

Abstract
Urban coasts receive watershed drainage from ecosystems that include highly developed lands with sewer and stormwater infrastructure. In these complex ecosystems, coastal waters are often contaminated with fecal pollution, where multiple delivery mechanisms that often contain multiple fecal sources make it difficult to mitigate the pollution. Here, we exploit bacterial community sequencing of the V6 and V6V4 hypervariable regions of the bacterial 16S rRNA gene to identify bacterial distributions that signal the presence of sewer, fecal, and human fecal pollution. The sequences classified to three sewer infrastructure-associated bacterial genera, Acinetobacter, Arcobacter, and Trichococcus, and five fecal-associated bacterial families, Bacteroidaceae, Porphyromonadaceae, Clostridiaceae, Lachnospiraceae, and Ruminococcaceae, served as signatures of sewer and fecal contamination, respectively. The human fecal signature was determined with the Bayesian source estimation program SourceTracker, which we applied to a set of 40 sewage influent samples collected in Milwaukee, WI, USA to identify operational taxonomic units (≥97 % identity) that were most likely of human fecal origin. During periods of dry weather, the magnitudes of all three signatures were relatively low in Milwaukee's urban rivers and harbor and nearly zero in Lake Michigan. However, the relative contribution of the sewer and fecal signature frequently increased to >2 % of the measured surface water communities following sewer overflows. Also during combined sewer overflows, the ratio of the human fecal pollution signature to the fecal pollution signature in surface waters was generally close to that of sewage, but this ratio decreased dramatically during dry weather and rain events, suggesting that nonhuman fecal pollution was the dominant source during these weather-driven scenarios. The qPCR detection of two human fecal indicators, human Bacteroides and Lachno2, confirmed the urban fecal footprint in this ecosystem extends to at least 8 km offshore.
Introduction
Human activity exerts a significant impact on coastal ecosystems. Since 1970, the number of people in the USA living in coastal watershed counties has increased by 50.9 million, so that now approximately 52 % of the population resides in close proximity to coastal waters [1]. The ecosystem services provided by coasts, which include the aesthetic, health, social, and economic benefits of recreational areas, are among the most visible and easily disrupted by anthropogenic pollutants. Untreated sewage poses one of the greatest of these pollution concerns. Each year, more than four trillion liters of untreated sewage enter US waterways [2], and this number does not reflect the contribution from numerous but less conspicuous routes produced by urban environments (e.g., stormwater drainage, city runoff, leaking sewer pipes). Supporting this notion, other studies have concluded that increases in the density and land coverage of urbanized areas led to increased fecal pollution in waterways [3–5].
Untreated sewage presents several challenges to coastal ecosystem health including high nutrient loads [6, 7], chemicals and pharmaceuticals [8, 9], personal care products [9, 10], and fecal waste [2, 11]. Among these pollutants, fecal waste presents the most acute risk to human health. Fecal waste generally harbors enteric pathogens in addition to agents that cause skin, eye, ear, and respiratory illnesses [12–15]. The type of pathogens present depends upon the host source of the waste [16]. In urban environments, both combined and separated sanitary sewer overflows [2] and the release of stormwater contaminated with sanitary sewage [17, 18] serve as common delivery routes of fecal waste to waterways.
Conventionally, the cultivation of enterococci or Escherichia coli cells from environmental samples has been used to indicate the presence of fecal contamination [19]. In ecosystems containing numerous modes of fecal contamination, these culture-based methods cannot discriminate among sources. Without source identification, it is often difficult to assess the ambient human health risks or make decisions about the necessity or direction of efforts to mitigate the pollution. More recently, alternative fecal indicator assays using molecular methods have targeted organisms thought to be abundant in fecal waste, but specific to a particular host animal (e.g., Bacteroidales, Bifidobacterium, Methanobrevibacterium, Lachnospiraceae; see [16] and citations therein). These methods have proven useful, but are limited to providing information about an a priori targeted source [16, 20–23].
Recently, we [22, 24] and others [25, 26] have suggested that profiling the microbial community composition with next generation sequencing might have the capacity to identify complex mixtures of fecal pollution sources in contaminated waters. The microbial community sequence distribution in feces or composite fecal samples, as is found in sewage, could act as a unique signature that identifies a particular fecal source. In theory, profiling multiple markers as a signature instead of a single marker could provide the needed specificity to identify multiple fecal sources in complex environmental samples.
The Great Lakes, as a region that contains more than 500 public beaches, serves as a source of drinking water to more than 40 million people, and supports more than $1 billion in recreational and commercial fishing ventures [27], is particularly sensitive to fecal pollution impacts. Our laboratory previously demonstrated that molecular markers have greater sensitivity for detecting fecal pollution than conventional culture-based methods [28] in this ecosystem and identified chronic human fecal contamination in the urban waterways leading to Lake Michigan [22]. However, we do not know which fecal sources contribute to the ongoing fecal pollution issues in these coastal waterways or whether these sources differ during different weather-driven watershed scenarios. In this study, we explore using microbial community sequencing methods to identify signatures of sewage and fecal pollution. We then track these signatures in the coastal waters of Lake Michigan near Milwaukee, WI, USA, during a variety of weather scenarios and examine the extent of the fecal bacterial footprint imposed by this urban area.
Materials and Methods
Sample Collection and DNA Extraction
We collected 40 wastewater treatment plant influent samples from one of two facilities, South Shore (250 million gallons per day [MGD] maximum flow) or Jones Island (300 million MGD maximum flow) in Milwaukee, WI, USA. Samples were collected during 2005, 2007–2009, and 2011 and represented all seasons (see Supplementary Table 1 for details). Samples were collected and filtered, and the DNA extracted as described in [24].
We collected 114 surface water samples on various dates from 2006 to 2011 from 20 stations in Milwaukee's rivers, harbor, and Lake Michigan (primary stations illustrated in Fig. 1; see Supplementary Table 1 for sample details). For each sample, we collected surface water into a 2- or 4-l bottle. All samples were stored on ice, returned to the laboratory within 4 h, and filtered as described previously [22]. Supplementary Table 1 reports sample dates and weather conditions during collection. Additionally, we collected water samples from four Milwaukee county stormwater outfalls and filtered 200 ml from each onto a 0.22-μm pore size mixed cellulose esters filter (47 mm diameter; Millipore, Billerica, MA, USA). Filters were folded and placed in 2-ml screw cap tubes and then stored at −80 °C until further processing. All filters were stored for less than 3 years prior to DNA extraction.
Fig. 1. Map of Milwaukee, WI, USA waterways and nearshore Lake Michigan. Points indicate common sampling locations used in this study.

A uniform procedure was used to extract DNA from all water samples. We removed the frozen sample from the freezer and immediately crushed the filter with a sterile spatula. We then added the frozen filter pieces to a tube containing a bead-beating matrix and buffer according to the standard protocol for the Fast DNA spin kit for soil (MP Biomedicals, Solon, OH, USA). DNA extraction commenced according to the manufacturers' instructions. All samples for pyrosequencing underwent an additional DNA purification step using the MO BIO PowerClean DNA cleanup kit (MO BIO Laboratories Inc., Carlsbad, CA, USA).
All samples from the coastal waterways were classified by the location of collection and environmental conditions present during sampling. River samples were collected from the Milwaukee, Menomonee, and Kinnickinnic rivers. Harbor samples include the following sample sites: Junction, Pierheads, Off JI, Main Gap, North Gap, and South Gap (Fig. 1). Lake samples include Atwater, Linwood, Bradford, ½ Linwood, McKinley ½ Green Can, Green Can, 0.5 Mile, 2 Mile, 3.5 Mile, and 5 Mile. No rain/dry weather samples and rain samples were those collected after a 48-h rainfall total of <1.2 cm and ≥2.5 prior to collection. Combined sewer overflow (CSO) samples were those samples collected during or directly following combined or sanitary sewer overflows in the Milwaukee county wastewater treatment system.
454 Pyrosequencing of Bacterial 16S rRNA Genes
In total, the bacterial communities from 97 water samples, including rivers, harbor, Lake Michigan, stormwater, and sewage influent, were characterized with pyrosequencing. Of these samples, 76 were sequenced by amplifying the V6 hypervariable region of the 16S rRNA gene from bacteria using a mixture of five fused primers at the 5′ end of the region (E. coli positions 967–985) and four primers at the 3′ end (E. coli positions 1046–1064) according to the procedures previously described by McLellan and coauthors [24]. Amplicons were prepared and sequenced using a Roche genome sequencer GS-FLX and then trimmed, quality-controlled, and aligned as described previously ([24]; see Supplementary Table 1 for sample details). Operational taxonomic units (OTUs) were created for the V6 dataset using ≥97 % identity groupings according to the single linkage preclustering methods described by [29]. Pyrosequencing profiles from a subset of these samples have been reported previously [24, 30].
For the remaining 21 samples, we constructed amplicon libraries for the V6 through V4 hypervariable domains (amplification in reverse DNA strand orientation). To set up the amplification procedure, amplification fusion primers contained either the A or B 454 Titanium amplicon adapter for GS-FLX sequencing (Roche Diagnostics; Indianapolis, IN, USA) followed by a 5-nt multiplex identifier (MID) and ending with a 16S-specific sequence. The 16S sequences used were 518 F, 5′CCAGCAGCYGCGGTAAN and 1064R, 5′CGACRRCCATGCANCACCT. The PCR mixture contained 1X Platinum HiFi Taq polymerase buffer, 1.6 units Platinum HiFi polymerase (Life Technologies, Carlsbad, CA, USA),3.7 mM MgSO4, 200 μM dNTPs (PurePeak polymerization mix, ThermoFisher, E. Providence, RI, USA), and 50 nM combined primers. Five to 25 ng of sample DNA was added to the PCR master mix to a final volume of 100 μl, and this was divided into three replicate 33-μl reactions. We included a no-template negative control for each MID. PCR conditions included an initial denaturation at 94 °C for 3 min; 30 cycles of 94 °C for 30 s, 60 °C for 45 s, and 72 °C for 1 min; and a final extension at 72 °C for 2 min using an Applied Biosystems 2720 or 9700 cycler (Life Technologies). We cleaned the reaction and removed products under 300 bp using Ampure beads at 0.75× volume (Beckman Coulter, Brea, CA, USA). The final products were resuspended in 100 μl of 10mM Tris-EDTA. The emulsion PCR, enrichment, and sequencing were done according to current Roche Titanium amplicon sequencing protocols (Lib-A emPCR reagents, XLR sequencing reagents, two region PicoTitre plate). Image processing and signal calling were done using the Roche amplicon-processing pipeline (version 2.53) with recursive phase correction algorithm to maximize the number of long reads.
Following sequencing, we quality-filtered the V6V4 reads by removing reads without exact matches to the 1064R sequence and the MID, reads containing ambiguous bases, and reads that lacked the conserved sequence 5′-TGGGCGTAAAG-3′ (position 565 F in E. coli) allowing two mismatches, or that had a quality score <30. Sequence reads were trimmed at the same evolutionarily homologous position proximal to the 565 F conserved sequence. UChime [31] using both a de novo and reference database (ChimerSlayer GOLD) eliminated chimeras. The Global Alignment for Sequence Taxonomy (GAST, [32]) assigned taxonomy. Unlike the V6 datasets, we used only taxonomic assignments produced by GAST to group data from the V6V4 datasets, so no OTUs were constructed for these data. All data, V6 and V6V4, were uploaded to the Visualization and Analysis of Microbial Population Structures website (http://vamps.mbl.edu). See Supplementary Table 1 for sample details.
Fecal Indicator Enumeration
Standard U.S. Environmental Protection Agency methods were used to enumerate enterococci (MEI medium) and E. coli (modified mTEC medium) in surface water samples [33, 34]. The volume of water filtered for each sample varied depending upon the expected level of contamination, but was generally 100 ml for Lake Michigan samples and 10–100 ml for the harbor and river samples.
Quantitative PCR Analyses
An ABI StepOne real-time PCR system with TaqMan hydrolysis probe chemistry was used to run qPCR assays for human Bacteroides [35] with the HF183 forward primer and conditions as described in [22] and the Lachno2 human marker [22]. Both assays detect bacteria primarily associated with human fecal waste. The human Bacteroides reaction targets bacterial taxa in the genus Bacteroides, while Lachno2 targets bacterial taxa in the genus Blautia. The qPCR setup and cycling conditions followed that of [22]. We report all qPCR data as copies per volume of sample water. These calculated values took into account the original water volume sampled, the resulting volume present following the DNA extraction, the volume of extracted DNA entering the qPCR reaction, and the relationship of the qPCR standard curve to the fluorescence product of the qPCR amplification in each well.
Identifying Sewer and Fecal Signatures
In this study, we define a “signature” for bacterial communities. The signature has two components: the relative magnitude of all sequence reads associated with the signature group and the distribution or profile of these sequence reads within the signature group. We report the signature magnitude as a relative ratio, calculated as the sum of the total sequence reads assigned to the signature divided by the total bacterial reads obtained in the sample. We report the signature profile as a distribution of sequence reads (presence or relative abundance) among sequence-based groups, created using taxonomic classification or sequence identity. Three bacterial genera, Acinetobacter, Arcobacter, and Trichococcus, represented a “sewer signature.” All sequence reads assigned to one of these groups contributed to the sewer signature observed. Likewise, five bacterial families, Bacteroidaceae, Porphyromonadaceae, Clostridiaceae, Lachnospiraceae, and Ruminococcaceae, represented a “fecal signature.” These families were chosen because of their common presence in fecal samples from many host animals [16, 36]. In addition to taxonomic classification, we also analyzed the fecal signature with a more refined resolution by using OTUs (≥97 % identity), where all OTUs classified within the aforementioned taxonomic groups comprised the signature.
We used SourceTracker [37], a program that employs a Bayesian approach, to calculate the probability that an OTU present in one community (the sink) is from another community (the source) to identify OTUs as being of human fecal origin (a human fecal signature). Human fecal sample V6 16S rRNA gene community data were collected from [38]and[39] and were processed in the same manner as all community sequencing datasets presented in this study. To set up SourceTracker [37], we considered the human fecal samples from [38] and [39] to be the source communities and in separate runs the sewage samples and then the harbor samples to be the sink communities (see Supplementary Table 1 for sink samples). We considered all OTUs that were identified with ≥10 % probability of being from the human fecal community as being part of the human fecal signature. A low probability of occurrence was selected because of the high variation in the human fecal sample communities [40, 41], which is one of the drivers for the Bayesian probability predictions. OTUs considered of human fecal origin in either the sewage or harbor communities or both communities contributed to the final human fecal signature community. In total, we identified 99 OTUs of human fecal origin. Of these 99 OTUs, 65 mapped to the five fecal signature bacterial families. To remain consistent with our fecal signature, these 65 OTUs comprised the human fecal signature examined in this study.
Statistical Analyses and Data Visualization
We used nonmetric multidimensional scaling (NMDS) to visualize the differences and similarities in the fecal signature (presence–absence only) among samples. NMDS calculations employed the R statistics suite of programs [42] using the metaMDS function in the vegan package [43]. We calculated Bray–Curtis similarities for the fecal signature among samples using the vegdist function in the vegan package [43]as input for the NMDS. Analysis of similarity (ANOSIM) tested the differences between a priori assigned groups (water samples collected during a CSO vs. samples collected during a dry weather period). The nonparametric ANOSIM technique allows statistical comparisons for multivariate datasets. All ANOSIMs were run with 1,000 permutations in R in the vegan package with the anosim function [43].
Heatmaps created in the gplots package with the heatmap.2 function [44] described the fecal signature of several location–environment sample groups. The heatmap represents the relative abundance of each OTU within the fecal signature as a color schematic calculated as the total sequences attributed to that OTU divided by the total sequences in the sample.
A human fecal signal, calculated as the sum of the total copies per volume of the amplified products from the human Bacteroides and Lachno2 qPCR assays, was mapped onto the corresponding sample collection station in Milwaukee's urban waterways and nearshore Lake Michigan. These data were then interpolated between sample points and smoothed for visualization using the smooth.ppp function in the R Spatstat package [45]. The statistical calculations for the Pearson's correlation coefficient and Student's t test (assuming equal variance between groups) were calculated in a standard manner using Microsoft® Excel.
Results
Defining Bacterial Communities as a Signature
Sequencing bacterial communities provides both the distribution of taxa and/or sequence types for groups of interest (the signature profile) and the relative abundance of these groups (the signature magnitude), which taken together (the signature) can be used to identify pollution sources. We defined three source signatures in this study: (1) a sewer signature, (2) a fecal signature, and (3) a human fecal signature. To define a sewer signature, we obtained sewage influent samples on more than 20 occasions spanning 5 years from two wastewater treatment facilities that service Milwaukee, WI, USA and used 454 pyrosequencing to characterize the bacterial community in each sample. The total number of sequences obtained per sample ranged from 16,931 to 41,531. Among these 40 samples, three bacterial genera, Acinetobacter, Arcobacter, and Trichococcus, consistently made up a large proportion of the community (20–50 %; Fig. 2a). Although these genera were very abundant in the sewage samples, they were not prevalent in human fecal samples (Fig. 2b); therefore, they were chosen to represent a sewer signature. Sequences classified to the three genera were recovered only 33 times out of the >1.2 million sequences (0.0036 %) present in the human fecal dataset.
Fig. 2.

Bar plots of the relative abundance of sequences recovered and assigned to the sewer-associated bacterial genera (sewer signature): Acinetobacter, Arcobacter, and Trichococcus (left); the fecal-associated bacterial families (fecal signature): Bacteroidaceae, Porphyromonadaceae, Clostridiaceae, Lachnospiraceae, and Ruminococcaceae (center); and the bacteria of the human fecal signature (n=65 OTUs, ≥97 % identity) from: a 40 sewage influent samples collected from influent at the South Shore and Jones Island Milwaukee wastewater treatment plants. Note: for the human fecal signature the V6V4 sequenced samples (38–40) are not included and b human fecal samples collected by [38] representing samples 1–15 and by [39], representing samples 16–48. The left axis corresponds the left plot and the right axis corresponds to the center and right plots
We chose the taxa/sequences assigned to five bacterial families, Bacteroidaceae, Porphyromonadaceae, Clostridiaceae, Lachnospiraceae, and Ruminococcaceae, to represent a fecal signature. These five families are prevalent in the feces of many animals [16, 19, 25, 36].Accordingly, these five taxa made up on average 85.3 % of the bacterial sequences present in the human fecal samples (Fig. 2a). The sequences from these five families also consistently occurred in sewage influent, typically representing 10–20 % of the total sequences in a sample (Fig. 2a). Together, the sewer and fecal signatures comprised 29–57 % of all sequences present in the sewage influent samples.
The human fecal signature (defined in the “Materials and Methods”) was generally present at ≤10 % of the total bacterial sequences in sewage influent, but on average represented 55 % of the fecal signature (Fig. 2). Among the human fecal samples, the human fecal signature generally represented 40–60 % of all bacterial sequences recovered (Fig. 2).
Tracking a Sewer and Fecal Signature in Coastal Waters
We tracked the relative magnitude of the sewer and fecal signatures in transects from Milwaukee's urban waterways to coastal Lake Michigan during three separate environmental scenarios: a CSO, a sewage blending event, and following at least 4 days of very little precipitation. During the CSO, we observed a strong sewer and fecal signature in nearshore waters, but they diminished as distance from the shore increased (Fig. 3a). We also observed this trend during a sewage blending event, but the magnitude of the signature was lower than in the CSO (Fig. 3b). In contrast, the magnitude of both the sewer and fecal signatures was low (<0.5 % of total sequences) during the dry weather transect (Fig. 3c). Likewise, in a series of samples collected across multiple dry weather and CSO events, the magnitude of the sewer and fecal signatures was regularly >1 % of the community during CSOs but never >0.4 % during dry weather periods (Table 1). Despite the magnitude of the sewer signature being much larger than the fecal signature in sewage influent (Fig. 2a), the fecal signature was often greater than the sewer signature in the surface water samples. Except during CSOs, the fecal and sewer signatures appeared rarely in the Lake Michigan samples but occurred consistently in the harbor and river samples (Table 1).
Fig. 3.

Bar plots of the relative abundance of sequences recovered and assigned to the sewer-associated bacterial genera (sewer signature), Acinetobacter, Arcobacter, and Trichococcus, and the fecal-associated bacterial families (fecal signature), Bacteroidaceae, Porphyromonadaceae, Clostridiaceae, Lachnospiraceae, and Ruminococcaceae from a a sampling transect collected during a combined sewer overflow on June 9, 2008, b a sampling transect collected during a wastewater treatment sewage blending event on June 23, 2010, and c a sampling transect collected following a 4-day period without rain. Samples for each plot include surface water collected at: 1 Junction, 2 Main Gap, 3 2 Mile, 4 3.5 Mile [in river plume], 5 3.5 Mile [out of river plume], 6 5 Mile, 7 Kinnickinnic River, 8 Menomonee River, 9 Milwaukee River, 10 McKinley, 11 Bradford, and 12 Linwood. See Fig. 1 for sample map and Supplementary Table 1 for more detailed sample descriptions
Table 1. Magnitude of sewer and fecal signatures.
| Location | Sample type | Environmental conditions | Sample date | % Sewer signaturea | No. of fecal signature OTUs identified | % Fecal signaturea | No. of human fecal signature OTUs identified | % Human fecal signature in fecal signatureb | Enterococci CFUs (per 100 ml) | E. coli CFUs (per 100 ml) |
|---|---|---|---|---|---|---|---|---|---|---|
| Junction | Harbor | CSO | April 03, 2007 | 0.2 | 127 | 0.8 | 34 | 35.3 | 11,250 | 38,000 |
| Junction | Harbor | CSO | April 11,2008 | 6.7 | 316 | 4.6 | 26 | 2.7 | 1,860 | 1,680 |
| Junction | Harbor | CSO | June 09, 2008 | 0.7 | 226 | 0.6 | 53 | 37.4 | 4,000 | 6,100 |
| Main Gap | Harbor | CSO | June 09, 2008 | 2.5 | 186 | 1.4 | 56 | 37.6 | 2,955 | 4,100 |
| Junction | Harbor | CSO | June 11, 2008 | 0.9 | 219 | 0.5 | 19 | 1.6 | 500 | 430 |
| Pierheads | Harbor | CSO | April 27, 2009 | 1.3 | 258 | 2.1 | 42 | 8.4 | 1,350 | 3,700 |
| Pierheads | Harbor | CSO | April 28, 2009 | 0.7 | 279 | 1.1 | 21 | 1.4 | 450 | 590 |
| Pierheads | Harbor | CSO | June 19, 2009 | 1.0 | 414 | 2.3 | 45 | 3.5 | 18,200 | 5,400 |
| Pierheadsc | Harbor | CSO | June 19, 2009 | 1.3 | 337 | 1.5 | 39 | 3.2 | 15,400 | 6,600 |
| Pierheads | Harbor | CSO | June 20, 2009 | 2.1 | 301 | 1.0 | 47 | 5.9 | 18,700 | 22,900 |
| Pierheads | Harbor | CSO | June 22, 2009 | 1.1 | 256 | 0.6 | 22 | 2.1 | 2,300 | 3,100 |
| Main Gap | Harbor | Dry | June 12, 2006 | 0.1 | 22 | 0.1 | 1 | 0.4 | 0 | 4 |
| Pierheads | Harbor | Dry | June 24, 2008 | 0.2 | 114 | 0.2 | 2 | 0.7 | 59 | 110 |
| Pierheads | Harbor | Dry | July 01, 2008 | 0.1 | 159 | 0.4 | 4 | 0.8 | 24 | 69 |
| Junction | Harbor | Rain | June 19, 2006 | 0.2 | 62 | 0.1 | 2 | 1.2 | 910 | 7,210 |
| Main Gap | Harbor | Rain | June 19, 2006 | 0.0 | 8 | 0.0 | 0 | 0.0 | 283 | NA |
| Pierheads | Harbor | Rain | July 08, 2008 | 0.3 | 119 | 0.2 | 2 | 0.3 | 47 | 2,610 |
| Pierheads | Harbor | Rain | October 23, 2009 | 0.3 | 297 | 1.0 | 38 | 2.0 | 930 | 240 |
| Pierheads | Harbor | Rain | October 24, 2009 | 0.1 | 147 | 0.3 | 6 | 0.6 | 940 | 750 |
| Pierheadsc | Harbor | Rain | October 24, 2009 | 0.2 | 206 | 0.5 | 16 | 0.6 | 7,000 | 1,960 |
| 0.5 Mile | Lake | CSO | April 03, 2007 | 0.1 | 117 | 0.6 | 26 | 15.5 | 145 | 1,190 |
| 2 Mile | Lake | CSO | June 09, 2008 | 1.2 | 131 | 2.4 | 46 | 31.0 | 1,220 | 1,340 |
| 3.5 Mile in plume | Lake | CSO | June 09, 2008 | 0.7 | 81 | 0.4 | 35 | 27.1 | 595 | 880 |
| 3.5 Mile out plume | Lake | CSO | June 09, 2008 | 0.1 | 26 | 0.1 | 11 | 8.9 | 0 | 2 |
| 5 Mile | Lake | CSO | June 09, 2008 | 0.2 | 24 | 0.1 | 9 | 7.6 | 0 | 0 |
| Gareen Can | Lake | Dry | June 12, 2006 | 0.2 | 18 | 0.2 | 0 | 0.0 | 0 | 0 |
| Linwood | Lake | Dry | June 12, 2006 | 0.1 | 11 | 0.0 | 1 | 0.4 | 0 | 0 |
| Linwood | Lake | Dry | August 07, 2006 | 0.0 | 21 | 0.1 | 1 | 0.8 | NA | 1 |
| Gareen Can | Lake | Rain | June 19, 2006 | 0.0 | 8 | 0.0 | 0 | 0.0 | 0 | 3 |
| Linwood | Lake | Rain | June 19, 2006 | 0.0 | 10 | 0.0 | 0 | 0.0 | 1 | 2 |
| Kinnickinnic River | River | CSO | April 03, 2007 | 0.4 | 194 | 0.5 | 46 | 38.5 | 600 | 1,100 |
| Menomenee River | River | CSO | April 03, 2007 | 1.3 | 237 | 2.6 | 57 | 50.6 | NA | 2,100 |
| Milwaukee River | River | CSO | April 03, 2007 | 0.1 | 147 | 1.1 | 31 | 32.4 | NA | 100 |
| Milwaukee River | River | Rain | October 23, 2009 | 0.7 | 319 | 0.7 | 36 | 6.9 | 57,000 | 14,800 |
| Stormwater outfall | Stormwater | NA | May 08, 2008 | 2.1 | 85 | 0.2 | 2 | 0.2 | 8,100 | 5,200 |
| Stormwater outfall | Stormwater | NA | May 30, 2008 | 6.9 | 51 | 0.6 | 1 | 12.2 | 16,300 | 1,120 |
| Stormwater outfall | Stormwater | NA | July 03, 2008 | 9.0 | 99 | 0.5 | 3 | 0.6 | 49,000 | 11,900 |
| Stormwater outfall | Stormwater | NA | July 03, 2008 | 10.0 | 160 | 5.6 | 7 | 1.5 | 121,000 | 390,000 |
| Sewage average | Sewage | NA | NA | 29.2 ± 6.7d | 432 ± 99d | 13.2 ± 4.8d | 63 ± ld | 54.9 ± 12.3d | NA | NA |
Indicates percentage of sequences in signature group divided by total sequences in sample
Indicates percentage of the fecal signature that is attributed to the human fecal signature
A second sample collected at the listed sample site/date
Indicates standard deviation among 37 sewage influent samples
Profiling Fecal Taxa at the Sequence Level
We created profiles for the five fecal-associated families using OTUs comprised of sequences with ≥97 % identity. The distribution of these OTUs (i.e., the fecal signature profile) was similar in samples grouped by location/environmental condition (Fig. 4). Specifically, samples collected from Milwaukee's sewage influent had a consistent signature that was unique from all surface water samples (Fig. 4). The Lake Michigan and harbor samples collected during dry periods differed significantly from samples collected in Milwaukee's urban waterways or in Lake Michigan during CSOs (ANOSIM R=0.70, p<0.005). Further, nearly all samples collected during CSOs had a similar signature and were more similar to the sewage samples than the dry weather samples, suggesting a relatively consistent influence from sewage (Fig. 4). The fecal signature in the stormwater samples also was distinct from the surface water and sewage samples (Fig. 4).
Fig. 4.
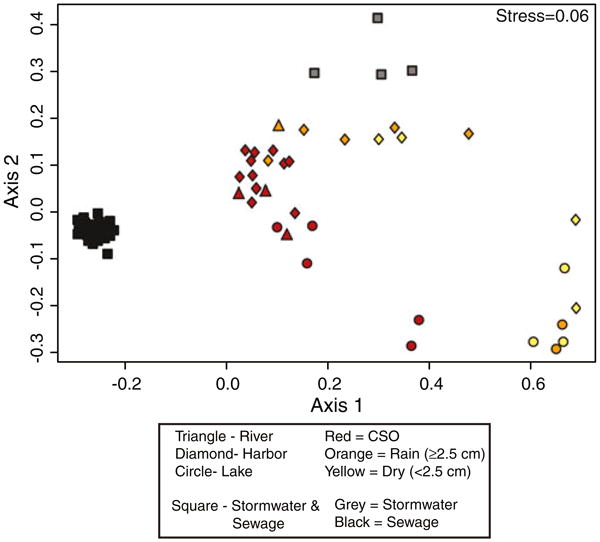
Nonmetric multidimensional scaling plot of the OTU (≥97 % identity) profile (presence or absence) from the fecal signature bacterial families Bacteroidaceae, Porphyromonadaceae, Clostridiaceae, Lachnospiraceae, and Ruminococcaceae present in either surface water or stormwater samples. See Table 1 for the list of samples included and descriptions of conditions during sample collection
The human fecal signature was one of the primary drivers of the clustering observed in Fig. 4 (Table 1). Samples collected during CSOs contained a significantly larger percentage of the human fecal signature than those samples collected during non-CSO periods (Table 1; t test, p value≤0.001), and at times, the human signature reached proportions near that of sewage influent. In the majority of receiving water samples, the relative abundance of the human fecal signature as a proportion of the fecal signature was less than that found in the average sewage sample (Table 1). The total number of fecal OTUs also widely varied among samples. In some samples, primarily lake samples during non-CSO conditions, less than 25 OTUs made up the fecal signature, whereas more than 100 OTUs typically comprised this signature in the harbor and river samples no matter the environmental conditions (Table 1).
In total, 1,008 OTUs associated with the fecal signature taxa were identified in the surface water samples and 2,736 were identified among all sewage samples (see Fig. 5 for a representation). In particular, the Lachnospiraceae and Ruminococcaceae families contained the largest number of OTUs associated with the fecal signature taxa, which amounted to 2,717 of the 3,554 total OTUs observed (Fig. 5). The distribution of OTUs also suggested potential origins for the fecal signature in each sample group. During CSOs, a large proportion of the fecal signature OTUs recovered in the surface water samples were also represented in the sewage influent samples (Fig. 5). Specifically, shared OTUs between the surface water and sewage samples made up 56.4, 26.6, and 30.2 % of the fecal signature in the river CSO, harbor CSO, and lake CSO samples, respectively. In contrast, the relative abundance of the fecal signature OTUs shared between sewage and surface water samples was greatly reduced in the non-CSO samples, comprising only 7.2, 8.9, and 1.9 % of the fecal signature in the harbor rain, harbor no rain, and lake no rain samples. The stormwater samples shared a moderate percentage (24.3 %) of their fecal signature (calculated as shared OTU relative abundance) with the sewage samples but a higher percentage with the surface water samples during CSOs (river 44.8 %, harbor 47.1 %, lake 31.3 %) and rain events (harbor, 63.3 %).
Fig. 5.

Heatmap illustrating the relative abundance of OTUs contributing to the fecal signature in each sample group. Samples contributing to the groupings are listed in Table 1, with the exception of the sewage group. The sewage influent sample information is listed in Supplementary Table 1. The number of samples contributing to each group is as follows: sewage (37), stormwater (4), river CSO (3), harbor CSO (11), harbor rain (6), harbor no rain (3), lake CSO (5), and lake no CSO (5). “Rain” samples are those that were collected following >2.5 cm of rain over a 48-h period prior to collection. For illustrative purposes, only the OTUs among the top 50 % most abundant in sewage for each fecal family and all OTUs not present in sewage are shown. The number of OTUs for each bacterial family illustrated in this figure is shown next to the family name
A comparison between the sewer, fecal, and human fecal signatures in the surface water samples and the enterococci and E. coli colony counts revealed two relevant trends. The enterococci counts showed a moderate but significant correlation to the magnitude of both the sewer and fecal signatures across all samples (Table 1; Pearson's R=0.70 and 0.58; p≤0.05). This trend was also similar for the E. coli counts (Table 1; Pearson's R=0.59 and 0.64; p≤0.05). Neither the enterococci nor the E. coli counts exhibited a significant relationship (p≥0.05) to the magnitude of the human fecal signature. In fact, the correlative relationship between the two measures was slightly negative in both cases (enterococci Pearson's R=−0.11, E. coli Pearson's R=−0.08); however, this relationship may be influenced by the wide variety of samples included in the study (Table 1). For example, stormwater had several of the highest concentrations of E. coli and enterococci, but contained a comparatively low human fecal signature, whereas residual CSOs in the harbor at times had relatively low levels of E. coli and enterococci but a large human component (Table 1).
The Human Fecal Footprint from an Urban Area
We detected the fecal signature following a CSO 8 km from shore in Lake Michigan (Fig. 3). However, community sequencing provides a relative abundance of the signature, but not a concentration. In order to more accurately identify and quantify the fecal signature in nearshore Lake Michigan, we used qPCR assays for two human fecal indicators on samples collected from the coastal zone surrounding Milwaukee, WI. During dry weather flows, the concentration of these human indicators was very low (0.0–2.5E4 copies/l; Fig. 6) and primarily contained to the harbor area. However, during rain events and especially during a large CSO, the human indicators were abundant several kilometers from the shore (>1.5E5 copies/l; Fig. 6). At this distance from the shore, fecal pollution was not detected using traditional culture methods for enterococci and E. coli (Table 1).
Fig. 6.

Interpolated mapping of the total human Bacteroides+Lachno2 (human fecal signal) copies per liter of sample collected during four weather scenarios (from left to right: dry weather, heavy rain, heavy rain/blending event, heavy rain/combined and sanitary sewer overflow. Sample points on each map indicate the samples used for interpolating data in the map. The dry weather map is an average concentration of the markers from four separate transects while the remaining three maps indicate a single sampling event
Discussion
Fecal pollution of coastal waters remains a serious environmental and public health threat [46, 47]. Despite improving water quality following the Clean Water Act mandated elimination of many sewage discharge sources, the regular identification in coastal waters of sewage-borne human pathogens suggests that sewage contamination remains a problem [11, 47, 48]. However, the complexity of coastal watersheds, which often drain upstream rural and agricultural lands and downstream urban regions, makes it difficult to identify and estimate the relative contributions of various fecal pollution sources in these ecosystems.
Ideally, indicator assays for the presence of fecal pollution would be sensitive (able to detect low concentrations) and source-specific [16]. The majority of fecal indicator methods created for source identification rely upon a single taxonomically narrow group of target organisms or genetic markers ([16] and references therein). However, single target assays are complicated by cross reactivity with other sources, geographic relevance, and differential distribution of the target in host populations [19, 49, 50]. It is unlikely that any single indicator will be able to meet all of the criteria desired for tracking fecal pollution. In this study, we used microbial community sequencing as a new approach for fecal source tracking. Sequencing a large portion of the bacterial community by targeting the universal 16S rRNA gene allowed us to identify signatures, i.e., subset distributions of sequences within the community profile, which indicated the presence of sewer, fecal, and human fecal bacteria and suggested that nonhuman fecal-associated bacteria were also present. With the appropriate source bacterial signatures identified, a profiling approach would be capable of identifying multiple fecal sources in a single sample and, therefore, would provide an advantage over traditional single indicator approaches when the pollution sources are not known or many sources are mixed into a single environment.
Initial studies involving sequence profiling of bacterial communities from various environments provide encouraging results that argue for the feasibility of the signature approach. Coastal ocean [51, 52] and Great Lake [53, 54] waters exhibit vastly different community compositions than that of sewage [24], or human and animal fecal waste [38, 39, 41, 55, 56]. To date, the fecal community surveys from animals suggest that each species harbors a unique fecal microbial community [25, 36]. If this trend holds, then sequence signatures from multiple fecal sources may be identified from environmental samples.
The consistency of the sewage influent community [24, 30] and in particular the taxa chosen for our signature approach (Fig. 2a) suggests that the relatively small number of taxa included adequately represented the source environment. If large variations in the source community composition had been observed, then a signature encompassing more taxa may have been warranted. Using our sewer and fecal signature approach, we tracked these taxa in the coastal waters of Lake Michigan. The magnitude of the signatures in three transects from the urban waterways to Lake Michigan suggests that this method is sufficiently sensitive to detect sewer and fecal pollution. The correlation to CSOs and low magnitudes in lake samples furthest from the urban pollution sources suggests the signatures accurately detected pollution presence in this coastal environment. During a dry weather period, the sewer signature recovered from the surface waters consisted almost entirely of sequences assigned to the genus Acinetobacter. This genus might represent a more sensitive portion of the signature because it is the most abundant component in sewage. Alternatively, it could be that Acinetobacter survives longer in the environment than the other components of the signature or that it is a residual sustaining member of the lake community. Further research will be needed to determine whether a subset of Acinetobacter taxa represent the trailing edge of the sewer signature or persist as members of the coastal lake community.
Since the signature approach was sensitive enough to detect fecal pollution in this ecosystem, we examined the fecal signature with a higher resolution (the distribution of OTUs) as a way to identify sources contributing to the fecal pollution in Milwaukee's waterways. We estimated the human fecal signature to be ∼55 % of the total fecal signature in sewage, but this is likely an underestimate. SourceTracker estimates the probability an OTU identified in a sink community was from a source community based in part on the distribution among source samples [37]. Large variations in and/or a small sample size for the source community would tend to decrease the likelihood an OTU would be attributed to that source. In this study, only 48 human fecal samples contributed to the human fecal source community, and it is known that fairly large variations occur among human fecal samples [40, 41]. Improved identification of a human fecal signature could be achieved with a larger dataset of human fecal microbial communities.
CSOs offer an opportunity to study a known pollution event, and as expected, during CSO events, sewage was a primary contaminating source in the coastal waterways. Less anticipated was that the fecal signature in the urban waterways during rain events had a fecal signature that was relatively similar to that of CSOs and sewage (Figs. 3 and 4), with differences primarily occurring among lower abundance OTUs. This pattern indicated to us that sewage was a source during rain events, but at a lower magnitude than during CSOs, thereby causing the limit of detection to be reached for the less significant components of the signature. Stormwater outfall runoff harboring nonhuman fecal waste mixed with sewage could produce these observed patterns. In a previous study, we found that Milwaukee's stormwater is commonly infiltrated with sewage [17], and here, we again observe a human fecal contribution to the fecal signature coming from stormwater outfalls. Although a human fecal component was apparent, the major fecal component in the stormwater samples was from OTUs that had never been detected in sewage. This nonsewage and therefore presumably nonhuman fecal signature was consistently present during rain and CSO events. Since we did not have the fecal signatures for nonhuman animal fecal sources, we cannot identify the other polluting sources being delivered to the coastal waters by stormwater, but it is apparent that stormwater is a primary route for both significant human and nonhuman fecal contamination in this coastal system.
In a previous study, we noted that human fecal pollution was chronic in Milwaukee's urban waterways [22]. Our fecal signature approach also identified chronic fecal pollution, including human and nonhuman fecal pollution in the harbor and both human and nonhuman fecal pollution out to 8 km from shore. Although the community sequencing approach facilitates fecal source identification, its reliance on relative abundance measures means it is not well suited for quantifying the fecal signatures observed. Using qPCR assays for human Bacteroides and Lachno2, both human fecal indicators, we provided complementary evidence of the human fecal signature during rain events and CSOs and quantified a human fecal signal in Lake Michigan's coastal waterways. The qPCR assays confirmed that human fecal pollution does impact an area a significant distance from the Lake Michigan shoreline.
Given the complexity of the data produced by community sequencing and the complexity of identifying mixed fecal pollution signals, this study provides only a glimpse at the potential of a microbial signatures approach. The primary strength of a signature-based approach and the facet distinguishing it from most currently applied approaches is that it is capable of deciphering the presence of multiple pollution sources in a single sample without the need for a priori decisions about what sources to target. Generally speaking, this facet of the signature approach has practical implications for resource managers, who often have limited funds to mitigate fecal pollution issues. If multiple fecal pollution sources could be identified from a single or a few samples, then the resource manager would have with limited effort or cost the capability to identify the largest and/or most problematic pollution sources in her/his system and direct funding toward those issues. Although offering significant potential, signature-based approaches are not the ideal method for all pollution tracking scenarios. Current signature-based approaches are not quantitative, so when pollution sources are known or quantification is needed, single target quantification may be more appropriate.
The signature approach also has unique drawbacks. Particularly, source identification using communities is only as good as the sequence databases that describe the source communities. Much larger microbial community databases that include fecal samples from diverse source animals and urban infrastructure are needed. Procedures governing consistent DNA extraction and amplification procedures to minimize molecular biases must be created and implemented. Further refinement of fecal signatures is also necessary and could be provided with methods like SourceTracker [37] that identify discriminatory signatures out of complex data matrices.
Conclusions
Despite marked improvements, fecal pollution still represents a major impairment to coastal waterways and a threat to human health. Pollution mitigation often depends upon being able to identify the major contributing sources; however, along urban coasts, the pollution sources and delivery routes are numerous, thereby rendering traditional fecal indicator assays insufficient for source identification. Source identification facilitated by our use of a microbial community sequencing approach to identify source signatures suggested that significant proportions of the coastal surface water bacterial community were made up of bacteria associated with fecal waste and urban infrastructure. The signature approach also revealed that human fecal contamination was entering via the stormwater outfall system and that nonhuman fecal sources were the largest contributors of fecal pollution during dry weather and non-CSO producing rain events. Our approach used signatures that could be used to target both the fecal and sewer (nonfecal) component of sewage. The sewer signature is present at consistently high levels in sewage influent, but has garnered little attention as a potential indicator. Although there is still much to be deciphered and refined, microbial community sequencing appears to be a promising method for identification of fecal sources in complex ecosystems and could be used in other avenues of microbial community research such as identifying sample contamination [37], environment mixing, or microbial immigration.
Supplementary Material
Acknowledgments
The authors would like to thank Jessica VandeWalle, Elizabeth Sauer, Colin Peake, and Sabrina Mueller-Spitz for assistance with sample collection and processing. We also thank Sharon Grim and Joseph Vineis for the DNA sequence library preparation and data curation. Special thanks go to Dan Knights who provided an unreleased version of SourceTracker, so we could identify source probabilities for individual OTUs. We also thank Linda Sackett for providing a supportive environment for discussion of this work. Finally, we would like to thank two anonymous reviewers whose comments greatly improved the clarity of this manuscript. Financial support for this work was provided by the University of Wisconsin Sea Grant Institute under grants from the National Sea Grant College Program, NOAA, the U.S. Department of Commerce, and the State of Wisconsin, grant #NA10OAR4170070 to SLM and the National Institutes of Health grant #1R01AI091829-01A1 to SLM and MLS.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00248-013-0200-9) contains supplementary material, which is available to authorized users.
Contributor Information
Ryan J. Newton, Great Lakes WATER Institute, School of Freshwater Sciences, University of Wisconsin—Milwaukee, 600 E. Greenfield Ave, Milwaukee, WI 53204, USA
Melinda J. Bootsma, Great Lakes WATER Institute, School of Freshwater Sciences, University of Wisconsin—Milwaukee, 600 E. Greenfield Ave, Milwaukee, WI 53204, USA
Hilary G. Morrison, Josephine Bay Paul Center, Marine Biological Laboratory, Woods Hole, MA 02543, USA
Mitchell L. Sogin, Josephine Bay Paul Center, Marine Biological Laboratory, Woods Hole, MA 02543, USA
Sandra L. McLellan, Email: mclellan@uwm.edu, Great Lakes WATER Institute, School of Freshwater Sciences, University of Wisconsin—Milwaukee, 600 E. Greenfield Ave, Milwaukee, WI 53204, USA.
References
- 1.NOAA. Communities: the U S population living in coastal watershed counties. Sepcial Projects Division, National Ocaeanic Service, National Oceanic and Atmospheric Administration, Department of Commerce USA; 2011. [Accessed 24 Sep 2012]. http://stateofthecoast.noaa.gov/population/welcome.html. [Google Scholar]
- 2.U.S. Environmental Protection Agency. Report to Congress: impacts and control of CSOs and SSOs. U.S. Environmental Protection Agency, Office of Water; Washington: 2004. [Google Scholar]
- 3.Graczyk TK, Sunderland D, Tamang L, Lucy FE, Breysse PN. Bather density and levels of Cryptosporidium, Giardia, and pathogenic microsporidian spores in recreational bathing water. Parasitol Res. 2007;101:1729–1731. doi: 10.1007/s00436-007-0734-1. [DOI] [PubMed] [Google Scholar]
- 4.Mallin MA, Williams KE, Esham EC, Lowe RP. Effect of human development on bacteriological water quality in coastal watersheds. Ecol Appl. 2000;10:1047–1056. [Google Scholar]
- 5.Walters SP, Thebo AL, Boehm AB. Impact of urbanization and agriculture on the occurrence of bacterial pathogens and stx genes in coastal waterbodies of central California. Water Res. 2011;45:1752–1762. doi: 10.1016/j.watres.2010.11.032. [DOI] [PubMed] [Google Scholar]
- 6.Anderson DM, Burkholder JM, Cochlan WP, Glibert PM, Gobler CJ, Heil CA, Kudela RM, Parsons ML, Rensel JEJ, Townsend DW, Trainer VL, Vargo GA. Harmful algal blooms and eutrophication: examining linkages from select coastal regions of the United States. Harmful Algae. 2008;8:39–53. doi: 10.1016/j.hal.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thevenon F, Graham ND, Herbez A, Wildi W, Poté J. Spatio-temporal distribution of organic and inorganic pollutants from Lake Geneva (Switzerland) reveals strong interacting effects of sewage treatment plant and eutrophication on microbial abundance. Chemosphere. 2011;84:609–617. doi: 10.1016/j.chemosphere.2011.03.051. [DOI] [PubMed] [Google Scholar]
- 8.Gros M, Petrovic M, Barceló D. Wastewater treatment plants as a pathway for aquatic contamination by pharmaceuticals in the Ebro river basin (Northeast Spain) Environ Toxicol Chem. 2007;26:1553–1562. doi: 10.1897/06-495r.1. [DOI] [PubMed] [Google Scholar]
- 9.Ternes TA, Joss A, Siegrist H. Scrutinizing pharmaceuticals and personal care products in wastewater treatment. Environ Sci Technol. 2004;38:392A–399A. doi: 10.1021/es040639t. [DOI] [PubMed] [Google Scholar]
- 10.Brausch JM, Rand GM. A review of personal care products in the aquatic environment: environmental concentrations and toxicity. Chemosphere. 2011;82:1518–1532. doi: 10.1016/j.chemosphere.2010.11.018. [DOI] [PubMed] [Google Scholar]
- 11.Lipp EK, Farrah SA, Rose JB. Assessment and impact of microbial fecal pollution and human enteric pathogens in a coastal community. Mar Pollut Bull. 2001;42:286–293. doi: 10.1016/s0025-326x(00)00152-1. [DOI] [PubMed] [Google Scholar]
- 12.Cabelli VJ, Dufour AP, McCabe LJ, Levin MA. Swimming-associated gastroenteritis and water quality. Am J Epidemiol. 1982;115:606–616. doi: 10.1093/oxfordjournals.aje.a113342. [DOI] [PubMed] [Google Scholar]
- 13.Leclerc H, Schwartzbrod L, Dei-Cas E. Microbial agents associated with waterborne diseases. Crit Rev Microbiol. 2002;28:371–409. doi: 10.1080/1040-840291046768. [DOI] [PubMed] [Google Scholar]
- 14.Pruss A. Review of epidemiological studies on health effects from exposure to recreational water. Int J Epidemiol. 1998;27:1–9. doi: 10.1093/ije/27.1.1. [DOI] [PubMed] [Google Scholar]
- 15.Wade TJ, Calderon RL, Brenner KP, Sams E, Beach M, Haugland R, Wymer L, Dufour AP. High sensitivity of children to swimming-associated gastrointestinal illness: results using a rapid assay of recreational water quality. Epidemiol. 2008;19:375–383. doi: 10.1097/EDE.0b013e318169cc87. [DOI] [PubMed] [Google Scholar]
- 16.McLellan SL, Boehm AB, Shanks OC. Marine and freshwater fecal indicators and source identification. In: Meyers RA, editor. Encyclopedia of sustainability science and technology. Springer; New York: 2012. [Google Scholar]
- 17.Sauer EP, VandeWalle JL, Bootsma MJ, McLellan SL. Detection of the human specific Bacteroides genetic marker provides evidence of widespread sewage contamination of stormwater in the urban environment. Water Res. 2011;45:4081–4091. doi: 10.1016/j.watres.2011.04.049. [DOI] [PubMed] [Google Scholar]
- 18.Sercu B, Van De Werfhorst LC, Murray J, Holden PA. Storm drains are sources of human fecal pollution during dry weather in three urban southern California watersheds. Environ Sci Technol. 2009;43:293–298. doi: 10.1021/es801505p. [DOI] [PubMed] [Google Scholar]
- 19.Field KG, Samadpour M. Fecal source tracking, the indicator paradigm, and managing water quality. Water Res. 2007;41:3517–3538. doi: 10.1016/j.watres.2007.06.056. [DOI] [PubMed] [Google Scholar]
- 20.Bernhard AE, Goyard T, Simonich MT, Field KG. Application of a rapid method for identifying fecal pollution sources in a multi-use estuary. Water Res. 2003;37:909–913. doi: 10.1016/s0043-1354(02)00384-6. [DOI] [PubMed] [Google Scholar]
- 21.Boehm AB, Fuhrman JA, Mrse RD, Grant SB. Tiered approach for identification of a human fecal pollution source at a recreational beach: case study at Avalon Bay, Catalina Island, California. Environ Sci Technol. 2003;37:673–680. doi: 10.1021/es025934x. [DOI] [PubMed] [Google Scholar]
- 22.Newton RJ, Vandewalle JL, Borchardt MA, Gorelick MH, McLellan SL. Lachnospiraceae and Bacteroidales alternative fecal indicators reveal chronic human sewage contamination in an urban harbor. Appl Environ Microbiol. 2011;77:6972–6981. doi: 10.1128/AEM.05480-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shanks OC, Nietch C, Simonich M, Younger M, Reynolds D, Field KG. Basin-wide analysis of the dynamics of fecal contamination and fecal source identification in Tillamook Bay, Oregon. Appl Environ Microbiol. 2006;72:5537–5546. doi: 10.1128/AEM.03059-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McLellan SL, Huse SM, Mueller-Spitz SR, Andreishcheva EN, Sogin ML. Diversity and population structure of sewage-derived microorganisms in wastewater treatment plant influent. Environ Microbiol. 2010;12:378–392. doi: 10.1111/j.1462-2920.2009.02075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee JE, Lee S, Sung J, Ko G. Analysis of human and animal fecal microbiota for microbial source tracking. ISME J. 2011;5:362–365. doi: 10.1038/ismej.2010.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu CH, Sercu B, Van de Werfhorst LC, Wong J, DeSantis TZ, Brodie EL, Hazen TC, Holden PA, Andersen GL. Characterization of coastal urban watershed bacterial communities leads to alternative community-based indicators. PLoS One. 2010;5(6):e11285. doi: 10.1371/journal.pone.0011285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Swackhamer DL. The past, present, and future of the North American Great Lakes: what lessons do they offer? J Environ Monit. 2005;7:540–544. doi: 10.1039/b506709j. [DOI] [PubMed] [Google Scholar]
- 28.Bower PA, Scopel CO, Jensen ET, Depas MM, McLellan SL. Detection of genetic markers of fecal indicator bacteria in Lake Michigan and determination of their relationship to Escherichia coli densities using standard microbiological methods. Appl Environ Microbiol. 2005;71:8305–8313. doi: 10.1128/AEM.71.12.8305-8313.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huse SM, Welch DM, Morrison HG, Sogin ML. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ Microbiol. 2010;12:1889–1898. doi: 10.1111/j.1462-2920.2010.02193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.VandeWalle JL, Goetz GW, Huse SM, Morrison HG, Sogin ML, Hoffmann RG, Yan K, McLellan SL. Acinetobacter, Aeromonas, and Trichococcus populations dominate the microbial community within urban sewer infrastructure. Environ Microbiol. 2012;14(9):2538–2552. doi: 10.1111/j.1462-2920.2012.02757.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huse SM, Dethlefsen L, Huber JA, Mark Welch D, Relman DA, Sogin ML. Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS Genet. 2008;4:e1000255. doi: 10.1371/journal.pgen.1000255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.U.S. Environmental Protection Agency. Method 1603: Escherichia coli(E coli) in water by membrane filtration using modified membrane-thermotolerant Escherichia coli agar (modified mTEC) U.S. Envrionmental Protection Agency, Office of Water; Washington: 2002. [Google Scholar]
- 34.U.S. Environmental Protection Agency. Method 1600: enterococci in water by membrane filtration using membrane-Enterococus indoxyl-B-D-glucoside agar (mEI) U.S. Environmental Protection Agency, Office of Water; Washington: 2002. [Google Scholar]
- 35.Kildare BJ, Leutenegger CM, McSwain BS, Bambic DG, Rajal VB, Wuertz S. 16S rRNA-based assays for quantitative detection of universal, human-, cow-, and dog-specific fecal Bacteroidales: a Bayesian approach. Water Res. 2007;41:3701–3715. doi: 10.1016/j.watres.2007.06.037. [DOI] [PubMed] [Google Scholar]
- 36.Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, Schlegel ML, Tucker TA, Schrenzel MD, Knight R, Gordon JI. Evolution of mammals and their gut microbes. Science. 2008;320:1647–1651. doi: 10.1126/science.1155725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Knights D, Kuczynski J, Charlson ES, Zaneveld J, Mozer MC, Collman RG, Bushman FD, Knight R, Kelley ST. Bayesian community-wide culture-independent microbial source tracking. Nature Methods. 2011;8:761–763. doi: 10.1038/nmeth.1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dethlefsen L, Huse S, Sogin ML, Relman DA. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008;6:2383–2400. doi: 10.1371/journal.pbio.0060280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, Egholm M, Henrissat B, Heath AC, Knight R, Gordon JI. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huse SM, Ye Y, Zhou Y, Fodor A. A core human microbiome as viewed through 16S rRNA sequence clusters. PLoS One. 2012;7:e34242. doi: 10.1371/journal.pone.0034242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, Heath AC, Warner B, Reeder J, Kuczynski J, Caporaso JG, Lozupone CA, Lauber C, Clemente JC, Knights D, Knight R, Gordon JI. Human gut microbiome viewed across age and geography. Nature. 2012;486:222–228. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.R Core Team. R: a language and environment for statistical computing. R Foundation for Statistical Computing; Vienna: 2012. http://www.R-project.org. [Google Scholar]
- 43.Oksanen J, Blanchet GF, Kindt R, Legendre P, Minchin PR, O'Hara RB, Simpson GL, Solymos P, Henry M, Stevens H, Wagner H. vegan: community ecology package. R package version 2.0-2 2011 [Google Scholar]
- 44.Warnes GR. gplots: various R programming tools for plotting data version 2.10.1 2011 [Google Scholar]
- 45.Baddeley A, Turner R. Spatstat: an R package for analyzing spatial point patterns. J Stat Softw. 2005;12:1–42. [Google Scholar]
- 46.NRC. Indicators for waterborne pathogens. National Research Council of the National. Academies; Washington: 2004. [PubMed] [Google Scholar]
- 47.Stewart JR, Gast RJ, Fujioka RS, Solo-Gabriele HM, Meschke JS, Amaral-Zettler LA, Del Castillo E, Polz MF, Collier TK, Strom MS, Sinigalliano CD, Moeller PD, Holland AF. The coastal environment and human health: microbial indicators, pathogens, sentinels and reservoirs. Environ Health. 2008;7(Suppl 2):S3. doi: 10.1186/1476-069X-7-S2-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Noble RT, Fuhrman JA. Enteroviruses detected by reverse transcriptase polymerase chain reaction from the coastal waters of Santa Monica Bay, California: low correlation to bacterial indicator levels. Hydrobiologia. 2001;460:175–184. [Google Scholar]
- 49.Shanks OC, Kelty CA, Sivaganesan M, Varma M, Haugland RA. Quantitative PCR for genetic markers of human fecal pollution. Appl Environ Microbiol. 2009;75:5507–5513. doi: 10.1128/AEM.00305-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shanks OC, White K, Kelty CA, Sivaganesan M, Blannon J, Meckes M, Varma M, Haugland RA. Performance of PCR-based assays targeting Bacteroidales genetic markers of human fecal pollution in sewage and fecal samples. Environ Sci Technol. 2010;44:6281–6288. doi: 10.1021/es100311n. [DOI] [PubMed] [Google Scholar]
- 51.Gilbert JA, Steele JA, Caporaso JG, Steinbrück L, Reeder J, Temperton B, Huse S, McHardy AC, Knight R, Joint I, Somerfield P, Fuhrman JA, Field D. Defining seasonal marine microbial community dynamics. ISME J. 2012;6:298–308. doi: 10.1038/ismej.2011.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zinger L, Amaral-Zettler LA, Fuhrman JA, Horner-Devine MC, Huse SM, Mark Welch DB, Martiny JBH, Sogin ML, Boetius A, Ramette A. Global patterns of bacterial beta-diversity in sea-floor and seawater ecosystems. PLoS One. 2011;6:e24570. doi: 10.1371/journal.pone.0024570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mueller-Spitz SR, Goetz GW, McLellan SL. Temporal and spatial variability in nearshore bacterioplankton communities of Lake Michigan. FEMS Microbiol Ecol. 2009;67:511–522. doi: 10.1111/j.1574-6941.2008.00639.x. [DOI] [PubMed] [Google Scholar]
- 54.Newton RJ, Jones SE, Eiler A, McMahon KD, Bertilsson S. A guide to the natural history of freshwater lake bacteria. Microbiol Mol Biol Rev. 2011;75:14–49. doi: 10.1128/MMBR.00028-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shanks OC, Kelty CA, Archibeque S, Jenkins M, Newton RJ, McLellan SL, Huse SM, Sogin ML. Community structures of fecal bacteria in cattle from different animal feeding operations. Appl Environ Microbiol. 2011;77:2992–3001. doi: 10.1128/AEM.02988-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.