

Abstract
Protease-activated receptors (PARs) are a family of G protein-coupled receptors (GPCRs) that are uniquely activated by proteolysis. There are four members of the PAR family including: PAR1, PAR2, PAR3 and PAR4. PARs are expressed primarily in cells of the vasculature and elicit cellular responses to coagulant and anti-coagulant proteases. PAR1 exemplifies the unusual proteolytic mechanism of receptor activation. Thrombin binds to and cleaves the N-terminal exodomain of PAR1 generating a new N-terminus that functions as a tethered ligand. The N-terminal tethered ligand domain of PAR1 binds intramolecularly to the receptor to trigger transmembrane signaling and cannot diffuse away. Similar to other GPCRs, activation of PARs promotes coupling to heterotrimeric G proteins at the plasma membrane. After activation, PARs are rapidly internalized to endosomes and then sorted to lysosomes and degraded. Internalization functions to uncouple PARs from heterotrimeric G proteins at the cell surface. However, recent studies indicate that activated internalized PARs signal from endosomes through the recruitment of β-arrestins and potentially other pathways. Here we provide an overview of methods and strategies used to examine endosomal signaling by PARs.
Keywords: β-arrestins, thrombin, GPCR, p38, MAPK, trafficking
1. Introduction
The idea that GPCRs can signal from endosomes was substantiated by studies showing that β-arrestins function as scaffolds to facilitate activation of MAPK signaling cascades on endosomes (Lefkowitz & Shenoy, 2005). Defea et al. was the first to show that activation of PAR2 results in β-arrestin-mediated recruitment of a Raf-1 and ERK1/2 signaling complex on endosomes (DeFea et al., 2000; Dery, Thoma, Wong, Grady, & Bunnett, 1999). In subsequent work, we demonstrated that phosphorylation of the PAR2 C-tail is critical for stabilizing β-arrestin association and kinetics of ERK1/2 activation, but is not essential for receptor desensitization nor internalization (Ricks & Trejo, 2009; Stalheim et al., 2005). In examining PAR1 signaling, it has become clear that β-arrestins transiently associate with the receptor (Chen, Paing, & Trejo, 2004) and are unlikely to mediate signaling from endosomes, raising the possibility that other mechanisms exist. We have shown that activated PAR1 is internalized and sorted to early endosomes at a time that coincides with p38 activation (Fig. 1) (Dores et al., 2012; M. M. Paing, Johnston, Siderovski, & Trejo, 2006), suggesting that p38 signaling may be initiated or sustained on endosomes. The majority of published studies have focused on PAR1 and PAR2 signaling and there is limited knowledge with regards to endosomal signaling by PAR3 or PAR4.
Figure 1.
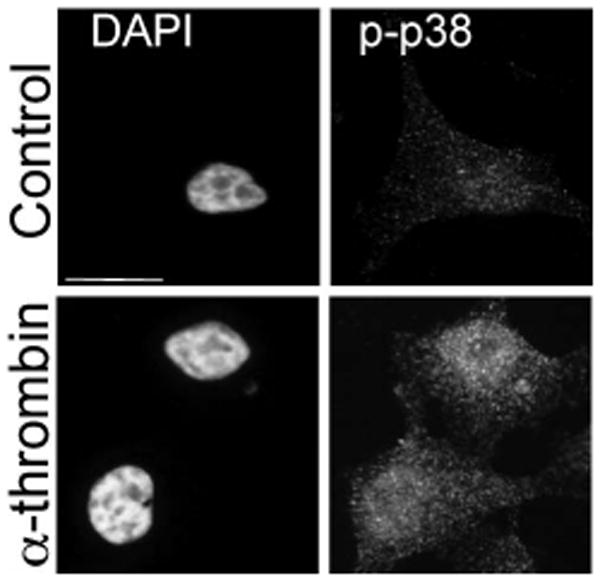
Thrombin-induced p38 phosphorylation. HeLa cells were serum starved and either left untreated (Control) or treated with 10 nM α-thrombin for 7 min at 37°C. Cells were fixed, processed and immunostained with anti-phospho p38 antibody and imaged by confocal microscopy. Cells were counter stained with DAPI to image nuclei. Scale bar, 10 μm.
While previous publications have provided detailed methodologies for examining growth factor receptor endosomal signaling, here we will provide an overview of methods used to examine endosomal signaling by PARs including imaging of p38, ERK1/2 and β-arrestins on endosomes as well as biochemical approaches to examine signaling complexes associated with PARs.
2. Imaging of p38, ERK1/2 and PAR1 on Endosomes
To investigate the potential activation of p38 and ERK1/2 on endosomes following stimulation of PAR1, we have used immunofluorescence microscopy. In these assays, endogenous p38 and ERK1/2 are detected with antibodies. The co-localization with early endosomal antigen-1 (EEA1), a marker of early endosomes, and/or PAR1 is used to assess recruitment to endosomes. While we outline approaches for PAR1, similar strategies can be used to examine p38 and ERK1/2 activation following stimulation of other PARs. We describe procedures for HeLa cells, which are commonly used to examine endocytic trafficking and endosomal signaling, and human umbilical vein-derived EA.hy926 endothelial cells, which express endogenous PAR1 and PAR2.
2.1. Detection of p38 MAPK by fluorescence microscopy
Glass coverslips (12 mm circular No.1, Chemglass Life Sciences #1760-012) are submerged in 100% ethanol, air dried, autoclaved and then placed in each well of a 24-well plate. Coverslips are coated with 0.4 ml of 0.33 μg/ml fibronectin (Sigma Cat. #F1141) diluted in phosphate buffered saline (PBS), pH 7.4 for 30 min at room temperature (RT) before cells are plated.
HeLa cells expressing PAR1 are seeded at 3 × 104 cells/well of a 24-well plate in DMEM supplemented with 10% fetal bovine serum (FBS) on fibronectin-coated coverslips. Human EA.hy926 endothelial cells are seeded at 1.5 × 105 cells/well in 24-well plates with DMEM containing 10% FBS as described (Edgell, McDonald, & Graham, 1983). Cells are grown for 48 h to reach ∼80% confluence.
Cells are then serum-starved by replacing growth medium with DMEM containing 10 mM HEPES, 1 mg/ml BSA and 1 mM CaCl2 (added just prior to use) (HeLa cells) or DMEM supplemented with 0.2% FBS (EA.hy926 cells).
After 24 h of serum starvation, cells are washed and incubated for an additional 3 h at 37°C with starvation media and then stimulated with 10 nM α-thrombin (Enzyme Research Laboratories Cat. #HT 1002a) for various times at 37°C.
After stimulation, cells are placed on ice, gently washed twice with cold PBS and fixed in 4% paraformaldehyde (PFA) for 5 min on ice, followed by 12 min at RT and then washed three times with PBS.
Coverslips are removed and placed with cells facing upwards onto a clean surface and then incubated with 100 μl of blocking buffer (5% goat serum + 0.3% Triton™ X-100 diluted in PBS) for 60 min at RT and then washed twice with PBS at RT.
Coverslips are then incubated with a 100 μl of either anti-phospho-p38 rabbit antibody (Cell Signaling Technology Cat. #4511) at 1:800 dilution, anti-p38 rabbit antibody (Cell Signaling Technology Cat. #9212) at 1:100 dilution or with anti-EEA1 monoclonal antibody (BD Biosciences Cat. #610457) at 1:1000 dilution in antibody dilution buffer (1% (w/v) BSA, 0.3% Triton™ X-100 and 0.5 mg/ml 4,6-diamidino-2-phenylindole (DAPI) reconstituted in PBS) and incubated overnight at 4°C in a sealed humidified container.
Cells are washed three times with PBS at RT and then incubated with 100 μl of Alexa Fluor®594 conjugated goat anti-rabbit antibody (Life Technologies Cat. #A-11012) or Alexa ®Fluor 488 goat anti-mouse antibody (Life Technologies Cat. #A-11001) diluted at 1:500 in antibody dilution buffer for 1 h at RT in a humidified chamber kept in the dark.
Cells are then washed three times with 100 μl of PBS at RT with each wash left on the cells for 5 min before removing.
After the last wash, 14 μl of FluorSave™ Reagent (EMD Millipore Cat. #345789) is gently layered on the coverslip. The coverslip is then mounted on a glass microscope slide (Fisher Cat. #12-550-123) and allowed to dry overnight. Mounted coverslips can be stored for months at 4°C
Mounted coverslips are warmed to room temperature before imaging on an Olympus IX81 Spinning Disc Unit Confocal Imaging system fitted with a PlanApo 60× oil objective (1.4 NA; Olympus) and a digital ORCA-ER Hamamatsu Photonics camera.
2.2. Detection of ERK1/2 by fluorescence microscopy
To detect the presence of ERK1/2 by fluorescence microscopy follow Steps 1 to 4 in Section 2.1. After fixation with 4% PFA for 5 min on ice (Step 5 in 2.1), wash cells three times with ice-cold PBS.
Then add ice-cold 100% methanol (use enough to cover the cells completely) and incubate at -20°C for 10 min. Cells are then washed twice with cold PBS.
After the second wash, the cells are incubated with 500 μl of blocking buffer containing 0.3% Triton™ X-100 and 5% goat serum diluted in PBS for 60 min at RT.
Cells are then washed twice with PBS at RT and incubated with anti-phospho-ERK1/2 mouse antibody (Cell Signaling Technology Cat. #4372) or anti-ERK1/2 rabbit antibody (Cell Signaling Technology Cat. #9102) diluted 1:200 in antibody dilution buffer described in Step 7 of Section 2.1.
After primary ERK1/2 antibody incubation, follow Steps 8-11 in Section 2.1.
2.3 Detection of PAR1 and PAR2 on endosomes by fluorescence microscopy
To assess PAR1 or PAR2 co-localization with p38 and ERK1/2, cells are pre-labeled with antibodies directed against epitope-tagged PARs expressed in HeLa cells or endogenous PARs expressed in endothelial cells. This approach can be used to determine if activated receptors co-localize with p38 and/or ERK1/2 signaling complexes on endosomes.
Follow Steps 1 to 3 of Section 2.1.
After 24 h of serum starvation, HeLa cells or EA.hy926 cells are washed and incubated for 3 h at 37°C with DMEM containing 10 mM HEPES, 1 mg/ml BSA and 1 mM CaCl2 and then chilled on ice.
HeLa cells expressing either PAR1 or PAR2 containing an N-terminal FLAG or HA epitope tag are then labeled with M1 anti-FLAG mouse antibody (Sigma Cat #F3040) or anti-HA mouse antibody (Covance Cat. # MMS-101R) respectively, diluted to 1:500 in DMEM starvation buffer for 1 h on ice. Endothelial cells expressing endogenous PAR1 can be labeled with anti-PAR1 WEDE mouse antibody (Beckman Coulter Cat. #IM2085) diluted at 1:100 in DMEM starvation buffer. Endogenous PAR2 can be labeled with anti-PAR2 rabbit polyclonal antibody generously provided by Dr. Wolfram Ruf (The Scripps Research Institute, La Jolla, CA) diluted at 1:500 in DMEM starvation buffer.
Cells are then washed three times and stimulated with either 100 μM TFLLRNPNDK (PAR1 specific agonist peptide), 100 μM SLIGKV (PAR2 specific agonist), 10 nM α-thrombin or 10 nM trypsin (Sigma Cat. #T-1426) for various times at 37°C. Note that proteases cleave off the N-terminal FLAG and HA epitope tags so should be avoided if using anti-FLAG or anti-HA antibodies.
Cells are then placed on ice, washed with cold PBS and processed for either p38 or ERK1/2 immunostaining as described in Sections 2.1 and 2.2. Note that the p38 immunostaining procedure will result in loss of PAR1 from the cell surface due to the Triton™ X-100 detergent. However, internalized PAR1 on endosomal structures is preserved.
3. β-arrestin recruitment to endosomes
β-arrestins associate with activated GPCRs on endosomes and function as scaffolds to facilitate assembly of MAPK signaling complexes (Lefkowitz & Shenoy, 2005). The intracellular localization of β-arrestin-1-GFP or β-arrestin-2-GFP using microscopy is a simple method to assess GPCR-stimulated endosomal signaling. Using HeLa cells, we described methods to examine co-localization of β-arrestins with activated PAR2 (Stalheim, et al., 2005). A similar strategy can be used to assess β-arrestin-GFP recruitment to thrombin-activated PAR1-PAR2 heterodimer in HeLa cells (Lin & Trejo, 2013). COS7 cells express low levels of β-arrestins and can be used as an alternative if a more robust cell model system is needed. Note that β-arrestin association with activated PAR1 is transient (Chen, et al., 2004) and β-arrestins are not required for PAR1 internalization (M.M. Paing, Stutts, Kohout, Lefkowitz, & Trejo, 2002). Thus, this method cannot be used to detect β-arrestin association with activated PAR1.
3.1. Detection of β-arrestin-GFP by fluorescence microscopy
Glass coverslips (18 mm circular No.1, Fisher #12-545-100) are treated as described in Step 1 of Section 2.1 and placed in 12-well dishes.
HeLa cells stably expressing a human PAR2 containing an N-terminal FLAG epitope (Stalheim, et al., 2005) are seeded at 8 × 104 cells/well in 12-well dishes to achieve ∼ 40% confluency (higher confluency will reduce transfection efficiency) and grown overnight in anti-biotic free DMEM containing 10% FBS.
Media was removed and replaced with 1000 μL of antibiotic free DMEM containing 10% FBS.
For each well of a 12-well dish the following was prepared. An aliquot 1.8 μL of 1 mg/ml PEI (Polysciences, Inc., Cat. #23966) was added to 100 μl of reduced serum Opti-MEM (Life Technologies Cat. #11058-021), gently mixed and incubated for 10 min at RT. Then, 300 ng of cDNA plasmid encoding β-arrestin-1-GFP or β-arrestin-2-GFP was added to the PEI-Opti-MEM, gently mixed and incubated at RT for 20 min. The ratio of PEI to plasmid used was optimized for each batch of 1 mg/ml PEI. Typically ratios of 6:1 to 3:1 of PEI:plasmid results in ∼ 60-80% transfection efficiency of HeLa cells after 48 h.
The final PEI, plasmid and Opti-MEM mixture was then added drop-wise to each well. After 24 h, wells are washed twice and left overnight in DMEM starvation buffer containing 10 mM HEPES, 1 mg/ml BSA and 1 mM CaCl2.
The next day, cells at ∼80% confluency are washed twice with DMEM starvation buffer and then incubated for an additional 3 h at 37°C in starvation media. Each well was washed once with pre-chilled DMEM starvation buffer, placed on ice and then incubated for 1 h at 4°C on ice with anti-FLAG rabbit antibody (Sigma Cat #7425) diluted 1:1000 in starvation buffer.
Each well was then washed three times in DMEM starvation media and stimulated with or without 100 μM SLIGKV (PAR2-specific agonist peptide) diluted in pre-warmed starvation DMEM buffer for various times at 37°C.
After stimulation, cells are place on ice, each well was washed twice with cold PBS, fixed with 1 ml of 4% PFA for 10 min on ice, washed with cold PBS and permeabilized with 1 ml of ice-cold 100% methanol for 30 sec on ice and then washed twice with PBS.
Coverslips are then removed and placed on a flat surface with cells facing upwards and washed three times with 300 μl of quench buffer (PBS containing 1% (w/v) non-fat dry milk and 0.15 M sodium acetate, pH 7) with each wash incubated for 5 min.
Coverslips are then washed three times with 300 μl of wash buffer (PBS with 1% (w/v) non-fat dry milk). Each wash was incubated for 5 min at RT.
Coverslips are then incubated with 300 μl Alexa Fluor®594 goat anti-rabbit antibody diluted in wash buffer at 1:1000 for 1 h at RT in the dark, washed three times with PBS and then mounted in 20 μl of FluorSave™ Reagent as described in Steps 10 -11 Section 2.1.
3.2 Detection of endogenous β-arrestin on endosomes by fluorescence microscopy
We have adapted this method from Marchese et al. (Malik & Marchese, 2010) to examine the intracellular localization of endogenous β-arrestins with thrombin-activated PAR1-PAR2 heterodimer in HeLa cells and endothelial cells (Lin & Trejo, 2013). This method can be used to image endogenous β-arrestin recruitment to endosomes following thrombin-activated PAR1-PAR2 heterodimer or activated PAR2 protomer.
Coverslips (12 mm circular No.1) are prepared and coated with fibronectin as described in Step 1 of Section 2.1 and placed in 24-well dishes.
HeLa cells expressing PAR1 and PAR2 are seeded at 3 × 104 cells/well of 24-well dishes. EA.hy926 endothelial cells expressing endogenous PAR1 and low levels of PAR2 are seeded 1.5 × 105 cells/well of 24-well dishes. To increase expression of PAR2 endothelial cells can be pretreated with 10 ng/ml TNF-α (PeproTech Inc. Cat. #300-01A) for 18 h.
After 48 h, each well was washed twice with DMEM starvation buffer and then incubated for an additional 3 h at 37°C in starvation media.
Cells are then stimulated with 10 nM α-thrombin, which transactivates PAR1-PAR2 heterodimer or 100 μM SLIGKV (PAR2 agonist peptide) diluted in DMEM starvation buffer for various times at 37°C.
Cells are placed on ice, each well was then washed twice with cold PBS, fixed with 4% PFA for 10 min at RT, washed twice with PBS and then permeabilized with 500 μl of 0.05% (w/v) saponin diluted in PBS for 10 min at RT.
Coverslips are then incubated with 500 μl of blocking buffer (PBS containing 0.05% (w/v) saponin and 5% goat serum) for 30 min at 37°C.
Coverslips are then placed on a flat surface and incubated with 50 μl of anti-β-arrestin rabbit antibody generously provided by Dr. Jeffrey Benovic (Thomas Jefferson University, Philadelphia, PA) diluted at 1:50 in blocking buffer for 1 h at 37°C in a moist chamber.
After incubation, coverslips with adherent cells are placed back into 24-well plates, washed five times with 500 μl of 0.05% (w/v) saponin diluted in PBS with the last wash incubated for 15 min at 37°C.
Coverslips are incubated with Alexa Fluor®594 conjugated goat anti-rabbit antibody diluted at 1:200 in blocking buffer for 30 min at 37°C in a moist chamber.
Coverslips are then washed five times with 0.05% (w/v) saponin diluted in PBS and mounted in 14 μl of FluorSave™ Reagent as described in Steps 10 -11 of Section 2.1.
3. Rab5 Q79L Expansion of Early Endosomes
Rab5 is a small GTP-binding protein that regulates vesicle budding, transport, tethering and fusion (Zerial & McBride, 2001). Ectopic expression of the Rab5 Q79L constitutively active mutant perturbs vesicle fusion and results in enlarged endosomes (Stenmark et al., 1994). The expansion of the early endosomal compartment can be used to enhance visualization of p38, ERK1/2 and PAR1 localized on endosomes. A similar strategy can be used for other PARs.
Glass coverslips are prepared as described in Step 1 of Section 2.1. HeLa cells expressing PAR1 or PAR2 are then seeded at 4 × 105 cells/per well of 24-well plates to achieve ∼40% confluency and grown overnight in DMEM supplemented with 10% FBS. Note that a higher confluency will reduce transfection efficiency.
After 24 h, media is removed and replaced with 500 μl of antibiotic free DMEM containing 10% FBS.
For each well of a 24-well dish the following was prepared. An aliquot 0.6 μl of PEI at 1 mg/ml was added to 50 μl of reduced serum Opti-MEM, gently mixed and incubated for 10 min at RT. Then, 100 ng of cDNA plasmid encoding Rab5 Q79L tagged with GFP was added to the PEI-Opti-MEM solution, gently mixed and incubated at RT for 20 min. Transfection of plasmids encoding Rab5 wildtype and/or GFP only should be performed in parallel as controls.
The final PEI and plasmid mixture diluted in Opti-MEM is then added drop-wise to each well.
Cells in suspension can be used to increase the transfection efficiency. In this method, 50 μl of the PEI, plasmid and Opti-MEM mixture is added directly to ∼8 ×104 cells diluted in 500 μl of antibiotic-free DMEM containing 10% FBS and incubated for 5 min at RT and then plated on fibronectin coated glass coverslips in 24-well plates.
After transfections, cells are then treated with various agonists and p38, ERK1/2, β-arrestins and PAR1 can be detected by immunofluorescence microscopy as described above.
5. Immunoprecipitation of PAR1 Signaling Complexes
Activation of PAR1 results in rapid internalization with the majority of the receptor localized to early endosomes after 10 min of agonist stimulation (Dores, et al., 2012; M. M. Paing, et al., 2006). Immunoprecipitation of internalized PAR1 can be used as a method to determine the potential co-association of signaling effectors using HeLa cells stably expressing PAR1 or endothelial cells expressing endogenous PAR1. The detection of signaling effectors associated with activated PAR1 can then be assayed by immunoblotting. Similar approaches can be used to examine other PAR family members.
HeLa cells expressing PAR1 are seeded at 1 × 106 cells per 10 cm2 dish coated with 4 ml of 0.33 μg/ml of fibronectin. Endothelial EA.hy926 cells are plated at 1.4 × 106 cells per 10 cm2 dish without fibronectin coating. Cells are grown for 48 h in normal growth media.
Cells are then incubated with DMEM BSA starvation (HeLa cells) or DMEM supplemented with 0.2% FBS (EA.hy926 cells) for 24 h at 37°C.
Cells are washed twice with 8 ml of pre-warmed DMEM starvation buffer, incubated for additional 3 h at 37°C and then stimulated with 10 nM α-thrombin for various times at 37°C.
After agonist stimulation, cells are placed on ice, washed with 10 ml of cold PBS supplemented with 1 mM CaCl2 (added just prior to use) and then lysed with 750 μl of ice-cold lysis buffer (50 mM Tris-HCl, pH 7.4, 1% Triton™ X-100, 150 mM NaCl, 50 mM NaF, 10 mM NaPP, 25 mM β-glycerol phosphate containing freshly added protease inhibitors including: aprotinin 2 μg/ml, leupeptin 1 μg/ml, pepstatin A 1 μg/ml, benzamidine 10 μg/ml and soybean trypsin inhibitor 1 μg/ml.
Cells lysates are transferred to a 1.5 ml microcentrifuge tube, passed 10 times through a 22.5 gauge needle and syringe, and gently rocked for 20 min at 4°C.
Cell lysates are centrifuged at 16,000 × g for 30 min at 4°C, transferred to a new 1.5 ml centrifuge tube and 50 μl is removed to determine protein concentrations determined using the Pierce® BCA Protein Assay Kit (Thermo Scientific Cat. #23225). Note that it is important to save ∼50 μg of lysates to use as controls for expression of proteins in total cell lysates.
Protein A Sepharose CL-4B (GE Health Biosciences Cat. #17-0780-01) are prepared by washing twice using lysis buffer and centrifuged at 450 × g for 3 min. Protein A Sepharose beads are then pre-incubated with 1 mg/ml BSA to block non-specific binding sites 1 h at 4°C and then washed with twice with lysis buffer.
For each sample, 12.5 μl of Protein A Sepharose blocked with BSA was pre-incubated with 0.4 μg of anti-PAR1 WEDE mouse antibody or IgG antibody control for 2 h at 4°C and then washed four times with lysis buffer before addition of cell lysates.
To preclear, add ∼1000 μg of cell lysates from each sample to 1.5 ml microcentrifuge tubes containing 12.5 μl of Protein A Sepharose beads blocked with BSA and gently rock for 1 h at 4°C.
Cell lysates are then centrifuged at 450 × g for 5 min at 4°C and supernatant is transferred to a new tube containing 12.5 μl of antibody immobilized Protein A Sepharose beads from Step 8 and gently rocked for 2 h at 4°C.
Samples are centrifuged at 450 × g and 4 °C, the supernatant is removed and the beads are washed three more times.
After the final wash, lysis buffer is completely removed from the beads using 27-guage needle and syringe and then 30 μl of 2 × Sample buffer (125 mM Tris-HCl, pH 6.5, 5% SDS, 20% glycerol, 0.003% bromophenol blue diluted in H20) containing 200 mM dithiothreitol (DTT) (added just prior to use) was added immediately. Samples are heated to 50°C for 10 min (to reduce sample/protein aggregation) then 95°C for 5 min.
Samples are centrifuged at 450 × g for 5 min at RT and ∼15 μl is loaded on SDS-PAGE, transferred to PVDF membranes and immunoblotted with various antibodies. Membranes can be stripped with Restore Western Blot Stripping Buffer (Thermo Scientific Cat. # 46430) and then reprobed with an anti-PAR1 rabbit polyclonal antibody to detect PAR1. Total cell lysates (10 μg) from Step 6 above can be also be examined by immunoblotting to ensure that equivalent amounts of proteins were present in cell lysates before immunoprecipitations.
6. Summary
Protease-activated receptors have important functions in vascular biology and cancer progression (Arora, Ricks, & Trejo, 2007; Coughlin, 2005). The regulation of PAR signaling is critical for the fidelity of thrombin signaling (Trejo, Hammes, & Coughlin, 1998) and dysregulation of PAR signaling has been implicated in pathophysiological disease processes (Arora, et al., 2007; Leger, Covic, & Kuliopulos, 2006). While it is clear that activation of PARs at the plasma membrane results in coupling to heterotrimeric G proteins the signaling elicited by internalized receptors from endosomes has yet to be fully elucidated and represents a significant gap in our knowledge that is critical to understand.
Acknowledgments
This work was supported by National Institutes of Health grants GM090689 and HL073328 (JT). NG is supported by an American Heart Association Postdoctoral Fellowship.
References
- Arora P, Ricks TK, Trejo J. Protease-activated receptor signalling, endocytic sorting and dysregulation in cancer. J Cell Sci. 2007;120:921–928. doi: 10.1242/jcs.03409. [DOI] [PubMed] [Google Scholar]
- Chen CH, Paing MM, Trejo J. Termination of protease-activated receptor-1 signaling by β-arrestins is independent of receptor phosphorylation. J Biol Chem. 2004;279:10020–10031. doi: 10.1074/jbc.M310590200. [DOI] [PubMed] [Google Scholar]
- Coughlin SR. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J Thromb Haemost. 2005;3:1800–1814. doi: 10.1111/j.1538-7836.2005.01377.x. [DOI] [PubMed] [Google Scholar]
- DeFea KA, Zalevski J, Thoma MS, Dery O, Mullins RD, Bunnett NW. β-Arrestin-dependent Endocytosis of Proteinase-activated Receptor-2 Is Required for Intracellular Targeting of Activated ERK1/2. J Cell Biol. 2000;148:1267–1281. doi: 10.1083/jcb.148.6.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dery O, Thoma MS, Wong H, Grady EF, Bunnett NW. Trafficking of proteinase-activated receptor-2 and β-arrestin-1 tagged with green fluorescent protein. J Biol Chem. 1999;274:18524–18535. doi: 10.1074/jbc.274.26.18524. [DOI] [PubMed] [Google Scholar]
- Dores MR, Chen B, Lin H, Soh UJK, Paing MM, Montagne WA, Meerloo T, Trejo J. ALIX binds a YPX3L motif of the GPCR PAR1 and mediates ubiquitin-independent ESCRT-III/MVB sorting. J Cell Biol. 2012;197:407–419. doi: 10.1083/jcb.201110031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgell CJ, McDonald CC, Graham JB. Permanent cell line expressing human factor VIII-related antigen established by hybridization. Proc Natl Acad Sci USA. 1983;12:3734–3737. doi: 10.1073/pnas.80.12.3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefkowitz RJ, Shenoy SK. Transduction of Receptor Signals by β-arrestins. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- Leger AJ, Covic L, Kuliopulos A. Protease-activated receptors in cardiovascular diseases. Circulation. 2006;114(10):1070–1077. doi: 10.1161/CIRCULATIONAHA.105.574830. [DOI] [PubMed] [Google Scholar]
- Lin H, Trejo J. Transactivation of the PAR1-PAR2 heterodimer by thrombin elicits beta-arrestin endosomal signaling. J Biol Chem. 2013 doi: 10.1074/jbc.M112.439950. In revision. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik R, Marchese A. Arrestin-2 interacts with the endosomal sorting complex required for transport machinery to modulate endosomal sorting of CXCR4. Mol Biol Cell. 2010;21(14):2529–2541. doi: 10.1091/mbc.E10-02-0169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paing MM, Johnston CA, Siderovski DP, Trejo J. Clathrin Adaptor AP2 Regulates Thrombin Receptor Constitutive Internalization and Endothelial Cell Resensitization. Mol Cell Biol. 2006;28:3231–3242. doi: 10.1128/MCB.26.8.3231-3242.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paing MM, Stutts AB, Kohout TA, Lefkowitz RJ, Trejo J. β-arrestins regulate protease-activated receptor-1 desensitization but not internalization or down-regulation. J Biol Chem. 2002;277:1292–1300. doi: 10.1074/jbc.M109160200. [DOI] [PubMed] [Google Scholar]
- Ricks T, Trejo J. Phosphorylation of protease-activated receptor-2 differentially regulates desensitization and internalization. J Biol Chem. 2009;284:34444–34457. doi: 10.1074/jbc.M109.048942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalheim L, Ding Y, Gullapalli A, Paing MM, Wolfe BL, Morris DR, Trejo J. Multiple independent functions of arrestins in regulation of protease-activated receptor-2 signaling and trafficking. Mol Pharm. 2005;67:1–10. doi: 10.1124/mol.104.006072. [DOI] [PubMed] [Google Scholar]
- Stenmark H, Parton RG, Steele-Mortimer O, Lutcke A, Gruenberg J, Zerial M. Inhibition of rab5 GTPase activity stimulates membrane fusion in endocytosis. EMBO J. 1994;13:1287–1296. doi: 10.1002/j.1460-2075.1994.tb06381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trejo J, Hammes SR, Coughlin SR. Termination of signaling by protease-activated receptor-1 is linked to lysosomal sorting. Proc Natl Acad Sci USA. 1998;95:13698–13702. doi: 10.1073/pnas.95.23.13698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerial M, McBride H. Rab Proteins As Membrane Organizers. Nat Rev Mol Cell Biol. 2001;2:107–118. doi: 10.1038/35052055. [DOI] [PubMed] [Google Scholar]