans RW, Liotta DC, Plemper RK, Snyder JP. Nonpeptide inhibitors of measles virus entry. J Med Chem. 2006;49:5080–5092. doi: 10.1021/jm0602559. [DOI] [PubMed] [Google Scholar]
- 17.Doyle J, Prussia A, White LK, Sun AM, Liotta DC, Snyder JP, Compans RW, Plemper RK. Two domains that control prefusion stability and transport competence of the measles virus fusion protein. J Virol. 2006;80:1524–1536. doi: 10.1128/JVI.80.3.1524-1536.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Russell CJ, Jardetzky TS, Lamb RA. Membrane fusion machines of paramyxoviruses: capture of intermediates of fusion. EMBO J. 2001;20:4024–4034. doi: 10.1093/emboj/20.15.4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee JK, Prussia A, Snyder JP, Plemper RK. Reversible inhibition of the fusion activity of measles virus F protein by an engineered intersubunit disulfide bridge. J Virol. 2007;81:8821–8826. doi: 10.1128/JVI.00754-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Radecke F, Spielhofer P, Schneider H, Kaelin K, Huber M, Dötsch C, Christiansen G, Billeter MA. Rescue of measles viruses from cloned DNA. Embo J. 1995;14:5773–5784. doi: 10.1002/j.1460-2075.1995.tb00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spearman C. The method of right and wrong cases (constant stimuli) without Gauss’s formula. Br J Psychol. 1908;2:227–242. [Google Scholar]
- 22.Plemper RK, Hammond AL, Gerlier D, Fielding AK, Cattaneo R. Strength of envelope protein interaction modulates cytopathicity of measles virus. J Virol. 2002;76:051–5061. doi: 10.1128/JVI.76.10.5051-5061.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sutter G, Ohlmann M, Erfle V. Non-replicating vaccinia vector efficiently expresses bacteriophage T7 RNA polymerase. FEBS Lett. 1995;371:9–12. doi: 10.1016/0014-5793(95)00843-x. [DOI] [PubMed] [Google Scholar]
- 24.Cathomen T, Naim HY, Cattaneo R. Measles viruses with altered envelope protein cytoplasmic tails gain cell fusion competence. J Virol. 1998;72:1224–1234. doi: 10.1128/jvi.72.2.1224-1234.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31:3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.PRIME, version 1.5. Schrödinger, LLC; New York, NY: 2005. [Google Scholar]
- 27.Vriend G. What If – A Molecular Modeling And Drug Design Program. J Mol Graph. 1990;8:52–56. doi: 10.1016/0263-7855(90)80070-v. [DOI] [PubMed] [Google Scholar]
- 28.Berendsen HJC, Vanderspoel D, Vandrunen R. GROMACS: a message-passing parallel molecular-dynamics implementation. Comput Phys Commun. 1995;91:43–56. [Google Scholar]
- 29.Lindahl E, Hess B, van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J Mol Model. 2001;7:306–317. [Google Scholar]
- 30.Jorgensen WL, Tiradorives J. The OPLS potential functions for proteins – energy minimizations for crystals of cyclic-peptides and crambin. J Am Chem Soc. 1988;110:1657–1666. doi: 10.1021/ja00214a001. [DOI] [PubMed] [Google Scholar]
- 31.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 32.Sybyl 7.2. Tripos Inc; St Louis, MI: 2007. [Google Scholar]
- 33.Tripos Bookshelf 7.3. Tripos Inc; St Louis, MI: 2007. [Google Scholar]
- 34.Flores S, Echols N, Milburn D, Hespenheide B, Keating K, Lu J, Wells S, Yu EZ, Thorpe M, Gerstein M. The database of macromolecular motions: new features added at the decade mark. Nucleic Acids Res. 2006;34:D296–D301. doi: 10.1093/nar/gkj046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.MAESTRO, version 8.5. Schrödinger, LLC; New York, NY: 2008. [Google Scholar]
- 36.Harrison S. Viral membrane fusion. Nat Struct Mol Biol. 2008;15:690–698. doi: 10.1038/nsmb.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dunitz JD. The entropic cost of bound water in crystals and biomolecules. Science. 1994;264:670–670. doi: 10.1126/science.264.5159.670. [DOI] [PubMed] [Google Scholar]
- 38.Chothia C. Hydrophobic bonding and accessible surface-area in proteins. Nature. 1974;248:338–339. doi: 10.1038/248338a0. [DOI] [PubMed] [Google Scholar]
- 39.Chothia C. Principles that determine the structure of proteins. Annu Rev Biochem. 1984;53:537–572. doi: 10.1146/annurev.bi.53.070184.002541. [DOI] [PubMed] [Google Scholar]
- 40.Chothia C, Janin J. Principles of protein-protein recognition. Nature. 1975;256:705–708. doi: 10.1038/256705a0. [DOI] [PubMed] [Google Scholar]
- 41.Russell CJ, Kantor KL, Jardetzky TS, Lamb RA. A dual-functional paramyxovirus F protein regulatory switch segment: activation and membrane fusion. J Cell Biol. 2003;163:363–374. doi: 10.1083/jcb.200305130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reeves JD, Lee FH, Miamidian JL, Jabara CB, Juntilla MM, Doms RW. Enfuvirtide resistance mutations: impact on human immunodeficiency virus envelope function, entry inhibitor sensitivity, and virus neutralization. J Virol. 2005;79:4991–4999. doi: 10.1128/JVI.79.8.4991-4999.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kilby JM, Lalezari JP, Eron JJ, Carlson M, Cohen C, Arduino RC, Goodgame JC, Gallant JE, Volberding P, Murphy RL, Valentine F, Saag MS, Nelson EL, Sista PR, Dusek A. The safety, plasma pharmacokinetics, and antiviral activity of subcutaneous enfuvirtide (T-20), a peptide inhibitor of gp41-mediated virus fusion, in HIV-infected adults. AIDS Res Hum Retroviruses. 2002;18:685–693. doi: 10.1089/088922202760072294. [DOI] [PubMed] [Google Scholar]
- 44.Munoz-Barroso I, Durell S, Sakaguchi K, Appella E, Blumenthal R. Dilation of the human immunodeficiency virus-1 envelope glycoprotein fusion pore revealed by the inhibitory action of a synthetic peptide from gp41. J Cell Biol. 1998;140:315–323. doi: 10.1083/jcb.140.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee JK, Prussia A, Paal T, White LK, Snyder JP, Plemper RK. Functional interaction between paramyxovirus fusion and attachment proteins. J Biol Chem. 2008;283:16561–16572. doi: 10.1074/jbc.M801018200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.