

Abstract
Current genomic perspectives on animal diversity neglect two prominent phyla, the molluscs and annelids, that together account for nearly one-third of known marine species and are important both ecologically and as experimental systems in classical embryology1–3. Here we describe the draft genomes of the owl limpet (Lottia gigantea), a marine polychaete (Capitella teleta) and a freshwater leech (Helobdella robusta), and compare them with other animal genomes to investigate the origin and diversification of bilaterians from a genomic perspective. We find that the genome organization, gene structure and functional content of these species are more similar to those of some invertebrate deuterostome genomes (for example, amphioxus and sea urchin) than those of other protostomes that have been sequenced to date (flies, nematodes and flatworms). The conservation of these genomic features enables us to expand the inventory of genes present in the last common bilaterian ancestor, establish the tripartite diversification of bilaterians using multiple genomic characteristics and identify ancient conserved long- and short-range genetic linkages across metazoans. Superimposed on this broadly conserved pan-bilaterian background we find examples of lineage-specific genome evolution, including varying rates of rearrangement, intron gain and loss, expansions and contractions of gene families, and the evolution of clade-specific genes that produce the unique content of each genome.
Molluscs, annelids and numerous smaller phyla typically share stereotyped spiral cleavage patterns, cell-fate assignments and characteristic ciliated trochophore larvae, features that originated in the Precambrian era3–5. These spiralian phyla are included in the larger lophotrochozoan clade6 that is a sister group to the ecdysozoans (arthropods, nematodes and other related phyla) but whose internal branching remains controversial. However, so far the only deeply sequenced lophotrochozoan genomes are those of platyhelminth flatworms (two parasitic schistosomes7,8 and a free-living planarian9), whose comparatively rapid rates of genome evolution do not reflect a general condition of lophotrochozoans (see below). In this study, we explore spiralian diversity at the genomic level by comparative analysis of one mollusc and two annelid genomes (Supplementary Note 1).
We assembled the limpet, polychaete and leech genomes from approximately eight-fold random whole-genome shotgun coverage with Sanger dideoxysequencing reads (Supplementary Note 2). No geneticor physical maps were available for these systems, so we reconstructed each genome as scaffolds (gap-containing sequences). The three genomes reported here each encode an estimated 23,000 to 33,000 protein-coding genes (Table 1, Supplementary Table 2.2.2 and Supplementary Note 2.2. The repetitive landscape of these genomes is discussed in Supplementary Note 3.2).
Table 1.
Genome sequencing and annotation summary
| Species | Size of genome assembly (Mbp) | Scaffold N50 (Mbp) | Repetitive content(%) | GC (%) | Predicted number of genes | Number of genes in orthologous clusters with other species | Number of genes in ancestral bilaterian gene families | Mean number of exons per gene (with ≥2 orthologues) | Mean exon length (bp) | Mean intron length (bp) |
|---|---|---|---|---|---|---|---|---|---|---|
| Lottia gigantea | 348 | 1.87 | 21 | 33 | 23,800 | 16,183 | 10,681 | 8 | 213 | 787 |
| Capitella teleta | 324 | 0.19 | 31 | 40 | 32,389 | 20,537 | 11,911 | 7 | 221 | 291 |
| Helobdella robusta | 228 | 3.06 | 33 | 33 | 23,400 | 13,820 | 8,707 | 8 | 203 | 526 |
GC, fraction of guanine plus cytosine nucleobases; Scaffold N50, the length such that half of the assembled sequence is in scaffolds longer than this length; Mbp, megabase pairs.
Comparing the new genomes with other metazoan sequences, we characterized 8,756 modern bilaterian gene families as likely to have arisen from single progenitor genes in the last common bilaterian ancestor (Supplementary Note 3.4). As gene loss is common and highly diverged orthologues can be difficult to detect, this is a conservative lower bound on the number of genes encoded by the last common bilaterian ancestor. Of the 8,756 gene families, 763 were newly identified as being of bilaterian ancestry based on the new spiralian genomes (Supplementary Note 3.4). These newly identified bilaterian families belong to various functional categories (Supplementary Table 3.4.1), the most prominent being members of the G-protein-coupled receptor superfamily and epithelial sodium channels (see below) as well as various metabolic enzymes. Through subsequent gene duplication, the 8,756 ancestral bilaterian families conservatively account for 47 to 85% of genes in other bilaterian species (70% of human genes; Supplementary Note 3.4). Most of the remaining genes in extant bilaterian genomes share at least one domain with the bilaterian gene families, or have a significant BLAST (Basic Local Alignment Search Tool) hit when compared against sequences from bilaterian gene families, suggesting that they have arisen through descent with modification (Supplementary Note 3.5).
Exon–intron structures are highly conserved between spiralians and other animals; thus we infer that cis-splicing of intron-rich genes was the ancestral state of metazoans, bilaterians and protostomes (Supplementary Note 5.2). In most cases, exon boundaries in the newly sequenced spiralians are precisely conserved between orthologous genes in sequenced deuterostomes (vertebrates, sea urchin and amphioxus) and non-bilaterians (Trichoplax and starlet sea anemone). For example, 75% of human introns are present in one or more of the spiralians, whereas only 14% of the same introns are found in Drosophila10,11. However, intron gain or loss rates vary markedly among the three spiralians. In particular, H. robusta also has substantially more novel introns than do the other two sequenced spiralians (Supplementary Notes 5.2 and 5.3, and Supplementary Fig. 5.2.1), the first of several indicators of a notably dynamic genome in this lineage.
Collectively and individually, the spiralian genomes reported here retain most of the inferred ancestral bilaterian gene families (8,203 out of 8,756, corresponding to a 94% retention rate, compared to 7,553 or 86% retention rate in human). In contrast, the collective retention rate of only 65% for sequenced flatworms (53% for schistosomes and 60% for Schmidtea) reflects the absence (and presumed loss) of more than 3,018 ancestral bilaterian gene families in these flatworms. Similar losses are observed for introns (Supplementary Note 5.3), as well as synteny (see below), which indicate a higher rate of genomic turnover in platyhelminths than in the mollusc and annelid genomes reported here.
Against this background of conserved gene content and structure, we find several significantly (P <0.05) expanded gene families in specific spiralian clades (Supplementary Note 4.2). The sensory transduction and signalling genes of the G-protein-coupled receptor (GPCR) superfamily in C. teleta are a prime example. All six of the rhodopsin-like GPCRs represented in the KEGG (Kyoto Encyclopedia of Genes and Genomes) neuroactive ligand–receptor interaction pathway are expanded in C. teleta (but not in H. robusta or L. gigantea), as are several other GPCRs (Supplementary Figs 4.3.2 and 4.4.1). Moreover, the C. teleta genome encodes 372 putative GPCR receptors that are most similar to peptide-binding GPCR subfamilies according to the family classification in the GPCR database (http://www.gpcr.org/7tm/proteinfamily/). This number is considerably higher than that obtained for H. robusta (58), L. gigantea (113), Drosophila (32) or human (120) using the same methods (Supplementary Note 4.4). Most of these expansions occur as tandem duplicates. The C. teleta genome also shows an expansion of the calcium-signalling pathway downstream of GPCR (Supplementary Note 4.3). It is tempting to speculate that these expansions are related to the function of polychaete chemosensory structures such as antennae, palps and cirri (head sensory organs), and the nuchal organ12. Another notable feature of all three genomes is the presence of several atypical GPCRs with weak similarity to both vertebrate rhodopsin-like GPCRs and chemosensory receptors described previously in nematodes (Supplementary Fig. 4.4.1). Further studies are needed to determine whether these receptors can be classified as divergent members of previously described GPCR classes or whether they constitute novel groupings as described recently in planarians13.
We also find changes in gene content associated with sensory processing in the leech. These changes include expansion of the epithelial sodium channel (ENaC) receptor gene family that functions in the taste-transduction pathway (Supplementary Fig. 4.3.5), and the gap-junction-forming innexin gene family, as well as gene families involved in development (for example, homeobox genes (see below, and Supplementary Notes 4.5 and 4.6)). Both mollusc and annelid genomes are also enriched in specific metabolic enzymes and pathways of unknown relevance (for example, galactoside 2-α-L-fucosyltransferase; see Supplementary Notes 4.2 and 4.3). In general, lineage-specific gene family expansions seem to be the norm in the evolutionary diversification of modern taxa from the bilaterian ancestor, whereas the more ancient unicellular–metazoan14 and metazoan–bilaterian transitions are more notably marked by the acquisition of apparently novel (or highly divergent) gene families (Supplementary Table 3.5.1).
We identified 231 putative spiralian-specific gene families whose members are readily aligned across all three spiralians (indicating purifying selection), but which lack obvious orthologues by BLAST in non-spiralian genomes (Supplementary Note 3.6). However, nearly two-thirds of these (188 out of 231; 62%) showed residual similarity to non-spiralian genes using more sensitive Hidden Markov Model methods, which suggests that they belong to ancient bilaterian gene families (Supplementary Note 3.6) that diverged extensively on the stem lineage leading from the bilaterian ancestor to the mollusc–annelid ancestor (‘type II’ novelties15). The remaining 43 out of 231 novel gene families are without any significant (E values of less than 0.01) similarities outside of spiralians (‘type I’ novelties15). More than one-half of the 231 spiralian novelties are transcribed based on existing expressed sequence tag (EST) evidence, with enriched expression in adult rather than embryonic tissues (Supplementary Notes 2.4 and 3.7), hinting at roles in clade-specific adaptations beyond the early conserved stages of development.
The inference of deep phylogenetic relationships among animal phyla is controversial but has benefitted from the use of multiple orthologous genes as phylogenetic markers16,17. Recent EST-based studies provide broad taxonomic representation but rely on a limited number of available genes or are forced to accommodate a substantial amount of missing data6,18. In contrast, full genome sequences provide nearly complete sets of orthologues exempt from sampling bias, but can be more sensitive to long-branch attraction artefacts.
To strike a balance between the number of phylogenetically informative characters and possible long-branch artefacts we ranked 1,180 clusters of orthologous genes (from 22 complete genomes) by their evolutionary rates (Supplementary Fig. 5.1.1) and identified a set of 827 slowly evolving genes that include 299,129 aligned amino acid positions suitable for deep phylogenetic analysis. These characters strongly support the tripartite view of bilaterians and the monophyly of available lophotrochozoans (annelids, molluscs and platyhelminths) (Fig. 1a)19; the progressive addition of characters representing more rapidly evolving genes monotonically erodes support for this view, as expected under long-branch attraction (Supplementary Fig. 5.1.2). Although taxon sampling is generally considered critical to resolving deep phylogeny, our analyses show the importance of gene sampling. The rate-stratification approach introduced here could be used to place problematic taxa (for example, acoels, ctenophores and chaetognaths) when appropriate genome data becomes available.
Figure 1. Full-genome evidence resolves metazoan relationships and verifies the monophyly of lophotrochozoans and spiralians.
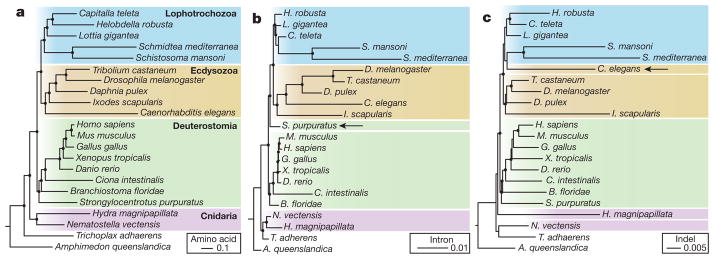
a, A protein tree inferred from 299,129 amino acid positions gathered from 827 slow-evolving orthologues using RAxML and modelling heterogeneity of substitution processes using a LG + Γ4 model with each gene partitioned. Strong support is obtained for the monophyly of lophotrochozoans. b, Intron tree obtained from a matrix of 5,377 introns analysed using MrBayes and an asymmetric binary model (probability of gain: 0.01). c, Indel tree reconstructed from a matrix of 1,928 indel sites using a regular binary model. Circles at nodes indicate a bootstrap support of >0.90 (a) or a posterior probability of >0.95 (b and c). In b and c, arrows indicate species that do not follow the protein family tree topology.
We also examined the phylogenetic signals in the gain and loss of introns, and insertions or deletions (indels) within coding sequences, incorporating spiralian sequences for the first time. Although few evolutionary reconstructions have been attempted with these characters, they have attractive properties for phylogenetic analysis as change is rare and generally irreversible20,21 (Supplementary Note 5.3). Phylogenetic reconstruction using binary matrices encoding intron and indel presence or absence recovered the backbone of metazoan phylogeny, and intron data provided strong support for grouping molluscs, annelids and platyhelminths (Fig. 1b). However, the analysis based on indels showed specific discrepancies relative to other data sets, notably the grouping of nematodes and platyhelminths (Fig. 1c). As this grouping is not consistent with either amino acid or intron analyses, we ascribe it to the accelerated genome evolution in these taxa and the low number of phylogenetically informative indel characters.
All three trees possess short internal branches near the base of bilateria (see ref. 22), which indicates that the diversification into separate lophotrochozoan, deuterostome and ecdysozoan lineages was relatively fast, taking perhaps 30 to 80 million years (Myr) (comparable to the diversification of mammals; Supplementary Note 5.5).
We also sought evidence for genome-wide functional diversification across metazoan genomes using principal component analysis (Supplementary Note 4.1). Remarkably, this phenetic approach grouped the newly sequenced mollusc and annelids with invertebrate deuterostomes (amphioxus, sea urchin and sea squirt) and non-bilaterian metazoan phyla (cnidarian, placozoan and demosponge) (Fig. 2). Given that this grouping includes both bilaterians and non-bilaterian metazoans, cladistic logic implies that these genomes approximate the ancestral bilaterian (and metazoan) genomic repertoire. In contrast, vertebrate genomes form a distinct cluster, and are thus functionally derived relative to this ancestral bilaterian state, partly owing to the diversification of genes related to the vertebrate innate and adaptive immune system that dominate the loadings of principal component 1 (PC1, Fig. 2) (Supplementary Table). The functional coherence of the genes that differentiate currently available ecdysozoan genomes through PC2 is unclear. Although this analysis may be skewed by the more complete functional annotation of vertebrates and classical model systems, other similar analyses less dependent on function confirm the clear separation of vertebrates from other metazoan genomes (Supplementary Note 4.1).
Figure 2. Clustering of metazoan genomes in a multidimensional space of molecular functions.

The first two principal components are displayed, accounting for 20% and 15% of variation, respectively. At least three clusters are evident, including a vertebrate cluster (far right), a non-bilaterian metazoan, invertebrate deuterostome or spiralian cluster (centre, top), and an ecdysozoan group (lower left). Drosophila and Tribolium (lower left) are outliers. Aqu, Amphimedon queenslandica (demosponge); Bfl, Branchiostoma floridae (amphioxus); Cel, Caenorhabditis elegans; Cte, Capitella teleta (polychaete); Cin, Ciona intestinalis (sea squirt); Dme, Drosophila melanogaster; Dpu, Daphnia pulex (water flea); Dre, Danio rerio (zebrafish); Isc, Ixodes scapularis (tick); Gga, Gallus gallus (chicken); Hsa, Homo sapiens (human); Hma, Hydra magnipapillata; Hro, Helobdella robusta (leech); Lgi, Lottia gigantea (limpet); Mmu, Mus musculus (mouse); Nve, Nematostella vectensis (sea anemone); Sma, Schistosoma mansoni; Sme, Schmidtea mediterranea (planarian); Spu, Strongylocentrotus purpuratus (sea urchin); Tad, Trichoplax adhaerens (placozoan); Tca, Tribolium castaneum (flour beetle); Xtr, Xenopus tropicalis (clawed frog).
The L. gigantea and C. teleta genomes show extensively conserved macrosynteny with each other, with chordates (including human; see Fig. 3 and Supplementary Note 6) and with several other extant metazoan lineages (sea anemone15, placozoan23 and demosponge14). In our analyses, conserved macrosynteny requires only conserved linkage between orthologous genes, and is independent of intra-chromosome rearrangements (that is, scrambling of gene order) that are typical in phylogenetically deep comparisons10,15,23. Conserved macrosynteny in L. gigantea and C. teleta involves nearly one-half of the conserved protein coding genes in these species (Supplementary Note 6 and Supplementary Table 6.3.1). In contrast, we found no significant conservation of macrosynteny between H. robusta and other species, implying extensive reorganization in the leech genome relative to the last common spiralian ancestor.
Figure 3. Macrosynteny between spiralians, humans and Trichoplax.

a, The location of genes in scaffolds of L. gigantea, C. teleta and Trichoplax (a non-bilaterian outgroup that conserves synteny) relative to the position of their orthologues in the human genome. The human chromosome segments have been grouped according to their ancestral linkage group (ALG); chromosome segment identifiers are also shown (see ref. 10). Human genes in ALG 2 have their orthologues co-located on a limited set of scaffolds in L. gigantea, C. teleta and Trichoplax, indicating conserved linkage of this group of genes across eumetazoan lineages. In contrast, although ALG 17 and ALG 9 are preserved separately in Trichoplax, scaffolds of L. gigantea and C. teleta have homologous gene content either with ALG 9 or with both ALG 9 and ALG 17, indicating a translocation of one or more chromosome segments from ALG 17 to ALG 9 in the common ancestor of molluscs and annelids, after the divergence of the spiralian and vertebrate lineages. Genes inferred to derive from this translocated segment are shown in red. Subsequent intra-chromosomal rearrangement has dispersed the translocated genes among the genes of ALG 9. b, The scenario in panel a represented schematically on a phylogenetic tree, with chromosomes of ancestral and living genomes represented as horizontal blue bars and the translocated segment represented in red. c, The positions of human genes and their L. gigantea, C. teleta and Trichoplax adhaerens orthologues compared in dot plots schematically (and in the real data; see panel a) for three ALGs.
The observed conserved macro-synteny demonstrates the persistence of 17 ancient bilaterian ancestral linkage groups (ALGs) in the common ancestor of L. gigantea and C. teleta. Independent fusions (two in L. gigantea, three in C. teleta) subsequently reduced the number of bilaterian ALGs that remain distinct in these genomes. The conservation of 17 bilaterian ALGs among L. gigantea, C. teleta and various deuterostomes implies that the last common protostome and deuterostome ancestors also had this organization. Some ecdysozoans like Caenorhabditis elegans (soil nematode), Tribolium castaneum (beetle) and Bombyx mori (moth) (Supplementary Note 6) also show clear evidence of conserved macrosynteny. However, the large number of chromosome fusions and rearrangement events make similar reconstruction of the ancestral ecdysozoan ALGs impossible with current data (Supplementary Note 6).
Remarkably, we can also use the L. gigantea and C. teleta genomes to infer ancient translocations between linkage groups (Supplementary Note 6). For example, L. gigantea and C. teleta share a translocation relative to the last common bilaterian ancestor (Fig. 3), indicating that this genomic rearrangement occurred on the stem lineage leading from the bilaterian to the mollusc–annelid node. As noted above, more recent translocations that are not shared between L. gigantea and C. teleta are also evident (Supplementary Note 6). It remains unclear whether these genome reorganizations were causally involved in the radiation of diverse bilaterian lineages, or were simply neutral changes.
The most famous example of conserved microsynteny—conserved tight linkages between orthologous genes—is the Hox complex, an ancient cluster of homeodomain-containing transcription factors with conserved roles in patterning the anteroposterior body axis of animals24. In L. gigantea, 11 Hox genes occur as a single cluster that is structurally collinear with intact Hox clusters found in other genomes, and is the first intact cluster found in a lophotrochozoan (Fig. 4). C. teleta Hox genes occur in one-to-one correspondence with their L. gigantea counterparts but lie on three scaffolds, with the scaffold harbouring the posterior class gene post1 clearly disconnected from the main cluster25. We therefore infer that the last common mollusc–annelid ancestor had a single 11-gene Hox cluster (Supplementary Note 8) with 3 anterior-and 6 central-class genes, plus 2 posterior-class genes (post1 and post2) that arose by duplication along the spiralian (or lophotrochozoan) stem lineage26. In contrast, the Hox complex of H. robusta has fragmented extensively, consistent with the general loss of synteny conservation in H. robusta, and there have been multiple duplications and loss of two mollusc–annelid paralogy groups (the orthologues of the anterior-class proboscipedia and post1). Intriguingly, although the gene rearrangements observed in H. robusta are as extreme as in C. elegans, the leech is not particularly derived with respect to other genomic characters. This lineage may therefore be an interesting model for focused studies on rapid evolution of gene order. We also find other tightly linked groups of anciently duplicated (that is, paralogous) genes, including clusters of deeply diverged gene superfamilies such as the homeodomain25, forkhead box27 and wingless28 gene families that duplicated extensively before the bilaterian radiation but have remained linked (Supplementary Note 7.4).
Figure 4. The Hox gene complement and linkage in the three lophotrochozoan genomes and selected bilaterians.

Arrows indicate direction of transcription (orientation between scaffolds is arbitrary). Scaffolds with ends marked by black dots may be part of a larger Hox complex because the Hox gene is at the end of the scaffold. B. floridae, Branchiostoma floridae. Colours indicate unambiguously assigned paralogy groups (Hox1, Hox2, Hox3, Hox4, central class and posterior class).
Overall, we found hundreds of other examples of conserved microsyntenic blocks involving thousands of genes in L. gigantea, C. teleta and other metazoan genomes (Supplementary Note 7.1). We consider a microsyntenic block to be a group of three or more genes whose orthologues are tightly linked (that is, separated by no more than ten intervening genes) in two or more genomes. Microsyntenic blocks are often, but not always, embedded in a conserved macrosyntenic context. The count of 469 microsyntenic blocks that are putatively preserved from the bilaterian ancestor in at least one protostome and one deuterostome genome is substantially greater than the 157 blocks that would persist by chance in a simple model of genome rearrangement in which gene order is randomized within the macrosynteny blocks defined above (Supplementary Notes 6 and 7), implying either functional constraint on genome organization or intrinsically slow rates of rearrangement in some genomic regions. Considering the deeply diverged bilaterian lineages represented by L. gigantea, C. teleta and amphioxus (Supplementary Table), we found 77 conserved microsyntenic blocks (Supplementary Note 7.1), which in some cases are stably conserved across other metazoan genomes (Fig. 5). It is tempting to speculate that these conserved linkages are due to selection for preserving complex cis-regulatory landscapes (Supplementary Note 7.2).
Figure 5. Examples of conserved orthologous gene clusters.

a–c, Clusters of linked genes across diverse species. Within each panel, genes in the same colour are members of the same orthologous group, with the gene identifiers of defining members of the group indicated: C7orf42, (human) chromosome 7 open reading frame 42; Exoc7, exocyst complex component 7; FAM100, family with sequence similarity 100; FoxJ, forkhead box protein J; HDC, histidine decarboxylase; MGRN E3 Ub ligase, mahogunin ring finger E3 ubiquitin ligase; MORC, MORC family CW-type zinc finger; PSMG1, proteasome (prosome, macropain) assembly chaperone 1; ROX/MINT, Max-binding protein family member; SYAP1, synapse-associated protein 1; WBP11, WW domain binding protein 11; ZMPSTE24, zinc metallopeptidase, STE24 homologue. Scaffold positions for all displayed linkages are listed in Supplementary Note 7.2. d, Cumulative rates of microsynteny change plotted on a fixed metazoan tree topology. Branch lengths are proportional to the number of inferred genomic rearrangements. A. queenslandica, Amphimedon queenslandica; C. intestinalis, Ciona intestinalis; D. melanogaster, Drosophila melanogaster; D. pulex, Daphnia pulex; D. rerio, Danio rerio; G. gallus, Gallus gallus; H. magnipapillata, Hydra magnipapillata; I. scapularis, Ixodes scapularis; M. musculus, Mus musculus; N. vectensis, Nematostella vectensis; Ub, ubiquitin; X. tropicalis, Xenopus tropicalis.
Although molluscs and annelids are related to flies, nematodes and flatworms within the protostomes, we find that their genomes are in many ways more similar to those of invertebrate deuterostomes (such as amphioxus and sea urchin) as well as non-bilaterian metazoans (such as cnidarians, sponges and placozoans). These similarities reveal features of bilaterian and/or metazoan genomes that have been lost or diverged in many protostome genomes reported so far, and thus enable a more complete reconstruction of genomic features of the last common ancestors of protostomes, bilaterians and metazoans, including gene and chromosome structure and organization. Superimposed on these conserved features are evolutionary innovations—novel gene families and gene-family expansions and losses, as well as large- and small-scale genomic rearrangements—that make each clade unique. Nearly 20 other phylum-level taxa lack even a single genome sequence, and intra-phylum genomic variation can be extensive. Thus, for a comprehensive genomic understanding of the metazoan radiation a far larger sampling of genomes will be needed.
METHODS SUMMARY
Gene families and phylogeny
Orthology relationships were reconstructed for 22 metazoan genomes (Supplementary Fig. 3.3.1) using a phylogenetic clustering approach, which progressively examined reciprocal best scoring BLAST hits at decreasing phylogenetic nodes of a reference animal tree. We recovered 1,235 gene families with orthologous members in all genomes. To assess the effect of fast and slow evolving characters on the tree topology, several phylogenetic approaches were taken (see Supplementary Note 5).
Identification of 8,756 ancestral bilaterian genes
Gene families were considered to be ancestral bilaterian gene families when an orthologous group had at least two protostome and two deuterostome representatives (in-group) or two sequences from either in-group and two from basal (that is, non-bilaterian) metazoans (out-group).
Macrosynteny
Draft genome scaffolds were clustered into ancestral linkage groups (ALGs) based on the locations of orthologous genes in other metazoan genomes, as described previously10 We iteratively constructed a parsimonious scenario of chromosome evolution, and ancestral genes were assigned to ancestral ALGs when any other assignment would imply more hops between ALGs in the history of that gene family (Supplementary Note 6).
Microsynteny
Chromosomal locations of orthologous genes in two different species were compared. If another set of orthologous genes is identified within a maximal distance of 10 genes of the previous set, both sets were merged together into a microsyntenic block. Only syntenic blocks with at least three orthologues per species were considered.
Intron and indel identification and phylogeny
Gene families with a maximum of 2 missing species (out of 22) were included. Intron and indel positions were detected using conserved flanking sites (3 out of 8 amino acids), no gaps were allowed to flank introns. For indels, the flanking amino acids had to be conserved. Phylogenetic inference based on presence or absence was computed with MrBayes as described in Supplementary Note 5.3.
Supplementary Material
Acknowledgments
Work conducted by the US Department of Energy Joint Genome Institute is supported by the Office of Science of the US Department of Energy under contract no. DE-AC02-05CH11231. Early work on the analysis of these genomes was also supported by the Gordon and Betty Moore Foundation through a grant to the Center for Integrative Genomics at the University of California. D.S.R. thanks R. Melmon for a grant in support of this project. N.H.P. is supported by the National Science Foundation (NSF) under grant EF-0850294; D.A.W. is supported by the National Institutes of Health (NIH; RO1 GM 074619) and NSF (I0S-0922792); and O.S. is supported by Boehringer Ingelheim Fonds. E.E-G. was supported by a training grant from the US National Human Genome Research Institute. We thank A. Meyer and J. Gerhart for helpful discussions.
Footnotes
Author Contributions This study was conceived by J.L.B., D.R.L., D.A.W., E.C.S. and D.S.R. The project was led by D.S.R. with N.H.P. and O.S. The three genomes were assembled by H.S. and annotated by A.Y.T., R.P.O., A.A. and I.V.G. Comparative analyses of gene complements were carried out by N.H.P., P.H. and O.S. Phylogenomic analyses were carried out by F.M. and O.S. Macro- and microsynteny was analysed by N.H.P., O.S. and J.L. GPCR complements were analysed by T.L., O.S. and D.A. Innexin analysis was carried out by O.S., D.A.W. and D.-H.K. Hox gene complements were analysed by D.A.W. and D.-H.K., S.-J.C. and E.C.S. Additional analyses, data and materials were contributed by U.H., J.A.C. J.G., P.d.J., K.O., R.S. and E.E.-G. The main paper was written by O.S., F.M., N.H.P., D.A.W., E.C.S. and D.S.R. with input from other authors. O.S. wrote the Supplementary Information with input from other authors.
Author Information The Lottia gigantea whole-genome shotgun project has been deposited in GenBank under the accession AMQO00000000. The sequence version described in this paper is deposited under accession AMQO01000000 (PRJNA175706). The Helobdella robusta whole-genome shotgun project has been deposited in GenBank under the accession AMQM00000000. The sequence version described in this paper is the first version, AMQM01000000 (PRJNA175704). The Capitella teleta whole-genome shotgun project has been deposited in GenBank under the accession AMQN00000000. The sequence version described in this paper is the first version, AMQN01000000 (PRJNA175705).
Reprints and permissions information is available at www.nature.com/reprints.
The authors declare no competing financial interests.
Readers are welcome to comment on the online version of the paper.
Supplementary Information is available in the online version of the paper.
References
- 1.Wilson EB. The cell-lineage of Nereis. A contribution tothe cytogeny of the annelid body. J Morphol. 1892;6:361–480. [Google Scholar]
- 2.Conklin EG. The Embryology of Crepidula: a Contribution to the Cell Lineage and Early Development of Some Marine Gasteropods. Ginn & Company; 1897. [Google Scholar]
- 3.Henry JQ, Hejnol A, Perry KJ, Martindale MQ. Homology of ciliary bands in spiralian trochophores. Integr Comp Biol. 2007;47:865–871. doi: 10.1093/icb/icm035. [DOI] [PubMed] [Google Scholar]
- 4.Fedonkin MA, Waggoner BM. The Late Precambrian fossil Kimberella is a mollusc-like bilaterian organism. Nature. 1997;388:868–871. [Google Scholar]
- 5.Maloof AC, et al. The earliest Cambrian record of animals and ocean geochemical change. Geol Soc Am Bull. 2010;122:1731–1774. [Google Scholar]
- 6.Dunn CW, et al. Broad phylogenomic sampling improves resolution of the animal tree of life. Nature. 2008;452:745–749. doi: 10.1038/nature06614. [DOI] [PubMed] [Google Scholar]
- 7.Berriman M, et al. The genome of the blood fluke Schistosoma mansoni. Nature. 2009;460:352–358. doi: 10.1038/nature08160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.The Schistosoma japonicum Genome Sequencing and Functional Analysis Consortium. The Schistosoma japonicum genome reveals features of host–parasite interplay. Nature. 2009;460:345–351. doi: 10.1038/nature08140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robb SM, Ross E, Sanchez Alvarado A. SmedGD: the Schmidtea mediterranea genome database. Nucleic Acids Res. 2008;36:D599–D606. doi: 10.1093/nar/gkm684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Putnam NH, et al. The amphioxus genome and the evolution of the chordate karyotype. Nature. 2008;453:1064–1071. doi: 10.1038/nature06967. [DOI] [PubMed] [Google Scholar]
- 11.Raible F, et al. Vertebrate-type intron-rich genes in the marine annelid Platynereis dumerilii. Science. 2005;310:1325–1326. doi: 10.1126/science.1119089. [DOI] [PubMed] [Google Scholar]
- 12.Purschke G. Sense organs in polychaetes (Annelida) Dev Hydrobiology. 2005;179:53–78. [Google Scholar]
- 13.Zamanian M, et al. The repertoire of G protein-coupled receptors in the human parasite Schistosoma mansoni and the model organism Schmidtea mediterranea. BMC Genomics. 2011;12:596. doi: 10.1186/1471-2164-12-596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Srivastava M, et al. The Amphimedon queenslandica genome and the evolution of animal complexity. Nature. 2010;466:720–726. doi: 10.1038/nature09201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Putnam NH, et al. Sea anemone genome reveals ancestral eumetazoan gene repertoire and genomic organization. Science. 2007;317:86–94. doi: 10.1126/science.1139158. [DOI] [PubMed] [Google Scholar]
- 16.Telford MJ, Copley RR. Improving animal phylogenies with genomic data. Trends Genet. 2011;27:186–195. doi: 10.1016/j.tig.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 17.Delsuc F, Brinkmann H, Philippe H. Phylogenomics and the reconstruction of the tree of life. Nature Rev Genet. 2005;6:361–375. doi: 10.1038/nrg1603. [DOI] [PubMed] [Google Scholar]
- 18.Philippe H, et al. Phylogenomics revives traditional views on deep animal relationships. Curr Biol. 2009;19:706–712. doi: 10.1016/j.cub.2009.02.052. [DOI] [PubMed] [Google Scholar]
- 19.Adoutte A, et al. The new animal phylogeny: reliability and implications. Proc Natl Acad Sci USA. 2000;97:4453–4456. doi: 10.1073/pnas.97.9.4453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roy SW, Gilbert W. Resolution of a deep animal divergence by the patterns of intron conservation. Proc Natl Acad Sci USA. 2000;102:4403–4408. doi: 10.1073/pnas.0409891102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roy SW, Irimia M. Rare genomic characteris do not support Coelomata: intron loss/gain. Mol Biol Evol. 2008;25:620–625. doi: 10.1093/molbev/msn035. [DOI] [PubMed] [Google Scholar]
- 22.Rokas A, King N, Finnerty J, Carroll SB. Conflicting phylogenetic signals at the base of the metazoan tree. Evol Dev. 2003;5:346–359. doi: 10.1046/j.1525-142x.2003.03042.x. [DOI] [PubMed] [Google Scholar]
- 23.Srivastava M, et al. The Trichoplax genome and the nature of placozoans. Nature. 2008;454:955–960. doi: 10.1038/nature07191. [DOI] [PubMed] [Google Scholar]
- 24.Duboule D. The rise and fall of Hox gene clusters. Development. 2007;134:2549–2560. doi: 10.1242/dev.001065. [DOI] [PubMed] [Google Scholar]
- 25.Frobius AC, Seaver EC. Capitella sp I homeobrain-like, the first lophotrochozoan member of a novel paired-like homeobox gene family. Gene Expr Patterns. 2006;6:985–991. doi: 10.1016/j.modgep.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 26.de Rosa R, et al. Hox genes in brachiopods and priapulids and protostome evolution. Nature. 1999;399:772–776. doi: 10.1038/21631. [DOI] [PubMed] [Google Scholar]
- 27.Shimeld SM, Boyle MJ, Brunet T, Luke GN, Seaver EC. Clustered Fox genes in lophotrochozoans and the evolution of the bilaterian Fox gene cluster. Dev Biol. 2010;340:234–248. doi: 10.1016/j.ydbio.2010.01.015. [DOI] [PubMed] [Google Scholar]
- 28.Cho SJ, Valles Y, Giani VC, Jr, Seaver EC, Weisblat DA. Evolutionary dynamics of the wnt gene family: a lophotrochozoan perspective. Mol Biol Evol. 2010;27:1645–1658. doi: 10.1093/molbev/msq052. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.