

Abstract
Inhibition and aging of neuropathy target esterase (NTE) by neuropathic organophosphorus (OP) compounds triggers OP compound-induced delayed neuropathy (OPIDN), whereas inhibition of acetylcholinesterase (AChE) produces cholinergic toxicity. The neuropathic potential of an OP compound is defined by its relative inhibitory potency (RIP) toward NTE vs. AChE assessed by enzyme assays following dosing in vivo or after incubations of direct-acting compounds or active metabolites with enzymes in vitro. The standard animal model of OPIDN is the adult hen, but its large size and high husbandry costs make this species a burdensome model for assessing neuropathic potential. Although the mouse does not readily exhibit clinical signs of OPIDN, it displays axonal lesions and expresses brain AChE and NTE. Therefore, the present research was performed as a further test of the hypothesis that inhibition of mouse brain AChE and NTE could be used to assess neuropathic potential using mouse brain preparations in vitro or employing mouse brain assays following dosing of OP compounds in vivo. Excellent correlations were obtained for inhibition kinetics in vitro of mouse brain enzymes versus hen brain and human recombinant enzymes. Furthermore, inhibition of mouse brain AChE and NTE after dosing with OP compounds afforded ED50 ratios that agreed with RIPs assessed in vitro. Taken together, results with mouse brain enzymes demonstrated consistent correspondence between in vitro and in vivo predictors of neuropathic potential, thus adding to previous studies supporting the validity of a mouse model for biochemical assessment of the ability of OP compounds to produce OPIDN.
Keywords: acetylcholinesterase (AChE), mouse, organophosphorus compound-induced delayed neuropathy (OPIDN), organophosphorus (OP) compounds, neuropathy target esterase (NTE)
INTRODUCTION
Organophosphorus (OP) compounds inhibit serine esterases by organophosphylation of the active site serine (Aldridge and Davison, 1953; Richardson, 2010). The inhibited enzyme can undergo aging, which involves net loss of a side chain from the OP moiety to yield a stable negatively charged species still attached to the active site serine.
Inhibition of acetylcholinesterase (AChE) produces cholinergic toxicity regardless of whether or not aging has occurred. In contrast, both inhibition and aging of neuropathy target esterase (NTE) are required to cause OP compound-induced delayed neurotoxicity (OPIDN), a distal degeneration of long, large-diameter sensorimotor axons in spinal cord and peripheral nerves resulting in sensory loss and paralysis (Richardson et al., 2013).
Although there are many causes of peripheral neuropathy (Hughes, 2002), including chronological aging and diabetes (Callaghan et al., 2012; Singer et al., 2012), OPIDN is a specific chemically induced disorder that involves a combination of peripheral neuropathy and axonopathy in spinal cord tracts (Richardson, 2005). Moreover, whereas AChE inhibition, acute stress, and inflammatory stimuli can induce changes in AChE gene expression and microRNAs targeting AChE (Evron et al., 2007; Kaufer et al., 1998; Shaked et al., 2009), AChE inhibition is not involved in the etiology of OPIDN (Lotti, 1992).
When an OP compound is substantially more potent as an inhibitor of AChE than of NTE, cholinergic toxicity could result in lethality, thus obviating the development of OPIDN. However, when an OP compound is neuropathic, cholinergic toxicity will be mild, and sufficient NTE can be inhibited and aged to initiate OPIDN. Accordingly, earlier studies with enzymes from hen and human brain have shown that the neuropathic potential of an OP compound can be assessed by its relative inhibitory potency (RIP) against NTE vs. AChE (Kropp and Richardson, 2003; Lotti and Johnson, 1978).
Apart from inhibition and aging of NTE, the pathogenic mechanisms that transpire between exposure to a neuropathic OP compound and the emergence of OPIDN 1–4 weeks later are poorly understood. Elucidation of the pathogenesis and mechanism of OPIDN, as well as the assessment of OP compounds for their neuropathic potential, have been hampered by gaps in knowledge concerning the physiological function of NTE and deficiencies in available models, either in vivo or in vitro (Pomeroy-Black et al., 2007). Thus, it has been difficult to develop models that accommodate such features as the apparent need for axons of sufficient length and caliber, higher susceptibility of adult versus young animals, and species differences in the biotransformation of OP compounds and turnover rates of NTE and other nervous system proteins (Moretto and Lotti, 2002).
Currently, the most widely accepted animal model for the study of OPIDN or assessment of the neuropathic potential of OP compounds is the adult hen (Doherty, 2006; Emerick et al., 2012). However, compared to the usual laboratory rodents (e.g., rats or mice), hens are difficult to acquire and maintain for laboratory studies, and their substantially larger size requires considerably greater amounts of test materials for dosing. Consequently, it would be of interest to develop a rodent model for studying at least some aspects of OPIDN and/or for assessing the neuropathic potential of OP compounds.
Among rodents, rats have been thought to be resistant to OPIDN, because they do not readily display clinical signs of hind limb paralysis, despite exposure to high levels of neuropathic compounds (Abou-Donia, 1981). Mice have been considered even less suitable than rats as models for OPIDN, because of the difficulty in producing clinical signs and achieving suprathreshold (> 70%) inhibition of neural NTE in vivo following dosing with neuropathic OP compounds (Veronesi et al., 1991). Nevertheless, mice develop axonal lesions and express brain AChE and NTE activities that are inhibited in a dose-related manner by OP compounds (Lapadula et al., 1985; Read et al., 2010). Moreover, owing to the rapid turnover of mouse brain NTE (Meredith and Johnson, 1988), higher levels of inhibition can be seen by assaying the enzyme at shorter intervals than 24–72 h after dosing (Read et al., 2010), as was the practice in earlier studies using mice or hens (Malygin et al., 2003; Veronesi et al., 1991).
The present work was carried out as a further test of the hypothesis that inhibition of mouse brain AChE and NTE in vitro and in vivo could be used to assess neuropathic potential of OP compounds. Using OP compounds spanning several orders of magnitude in inhibitory potency toward each enzyme, we determined bimolecular rate constants of inhibition (ki) for mouse brain AChE and NTE, calculated RIPs, and examined the correlations of these values with those obtained for hen brain AChE and NTE as well as human recombinant AChE and NTE esterase domain (NEST). In addition, in order to evaluate the capability of the mouse model for assessing neuropathic potential in vivo, we determined 24-h LD50 values as well as ED50 values from dose-response curves for mouse brain AChE and NTE inhibition soon (1 h) after dosing by three OP compounds that differed in neuropathic potential as assessed in vitro. The structures of the OP compounds used in the present study are shown in Fig. 1.
Figure 1.

Structures of test compounds.
MATERIALS AND METHODS
Chemicals
Phenyl valerate (PV), N,N′-di-2-propylphosphorodiamidofluoridate (mipafox, MIP), O,O-di-1-propyl-O-2,2-dichlorovinyl phosphate (PrDChVP), O,O-diethyl-O-(1-trifluoromethyl-2,2,2-trifluoroethyl) phosphate (diEt-PFP); O,O-dibutyl-O-(1-trifluoromethyl-2,2,2-trifluoroethyl) phosphate (diBu-PFP) were synthesized and characterized in the Institute of Physiologically Active Compounds, Russian Academy of Sciences (IPAC, RAS, Chernogolovka, Russia) and kindly furnished by Dr. Alexey Aksinenko. Syntheses have also been previously described for PV and MIP (Johnson, 1977), PrDChVP (Allen and Johnson, 1955), and diEt-PFP and diBu-PFP (Makhaeva et al., 2008, 2009a). The purity of all substances produced by IPAC, RAS was > 99% (by spectral and chromatographic analysis). O,O-diisopropylphosphorofluoridate (DFP; > 90%, GC/MS) and O,O-diethyl-4-nitrophenyl phosphate (paraoxon, PO; > 90%, GC/MS) were purchased from Sigma-Aldrich (St. Louis, MO); O,O-diethyl-O-(3,4,5-trichloro-2-pyridyl) phosphate (chlorpyrifos oxon, CPO; > 96%, GC), was kindly furnished by Dow AgroSciences (Indianapolis, IN); O,O-diethyl-O-(3,4,5-trichloro-2-pyridyl) phosphorothioate (chlorpyrifos, CPS; > 99%, GC), was provided by Dow Chemical (Midland, MI). All other chemicals were analytical grade or the highest grade commercially available and used without further purification. Aqueous solutions were prepared using deionized water.
Animals
All experiments with animal subjects were carried out according to protocols for the use and care of laboratory animals approved by the Institute of Physiologically Active Compounds, RAS (Chernogolovka, Russia) or the University Committee on Use and Care of Animals (Ann Arbor, MI). Adult white Leghorn hens were from the Department of Animal Science, Michigan State University (East Lansing, MI) or Noginsk poultry farm (Noginsk, Russia). Adult male and/or female C57BL mice (18–28 g, 3 mo of age) from the University of Michigan Department of Neurology colony (Ann Arbor, MI) or outbred male CD-1 albino mice (20–24 g, 2 mo of age) (Pushchinko, Russia) were used. Hens were housed 3 per stainless steel cage at 20–23 °C and fed laying mash (Purina, St. Louis, MI; or Agro-range, Russia) and tap water ad libitum. Mice were housed 6–8 per plastic shoebox cage at 22–24 °C and fed mouse chow (Purina, St. Louis, MO; or Agro-range, Russia) and tap water ad libitum.
Hen Brain and Mouse Brain AChE and NTE Preparations
Animals were euthanized via CO2 asphyxiation. Brains were immediately removed, weighed, frozen in dry ice or liquid nitrogen, and stored at −80 °C until use. Frozen brains were thawed and homogenized in 0.32 M sucrose at 4 °C. Homogenates were centrifuged 9000 × g for 20 min at 4 °C. Aliquots of the supernatants (brain 9S fraction) were stored at −80 °C until use. For some experiments with hen brain NTE, a lyophilized membrane fraction consisting of combined mitochondrial/synaptosomal and microsomal pellets (P2 + P3) (Richardson et al., 1979) preinhibited with PO (40 μM at 25°C for 45 min) was used (Makhaeva and Malygin, 1999; Makhaeva et al., 1998). For a given enzyme and species, no significant differences in ki values for a specific inhibitor were found between enzyme sources (data not shown).
Human Recombinant AChE and NTE Esterase Domain (NEST)
In order to avert the logistical and biohazard issues accompanying the use of human tissues and to take advantage of the availability of pure human enzymes, human recombinant enzymes were used. Human recombinant AChE was obtained from Sigma-Aldrich (St. Louis, MO). Because full-length NTE is difficult to isolate or produce, human recombinant NTE esterase domain (NEST) was used as a surrogate. NEST was prepared and purified as previously described (Atkins and Glynn, 2000; Atkins et al., 2002) using a human brain plasmid kindly supplied by Dr. Paul Glynn of the MRC Toxicology Unit (Leicester, UK). Comprising the active site serine, NEST is the shortest segment of NTE that retains esterase activity; moreover, the catalytic properties of NEST, including its response to OP inhibitors, closely resemble those of full-length NTE (Kropp et al., 2004; Makhaeva et al., 2010; Van Tienhoven et al., 2002).
Protein Assay
Protein was assayed by the Coomassie blue dye-binding method, with bovine serum albumin as a standard (Bradford, 1976).
AChE Activity and Inhibition
AChE was inhibited by preincubation with OP compounds (Makhaeva et al., 2010) and its residual activity determined by a modification of a colorimetric assay (Ellman et al., 1961). Briefly, stock solutions of inhibitors in water-miscible organic solvents (acetone or DMSO) were serially diluted to appropriate working concentrations in 50 mM Tris-citrate buffer (pH 6.0 at 25 °C), so that the final organic solvent concentration in the preincubation mixture was ≤ 1% (v/v), a concentration range previously shown to have no significant effect on enzyme activity. Enzyme preparations in 100 mM sodium phosphate buffer pH 8.0 (250 μL) were preincubated with inhibitor working solutions (50 μL) at 37 °C for measured times, e.g., 0, 3, 6, 9, and 12 min. At the end of each preincubation interval, 50 μL of the preincubated enzyme mixture was added to 200 μL prewarmed substrate solution containing 1.25 mM acetylthiocholine and 0.4 mM 5,5′-dithio-bis(2-nitrobenzoic acid) in 96-well plates, and the activity of AChE at 37 °C was measured by the rate of change in absorbance at 412 nm using a SpectraMax 340 or Bio-Rad Benchmark Plus microplate reader.
NTE or NEST Activity and Inhibition
NTE or NEST was inhibited by preincubation with OP compounds (Makhaeva et al., 2010) as described above for AChE inhibition, and its residual activity determined by a modification of a colorimetric assay (Kayyali et al., 1991). All reactions were carried out at 37 °C for the entire assay. Homogenates were diluted in 50 mM Tris-HCl, 0.1 mM EDTA, pH 8.0 at 37 °C. NTE activity was defined as the PV hydrolase activity inhibited by preincubation for 20 min with 40 μM PO but not abolished by preincubation for 20 min with 40 μM PO plus 250 μM MIP. When the lyophilized hen brain NTE preparation was used, PO was omitted from the preincubation step, because this preparation was previously preincubated with PO. For NEST activity no preincubation with PO or MIP was required, because no other esterases are present (Kropp et al., 2004). Test inhibitors were prepared and diluted as described in the section above on measurement of AChE activity and inhibition. For NTE, test inhibitors were added at the end of the first preincubation interval and inhibition was continued for various measured times, usually between 0 and 20 min, before addition of substrate (Kropp and Richardson, 2003). For NEST, no preincubation with PO or PO plus MIP was required; therefore, test inhibitors were added to NEST preparations and inhibition allowed to progress for various measured times. At the end of the final preincubation interval, substrate solution (100 μL) consisting of 5.3 mM PV/N,N′-dimethylformamide diluted in 0.03% (w/v) Triton X-100 was added, and the reaction was allowed to proceed for 20 min, during which PV was hydrolyzed by active NTE or NEST to produce phenol. Stop solution (100 μL) of 5.0 mg/mL sodium dodecyl sulfate/1.23 mM 4-aminoantipurine (4-AAP) was added, and the reaction of 4-AAP with the phenol product was allowed to proceed for 3 min. The chromophore was produced by adding 50 μL of 12.1 mM K3Fe(CN)6, and color was allowed to develop and stabilize for 10 min. The endpoint absorbance was measured at 486 nm using a SpectraMax 340 or Bio-Rad Benchmark plus microplate reader.
Calculation of Bimolecular Rate Constants of Inhibition, ki
The apparent ki, values of OP inhibitors against esterases were determined as described previously (Doorn et al., 2003; Kropp and Richardson, 2003; Richardson, 1992). Briefly, slopes of primary linear kinetic plots of ln (% activity) vs. time gave the apparent first-order rate constants of inhibition (k′) for each [I], and the slope of the secondary linear kinetic plot of −k′ vs. [I] gave the ki for each inhibitor.
In Vivo Inhibition of AChE and NTE in Mouse Brain
In vivo experiments were carried out on outbred male white mice (18–25 g). PrDChVP, diEt-PFP, and diBu-PFP were dissolved in DMSO and injected i.p. in a volume of approximately 0.1 ml in 5–12 increasing doses of each tested compound. For each dose at least 6 animals were used. Control animals for diEt-PFP and diBu-PFP were injected only with DMSO. Because of the higher cholinergic toxicity of PrDChVP, mice in this group were given atropine sulfate, 20 mg/kg i.p. in water 20 min before injection with the OP compound; in this case, control animals received atropine sulfate and DMSO. After 1 h, mice were decapitated under CO2 anesthesia and brains removed for determination of NTE and AChE activities. Brains were weighed, frozen in liquid nitrogen, and stored at −80 °C until use. For assay, brains were thawed and each brain was homogenized at 4°C in 5 volumes of buffer (50 mM Tris-HCl, 0.2 mM EDTA, pH 8.0) with a Potter homogenizer. The homogenates were centrifuged (15 min at 9000 × g at 4°C) to prepare the 9S supernatant used for enzyme assay (Padilla and Veronesi, 1985). Aliquots of the supernatants (brain 9S fraction) were stored at −80 °C until use. Esterase activity in brain from mice treated with the OP compounds (OP compound plus atropine for PrDChVP) was determined and compared to activity in tissue samples from animals treated with DMSO or DMSO plus atropine.
Acute Toxicity Assessment
The 24-h acute i.p. toxicity of PrDChVP, diEt-PFP, and diBu-PFP was determined in outbred male white mice weighing 18–25 g, using 5–7 dose levels per compound and 6–8 animals per dose level.
Statistical Analysis
Data are expressed as mean ± SEM or mean and 95% CI. Plots, regressions, and correlations were carried out using Origin 6.1 software, OriginLab Corp. (Northampton, MA), Prism 6.0 for Windows, or Prism 6d for Mac OS X, GraphPad Software, Inc. (San Diego, CA). LD50 values were calculated by probit analysis using BioStat 2006 (AnalystSoft, Alexandria, VA).
RESULTS
Inhibition of AChE and NTE/NEST In Vitro
Tables 1 and 2 list the experimental ki and corresponding calculated IC50 values obtained in the present study for CPS, CPO, DFP, MIP, PrDChVP, diEt-PFP, and diBu-PFP against AChE and NTE or NEST from different sources (hen brain, mouse brain, and human recombinant enzymes). There was good agreement in inhibitory potency for a given inhibitor across species for AChE and for NTE or NEST. Moreover, correlations of log ki values for mouse brain with those for hen brain or human recombinant enzymes were excellent (r > 0.99), as was the correlation between log ki values for human and hen enzymes (Fig. 2 and 3).
Table 1.
Experimental ki and calculated 20-min IC50 values for OP inhibitors against different species of AChEa
| Inhibitor | Species | |||||
|---|---|---|---|---|---|---|
| Hen | Mouse | Human | ||||
| ki | IC50 | ki | IC50 | ki | IC50 | |
| CPS | NI | ND | NI | ND | NI | ND |
| CPO | 1.06 ± 0.07 × 107 | 3.27 ± 0.22 × 10−9 | 1.13 ± 0.03 × 107 | 3.07 ± 0.08 × 10−9 | 1.30 ± 0.08 × 107 | 2.66 ± 0.16 × 10−9 |
| DFP | 1.43 ± 0.05 × 105 | 2.42 ± 0.08 × 10−7 | 9.89 ± 0.61 × 104 | 3.50 ± 0.22 × 10−7 | 1.11 ± 0.11 × 105 | 3.12 ± 0.31 × 10−7 |
| MIP | 1.14 ± 0.07 × 103 | 3.03 ± 0.19 × 10−5 | 4.64 ± 0.60 × 102 | 7.46 ± 0.97 × 10−5 | 2.29 ± 0.06 × 103 | 1.51 ± 0.04 × 10−5 |
| PrDChVP | 3.60 ± 0.12 × 105 | 9.62 ± 0.32 × 10−8 | 3.70 ± 0.22 × 105 | 9.36 ± 0.56 × 10−8 | ND | ND |
| diEt-PFP | 1.11 ± 0.06 × 103 | 3.12 ± 0.17 × 10−5 | 7.22 ± 0.36 ×102 | 4.80 ± 0.24 × 10−5 | ND | ND |
| diBu-PFP | 1.80 ± 0.14 × 105 | 1.92 ± 0.15 × 10−7 | 7.12 ± 0.33 × 104 | 4.87 ± 0.22 × 10−7 | ND | ND |
Values are mean ± SEM (n = 3 or 4). Fixed-time (20-min) IC50 values were calculated from the relationship, IC50 = ln2/(ki × t), where IC50 = inhibitor concentration to produce 50% inhibition of the enzyme, ln2 = natural logarithm of 2 = 0.693, ki = bimolecular rate constant of inhibition, and t = preincubation time with inhibitor = 20 min. Units are M−1min−1 for ki and M for IC50. Hen and mouse AChE activity was assayed in 9S fractions from brain homogenates. Human AChE was assayed in preparations from commercial recombinant enzyme. NI = No inhibition at the highest concentration tested (0.1 mM). ND = Not determined.
Table 2.
Experimental ki and calculated 20-min IC50 values for OP inhibitors against different species of NTE or NESTa
| Inhibitor | Species | |||||
|---|---|---|---|---|---|---|
| Hen | Mouse | Human | ||||
| ki | IC50 | ki | IC50 | ki | IC50 | |
| CPS | NI | ND | NI | ND | NI | ND |
| CPO | 4.00 ± 0.76 × 105 | 8.66 ± 1.65 ×10−8 | 4.53 ± 1.09 × 105 | 7.65 ± 1.84 ×10−8 | 1.54 ± 0.33 × 105 | 2.25 ± 0.48 ×10−7 |
| DFP | 6.37 ± 0.26 × 104 | 5.44 ± 0.22 ×10−7 | 3.08 ± 0.39 × 104 | 1.12 ± 0.14 ×10−6 | 3.75 ± 0.24 × 104 | 9.24 ± 0.59 ×10−7 |
| MIP | 4.66 ± 0.49 × 103 | 7.44 ± 0.78 ×10−6 | 1.37 ± 0.13 × 103 | 2.53 ± 0.24 ×10−5 | 4.34 ± 0.14 × 103 | 7.98 ± 0.26 ×10−6 |
| PrDChVP | 1.25 ± 0.11 × 106 | 2.77 ± 0.24 ×10−8 | 1.44 ± 0.06 × 106 | 2.41 ± 0.10 ×10−8 | ND | ND |
| diEt-PFP | 2.50 ± 0.14 × 102 | 1.39 ± 0.08 ×10−4 | 1.22 ± 0.05 × 102 | 2.84 ± 0.12 ×10−4 | ND | ND |
| diBu-PFP | 1.27 ± 0.09 × 106 | 2.73 ± 0.19 ×10−8 | 5.11 ± 0.23 × 105 | 6.78 ± 0.30 ×10−8 | ND | ND |
Values are mean ± SEM (n = 3 or 4). Fixed-time (20-min) IC50 values were calculated from the relationship, IC50 = ln2/(ki × t), where IC50 = inhibitor concentration to produce 50% inhibition of the enzyme, ln2 = natural logarithm of 2 = 0.693, ki = bimolecular rate constant of inhibition, and t = preincubation time with inhibitor = 20 min. Units are M−1min−1 for ki and M for IC50. Hen NTE activity was assayed in 9S or lyophilized membrane fractions from brain homogenates. Mouse NTE activity was assayed in 9S fractions from brain homogenates. Human NEST activity was assayed in preparations from human recombinant NEST. NI = No inhibition at the highest concentration tested (0.1 mM). ND = Not determined.
Figure 2.
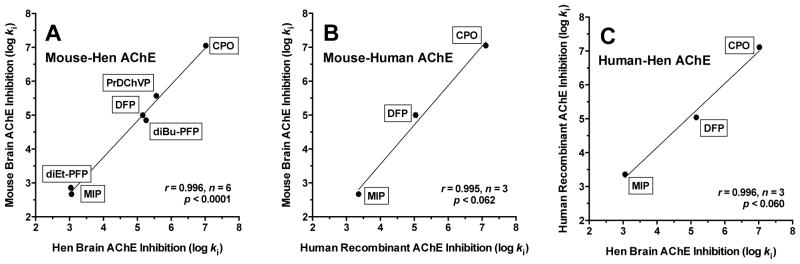
Correlations of log ki for inhibition of AChE. (A) mouse brain and hen brain AChE; (B) mouse brain and human recombinant AChE; (C) human recombinant AChE and hen brain AChE. ki = bimolecular rate constant of inhibition (M−1min−1).
Figure 3.
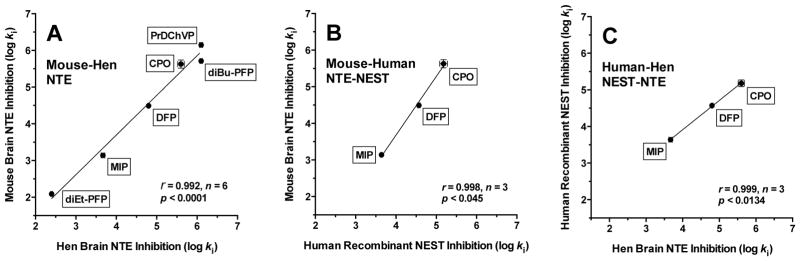
Correlations of log ki for inhibition of NTE or NEST. (A) mouse brain NTE and hen brain NTE; (B) mouse brain NTE and human recombinant NEST; (C) human recombinant NEST and hen brain NTE. ki = bimolecular rate constant of inhibition (M−1min−1).
Table 3 shows the RIP values calculated from the respective ki data given in Tables 1 and 2. The results predict that CPO is highly cholinergic (RIP ≪ 1), whereas DFP and diEt-PFP are slightly cholinergic (RIP <1), and MIP, PrDChVP, and diBu-PFP are neuropathic (RIP >1). In addition, there were strong correlations (r > 0.97) of log RIP values for mouse brain enzymes with those obtained from either hen brain or human recombinant enzymes, as well as between log RIP values for human and hen enzymes (Fig. 4).
Table 3.
Relative inhibitory potential (RIP) for OP inhibitors against AChE versus NTE or NEST from different speciesa
| Inhibitor | Species | ||
|---|---|---|---|
| Hen | Mouse | Human | |
| CPS | NI | NI | NI |
| CPO | 0.030 ± 0.006 | 0.036 ± 0.009 | 0.012 ± 0.003 |
| DFP | 0.45 ± 0.024 | 0.30 ± 0.02 | 0.34 ± 0.04 |
| MIP | 4.09 ± 0.51 | 2.98 ± 0.41 | 1.90 ± 0.08 |
| PrDChVP | 3.47 ± 0.33 | 3.90 ± 0.28 | ND |
| diEt-PFP | 0.23 ± 0.018 | 0.17 ± 0.011 | ND |
| diBu-PFP | 7.10 ± 0.74 | 7.30 ± 0.46 | ND |
Data are mean ± SEM. RIP = [ki(NTE or NEST)/ki(AChE)]; where ki = bimolecular rate constant of inhibition; n = 3 – 4 for each ki (from Tables 1 and 2). Hen and mouse AChE activity was assayed in 9S fractions from brain homogenates. Human AChE was assayed in preparations from commercial recombinant enzyme. Hen NTE activity was assayed in 9S or lyophilized membrane fractions from brain homogenates. Mouse NTE activity was assayed in 9S fractions from brain homogenates. Human NEST activity was assayed in preparations from human recombinant NEST. NI = No inhibition of either enzyme at the highest concentration tested (0.1 mM). ND = Not determined.
Figure 4.
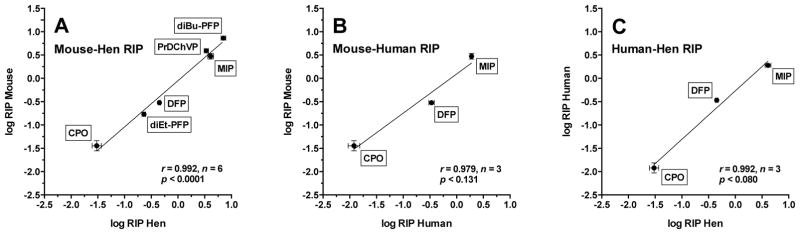
Correlations of log RIP. (A) mouse brain and hen brain; (B) mouse brain and recombinant human enzymes; (C) human recombinant enzymes and hen brain. RIP = [ki(NTE or NEST)/ki(AChE)].
Inhibition of AChE and NTE in Mouse Brain In Vivo
Inhibition of AChE and NTE in mouse brain was determined 1 h after i.p. administration of increasing doses of the known neuropathic OP compound PrDChVP and two new OP compounds possessing different neuropathic potentials according to in vitro data: diEt-PFP (slightly cholinergic) and diBu-PFP (neuropathic). The data obtained in three series of in vivo experiments are shown in Fig. 5.
Figure 5.
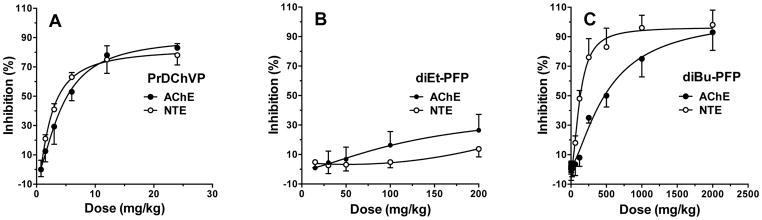
Inhibition of NTE and AChE activities in mouse brain 1 h after i.p. administration of increasing doses of (A) PrDChVP; (B) diEt-PFP; (C) diBu-PFP (C) Data are presented as % inhibition of the corresponding esterase in the control animals. Esterase activities in mouse brain of the control animals, nmol/(min × mg protein), (mean ± SEM): AChE = 69.20 ± 3.54 (n = 20), NTE =13.44 ± 0.52 (n = 10).
For PrDChVP (Fig. 5A), inhibition of both AChE and NTE in mouse brain was relatively potent and dose-dependent, yielding ED50 values of 4.34 ± 0.55 mg/kg for AChE and 2.17 ± 0.37 mg/kg for NTE. In contrast, diEt-PFP produced relatively low inhibition of AChE and especially NTE at 1 h after dosing, and unambiguous ED50 values could not be calculated. However, at the maximum dose of 200 mg/kg diEt-PFP, AChE activity was reduced to 26% and NTE to 14% of control (Fig. 5B). For diBu-PFP, both enzymes were inhibited in a dose-dependent manner (Fig 5C), yielding ED50 values of 516 ± 83.9 mg/kg for AChE and 127 ± 7.5 mg/kg for NTE.
Acute Toxicity Determination
LD50 values and their 95% CI for outbred white male mice (i.p. administration; 24-h observation period) were 15 (13.4–17.3) mg/kg for PrDChVP, 200 (148.3–251.7) mg/kg for diEt-PFP, and > 2000 mg/kg for diBu-PFP.
DISCUSSION
The objective of the present work was to provide corroborating and new data to help establish the use of mouse brain AChE and NTE in vitro and in vivo for biochemical assessment of the potential for OP compounds to produce OPIDN. In vitro assays could be used for toxicodynamic assessment of directly acting inhibitors, whereas dosing in vivo could be used to incorporate toxicokinetic factors of absorption, distribution, metabolism (activation and/or deactivation), and excretion into the assessment.
The OP compounds selected for the present study included CPS, CPO, DFP, MIP, PrDChVP, diEt-PFP, and diBu-PFP. Among these, CPS is a pro-toxicant that requires metabolic activation to its oxon, CPO, to inhibit serine esterases (Richardson, 1995). Thus, CPS served as an OP control for CPO in our in vitro experiments. CPO is a predominantly cholinergic OP compound, whereas DFP, MIP, and PrDChVP are intermediate between cholinergic and neuropathic, but each capable of producing OPIDN (Kropp and Richardson, 2003; Makhaeva et al., 1995, 2003, 2009b). PrDChVP was intensively studied in our previous work in experiments in vitro and in vivo using both hens and rats (Makhaeva et al., 1995, 2003, 2009b). Two new experimental compounds, diEt-PFP and diBu-PFP, were chosen for their differing hydrophobicity and demonstrated variations in neuropathic potential in our preliminary in vitro experiments using human erythrocyte AChE and hen brain NTE (Makhaeva et al., 2008).
Our choice of enzymes was based on the concept that the ability of an OP compound to produce OPIDN depends on its relative inhibitory potency (RIP) toward NTE, the target for initiating OPIDN, and AChE, the target for producing cholinergic toxicity. Although in some previous work we have defined the RIP as its inverse (Richardson, 1992; Richardson et al., 1993; Kropp and Richardson, 2003), in the present study, we define the ratio as it was originally proposed, RIP = ki(NTE)/ki(AChE = IC50(AChE)/IC50(NTE). By this definition, if the RIP is less than one, then the compound is predicted to produce cholinergic toxicity of sufficient severity that it cannot produce OPIDN at doses less than the LD50; however, if the RIP is greater than one, then the compound is predicted to produce OPIDN at doses less than the LD50 (Lotti and Johnson, 1978; Malygin et al., 2003).
Our results demonstrate that the neuropathic potential of OP compounds can be predicted from RIP values obtained by measuring inhibitory potencies against AChE and NTE activities in 9S fractions from hen or mouse brain homogenates or against preparations of human recombinant AChE and NEST. These in vitro findings confirm and extend previous work using mouse brain homogenates (Quistad et al., 2002), hen or human brain homogenates (Capodicasa et al., 1991; Emerick et al., 2012; Lotti and Johnson, 1978; Richardson et al., 1993) hen brain particulate fractions (Makhaeva et al., 2013; Malygin et al., 2003; Sogorb et al., 2010) or preparations from human or murine cell cultures (Ehrich et al., 1997; Li and Casida, 1997; Sogorb et al., 2010). In addition, the present work provides new in vitro and in vivo data on the neuropathic potential of PrDChVP and two new compounds, diEt-PFP and diBu-PFP.
Although inhibitory potencies of OP compounds can be estimated by directly measuring fixed-time IC50 in tissue preparations, extrinsic interactions with other proteins can introduce substantial error into determinations of intrinsic sensitivity of target enzymes to these inhibitors (Mortensen et al., 1998). Moreover, because fixed-time methods cannot follow the progressive nature of the inhibition or establish whether or not ideal pseudo-first-order conditions have been met (Fukuto, 1990; Richardson et al., 1993), we used kinetic methods in the present study to determine bimolecular rate constants of inhibition (ki) as measures of inhibitory potency. However, once ki values have been determined, it is valid to use the relationship IC50 = ln2/(ki × t) to calculate corresponding IC50 values for comparison with those obtained in other studies by fixed-time methods (Aldridge and Reiner, 1972; Richardson, 1992). Accordingly, the calculated 20-min IC50 values obtained from the ki determinations in the present study are listed in Tables 1 and 2. When making comparisons, it should be borne in mind that both ki and IC50 are temperature-dependent within the thermal limits of the enzyme, with ki increasing with increasing temperature and IC50 decreasing with increasing temperature, and that unlike the ki, the fixed-time IC50 is by definition time-dependent, decreasing in value with increasing time of incubation of enzyme with inhibitor.
The fixed-time IC50 values calculated for the hen brain enzymes in the present work are consistent with previously reported values by either falling within the range or being within one standard error for MIP (Richardson et al., 1993, Kropp and Richardson, 2003), DFP (Lotti and Johnson, 1978, Atkins and Glynn, 2000); CPO (Lotti and Johnson, 1978, Correll et al., 1987, Atkins and Glynn, 2000, Kropp and Richardson, 2000) and PrDChVP (Lotti and Johnson, 1978, Makhaeva et al., 1995, 2003).
Regarding mouse brain enzymes, our calculated IC50 values are again in good agreement for DFP (Ehrich et al., 1997; Quistad et al., 2002) and MIP (Ehrich et al., 1997; Quistad et al., 2001), but lower than those previously published for CPO (Ehrich et al., 1997; Quistad et al., 2002). However, Ehrich et al. (1997) used murine neuroblastoma cells rather than mouse brain tissue, and the incubation with OP compounds was for 1 h rather than 20 min; moreover, the authors stated that at the concentrations of inhibitors used, inhibition reached a plateau by 30 min. Quistad et al., (2001, 2002) determined the IC50 at 25 °C for 15 min rather than 37 °C for 20 min. These discrepancies serve to highlight the fact that IC50 values for progressive inhibition of esterases by OP compounds are by definition time-dependent and might not have been determined under ideal pseudo-first-order inhibition conditions.
Our calculated IC50 values for human recombinant enzymes aligned well with literature values for human or human recombinant enzymes for MIP and DFP (Atkins and Glynn, 2000; Ehrich et al., 1997; Kropp and Richardson, 2006; Kropp et al., 2004; Lotti and Johnson, 1978), as well as human neuroblastoma NTE inhibited by CPO (Ehrich et al., 1997). The apparent IC50 reported by Ehrich et al. (1997) for human neuroblastoma AChE inhibited by CPO was approximately tenfold lower than our value. In contrast, Amitai et al. (1998) used kinetic methods and obtained a ki for inhibition of human recombinant AChE by CPO at 25 °C that was about 70% of our value at 37 °C.
Overall, the fixed-time IC50 values calculated from kinetically determined ki values in this study agree well with literature values. In addition, as shown in Fig. 2 and 3, correlations of log ki values determined in the present study showed strong correlations (r > 0.99) across species. However, in order to make predictions about neuropathic potential, it is more important to examine ratios of ki or IC50 values (RIP values) rather than to scrutinize the individual ki or IC50 values. Thus, we found that the calculated RIP value of CPO is much less than 1, meaning that it is not likely to cause OPIDN at doses less than the LD50. Moreover, we found that the RIP of DFP is slightly less than 1, indicating neuropathicity at doses near the LD50, and that the RIP for MIP and PrDChVP is slightly higher than 1, which signifies that these compounds are neuropathic. These results correspond with previously published data (Kropp and Richardson, 2003; Lotti and Johnson, 1978; Makhaeva et al., 2003). Furthermore, earlier studies of other OP compounds in vitro have shown similar correlations of neuropathic potential with RIPs determined from human and hen brain tissues as well as neuroblastoma cell lines of both human and murine origin (Ehrich, 1997; Lotti and Johnson, 1978). In addition, as shown in Fig. 4, log RIP values obtained in the present study were strongly correlated across species (r > 0.979), despite not reaching the customary value for statistical significance (p < 0.05) in the comparisons with human enzymes for which only 3 data pairs were available. In particular, these strong correlations indicate that the same conclusions about neuropathic potential would be reached using human recombinant, hen brain, or mouse brain enzymes.
In order to model all aspects of OPIDN, >70% of neural NTE must be inhibited and aged in hens, and this can be induced in a single dose of a neuropathic OP compound (Johnson, 1982). Moreover, the clinical course and spatial-temporal distribution of axonal lesions in the hen closely mimics OPIDN in humans, making the hen the standard for testing OP compounds for delayed neuropathic potential (Doherty, 2006; Emerick et al., 2012). At the same time, in vivo studies of OPIDN in mice have proved to be more difficult; initially it appeared that OPIDN could be produced only if a neuropathic compound were administered chronically (Lapadula et al., 1985). Furthermore, a potent NTE inhibitor known to induce OPIDN in hens produces an acute neurotoxic syndrome with brain edema in mice (Winrow et al., 2003; Wu and Casida, 1995, 1996). Finally, although NTE inhibition and axonopathy could be produced in mice following a single dose of a neuropathic OP compound (Veronesi et al., 1991; Read et al., 2010), clinical signs of OPIDN are not apparent in this species. Nevertheless, the objective of the present study was not to produce a complete model of OPIDN in the mouse; rather, our intent was to demonstrate that the mouse could serve as a source of brain AChE and NTE for biochemical assessment of the neuropathic potential of OP compounds in vitro and in vivo.
For the in vivo work in the present study, we selected three compounds, PrDChVP and, diEt-PFP, and diBu-PFP. PrDChVP was intensively studied in our previous works in experiments in vivo both in hens and in rats (Makhaeva et al., 1995, 2003, 2009a, 2009b). In both species, this compound showed dose-dependent inhibition of brain NTE and AChE. In hen experiments, i.m. administration was used with subsequent determination of NTE and AChE activity 24 h after administration (Makhaeva et al., 1995, 2003, 2009b), and in rat experiments NTE and AChE activities were determined 1 h after i.p. administration (Makhaeva et al., 1995). Based on these results, in the present study in mice, we used i.p. administration of PrDChVP with assay of NTE and AChE 1 h after dosing.
As shown in Figure 4A, inhibition of NTE to the 70% threshold thought to be required for initiation of OPIDN (Johnson, 1982; Lotti, 1992; Moretto, 1998) was achieved by PrDChVP at a dose of 10 mg/kg, which is lower than the LD50 of 15 mg/kg. In hen brain, 70% NTE inhibition occurred at a dose of 0.6 mg/kg (Makhaeva et al., 1995, 2003), indicating that mice are less sensitive than hens to this neuropathic OP compound. In addition, the ED50 values calculated from the dose-response curves in Fig. 4A were 4.34 ± 0.55 mg/kg for AChE and 2.17 ± 0.37 mg/kg for NTE. From our previous work on hens (Makhaeva et al., 1995, 2003, 2009b), we have calculated the ED50 for AChE as 0.8 ± 0.1 mg/kg and 0.44 ± 0.01 mg/kg for NTE, again indicating that the mouse is less sensitive than the hen to inhibition of brain AChE and NTE by PrDChVP. Nevertheless, despite the species difference in sensitivity in vivo, the ratio ED50(AChE)/ED50(NTE), which is the in vivo counterpart of the RIP(Malygin et al., 2003; Richardson, 1992), is quite close for both species: 2.0 for mice and 1.8 for hens. This result serves to support the validity of the mouse model for biochemical assessment of the neuropathic potential of OP compounds.
Further in vivo assessment was carried out using diEt-PFP and diBu-PFP. These compounds were chosen based on their characteristics determined in vitro (Tables 1–3). The compounds differed markedly in RIP; diEt-PFP was less neuropathic than the more hydrophobic diBu-PFP compound. Additionally, there is abundant information on the differing neuropathic potential of OP compounds with diethyl versus dibutyl substituents (Davis et al., 1985; Johnson, 1988).
We found that diEt-PFP is a weak inhibitor of NTE and AChE in vivo (Fig. 5B). At a dose of 200 mg/kg, which corresponds to the LD50 of this compound, NTE activity was reduced only 10% and AChE was decreased by 26%. The fact that a higher dose than the LD50 would be required to reach the threshold of 70% NTE inhibition agrees with our in vitro data on this compound.
In contrast, as shown in Fig. 5C, 70% NTE inhibition was achieved at a dose 200 mg/kg, which was substantially lower than the estimated LD50 of > 2000 mg/kg. These results indicate that diBu-PFP, with its low acute toxicity, might initiate OPIDN at doses that do not produce warning signs of acute cholinergic poisoning. Indeed, the ED50 values for this compound were 516 ± 83.9 mg/kg for AChE and 127 ± 7.5 mg/kg for NTE, giving an ED50 ratio of 4.1. Thus, the in vivo result for diBu-PFP agrees with our in vitro predictions, which indicates distinct neuropathic potential for this compound.
In conclusion, although hens have been historically used as the standard for testing neuropathic OP compounds, the data from the present study demonstrate that relative inhibition of AChE and NTE in the 9S fraction of mouse brain homogenates may serve as an in vitro toxicodynamic model for neuropathic potential assessment of directly acting inhibitors. In addition, relative inhibition of AChE and NTE in mouse brain after dosing with OP compounds can provide a model for the biochemical assessment of neuropathic potential with the added benefit of incorporating toxicokinetic factors. Future work should include testing the mouse model with OP compounds that require metabolic activation in order to be AChE and NTE inhibitors. In addition, because aging of inhibited NTE appears to be required for initiation of OPIDN, assays for aging should be incorporated as has been done in other recent work (Sogorb et al., 2010).
Acknowledgments
The authors gratefully acknowledge the support of this work by the Russian Foundation for Basic Research (RFBR, grant 11-03-00581), Russian Academy of Sciences (Program “Medicinal Chemistry”), NATO Science for Peace and Security Program (grant SfP 984082), the United States National Institutes of Health grant R01 NS069700, a research grant from the Dow Chemical Company, and a research grant from the University of Michigan Risk Science Center. Finally, we thank Ms. K.M. Richardson for assistance with the references.
Footnotes
CONFLICT OF INTEREST
No conflict of interest is declared. Whereas the research was partially supported by a research grant from the Dow Chemical Company, which also produces chlorpyrifos, one of the compounds used in the study, neither Dow nor the other sponsors of the research had any involvement in the study design, collection, analysis and interpretation of data, writing of the manuscript, decision to submit the manuscript for publication, or approval of the manuscript.
References
- Abou-Donia MB. Organophosphorus ester-induced delayed neurotoxicity. Annu Rev Pharmacol Toxicol. 1981;21:511–548. doi: 10.1146/annurev.pa.21.040181.002455. [DOI] [PubMed] [Google Scholar]
- Aldridge WN, Davison AN. The mechanism of inhibition of cholinesterases by organophosphorus compounds. Biochem J. 1953;55:763–766. doi: 10.1042/bj0550763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldridge WN, Reiner E. Enzyme Inhibitors as Substrates. North Holland; Amsterdam: 1972. [Google Scholar]
- Allen JF, Johnson OH. The synthesis of monovinyl esters of phosphorus (V) acids. J Am Chem Soc. 1955;77:2871–2875. [Google Scholar]
- Amitai G, Moorad D, Adani R, Doctor BP. Inhibition of acetylcholinesterase and butyrylcholinesterase by chlorpyrifos-oxon. Biochem Pharmacol. 1998;56:293–299. doi: 10.1016/S0006-2952(98)00035-5. [DOI] [PubMed] [Google Scholar]
- Atkins J, Glynn P. Membrane association of and critical residues in the catalytic domain of human neuropathy target esterase. J Biol Chem. 2000;275:24477–24483. doi: 10.1074/jbc.M002921200. [DOI] [PubMed] [Google Scholar]
- Atkins J, Luthjens LH, Hom ML, Glynn P. Monomers of the catalytic domain of human neuropathy target esterase are active in the presence of phospholipid. Biochem J. 2002;361:119–123. doi: 10.1074/jbc.M210743200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Callaghan BC, Cheng HT, Stables CL, Smith AL, Feldman EL. Diabetic neuropathy: clinical manifestations and current treatments. Lancet Neurol. 2012;11:521–534. doi: 10.1016/S1474-4422(12)70065-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capodicasa E, Scapellato ML, Moretto A, Caroldi S, Lotti M. Chlorpyrifos-induced delayed polyneuropathy. Arch Toxicol. 1991;65:150–155. doi: 10.1007/BF02034943. [DOI] [PubMed] [Google Scholar]
- Correll L, Ehrich M. Comparative sensitivities of avian neural esterases to in vitro inhibition by organophosphorus compounds. Toxicol Lett. 1987;36:197–204. doi: 10.1016/0378-427(87)90185. [DOI] [PubMed] [Google Scholar]
- Davis CS, Johnson MK, Richardson RJ. Organophosphorus compounds. In: O’Donoghue JL, editor. Neurotoxicity of Industrial and Commercial chemicals. II. CRC Press; Boca Raton, FL: 1985. pp. 1–23. [Google Scholar]
- Doherty JD. Screening pesticides for neuropathogenicity. J Biomed Biotechnol. 2006;2006:1–13. doi: 10.1155/JBB/2006/70414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doorn JA, Thompson CM, Christner RB, Richardson RJ. Stereoselective interaction of Torpedo californica acetylcholinesterase by isomalathion: inhibitory reactions with (1R)- and (1S)-isomers proceed by different mechanisms. Chem Res Toxicol. 2003;16:958–965. doi: 10.1021/tx030026e. [DOI] [PubMed] [Google Scholar]
- Ehrich M, Correll L, Veronesi B. Acetylcholinesterase and neuropathy target esterase inhibitions in neuroblastoma cells to distinguish organophosphorus compounds causing acute and delayed neurotoxicity. Fundam Appl Toxicol. 1997;38:55–63. doi: 10.1006/faat.1997.2330. [DOI] [PubMed] [Google Scholar]
- Emerick GL, DeOliveira GH, Oliveira RV, Ehrich M. Comparative in vitro study of the inhibition of human and hen esterases by methamidophos enantiomers. Toxicology. 2012;292:145–150. doi: 10.1016/j.tox.2011.12.004. [DOI] [PubMed] [Google Scholar]
- Ellman G, Courtney KD, Andres V, Jr, Featherstone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol. 1961;7:88–95. doi: 10.1016/00006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- Evron T, Greenberg D, Mor TS, Soreq H. Adaptive changes in acetylcholinesterase gene expression as mediators of recovery from chemical and biological insults. Toxicology. 2007;233:97–107. doi: 10.1016/j.tox.2006.08.018. [DOI] [PubMed] [Google Scholar]
- Fukuto TR. Mechanism of action of organophosphorus and carbamate insecticides. Environ Health Perspec. 1990;87:245–254. doi: 10.1289/ehp.9087245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes RAC. Peripheral neuropathy. Brit Med J. 324:466–469. doi: 10.1136/bmj.324.7335.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MK. Structure-activity relationships for substrates and inhibitors of neurotoxic esterase. Biochem Pharmacol. 1975;24:797–805. doi: 10.1016/0006-2952(75)90123-9. [DOI] [PubMed] [Google Scholar]
- Johnson MK. Improved assay of neurotoxic esterase for screening organophosphates for delayed neurotoxicity potential. Arch Toxicol. 1977;37:113–115. doi: 10.1007/BF00293860. [DOI] [PubMed] [Google Scholar]
- Johnson MK. The target for initiation of delayed neurotoxicity by organophosphorus esters: biochemical studies and toxicological applications. In: Hodgson E, Bend JR, Philpot RM, editors. Reviews in Biochemical Toxicology. Vol. 4. Elsevier; Amsterdam: 1982. pp. 141–212. [Google Scholar]
- Johnson MK. Sensitivity and selectivity of compounds interacting with neuropathy target esterase: further structure-activity studies. Biochem Pharmacol. 1988;37:4095–4104. doi: 10.1016/0006-2952(88)90101-3. [DOI] [PubMed] [Google Scholar]
- Kayyali US, Moore TB, Randall JC, Richardson RJ. Neurotoxic esterase (NTE) assay: optimized conditions based on detergent-induced shifts in the phenol/4-aminoantipyrine chromophore spectrum. J Anal Toxicol. 1991;15:86–89. doi: 10.1093/jat/15.2.86. [DOI] [PubMed] [Google Scholar]
- Kaufer D, Friedman A, Seidman S, Soreq H. Acute stress facilitates long-lasting changes in cholinergic gene expression. Nature. 1998;393:373–377. doi: 10.1038/30741. [DOI] [PubMed] [Google Scholar]
- Kropp TJ, Glynn P, Richardson RJ. The mipafox-inhibited catalytic domain of human neuropathy target esterase ages by reversible proton loss. Biochemistry. 2004;43:3716–3722. doi: 10.1021/bi049960e. [DOI] [PubMed] [Google Scholar]
- Kropp TJ, Richardson RJ. Relative inhibitory potencies of chlorpyrifos oxon, chlorpyrifos methyl oxon, and mipafox for acetylcholinesterase versus neuropathy target esterase. J Toxicol Environ Health A. 2003;66:1145–1157. doi: 10.1080/15287390306360. [DOI] [PubMed] [Google Scholar]
- Kropp TJ, Richardson RJ. Aging of mipafox-inhibited human acetylcholinesterase proceeds by displacement of both isopropylamine groups to yield a phosphate adduct. Chem Res Toxicol. 2006;19:334–339. doi: 10.1021/tx0503420. [DOI] [PubMed] [Google Scholar]
- Li W, Casida JE. Actions of two highly potent organophosphorus neuropathy target esterase inhibitors in mammalian cell lines. Toxicol Lett. 1997;92:123–130. doi: 10.1016/s0378-4274(97)00047-7. [DOI] [PubMed] [Google Scholar]
- Lapadula DM, Patton SE, Campbell GA, Abou-Donia MB. Characterization of delayed neurotoxicity in the mouse following chronic oral administration of tri-o-cresyl phosphate. Toxicol Appl Pharmacol. 1985;79:83–90. doi: 10.1016/0041-008X(85)903709. [DOI] [PubMed] [Google Scholar]
- Lotti M. The pathogenesis of organophosphate-induced delayed polyneuropathy. Crit Rev Toxicol. 1992;21:467–487. doi: 10.3019/10408449209089884. [DOI] [PubMed] [Google Scholar]
- Lotti M, Johnson MK. Neurotoxicity of organophosphorus pesticides: predictions can be based on in vitro studies with hen and human enzymes. Arch Toxicol. 1978;41:215–221. doi: 10.1007/BF00354093. [DOI] [PubMed] [Google Scholar]
- Makhaeva GF, Malygin VV. A stable preparation of hen brain neuropathy target esterase for rapid biochemical assessment of neurotoxic potential of organophosphates. Chem Biol Interact. 1999;119–120:551–557. doi: 10.1016/s0009-2797(99)00069-1. [DOI] [PubMed] [Google Scholar]
- Makhaeva GF, Filonenko IV, Malygin VV. A comparative study of the interaction of phosphoric acid dichlorovinyl esters with a neurotoxic esterase from the brain of hens and rats. Zh Evol Biokhim Fiziol. 1995;4:396–403. Article in Russian. [PubMed] [Google Scholar]
- Makhaeva GF, Filonenko IV, Yankovskaya VL, Fomicheva SB, Malygin VV. Comparative studies of O,O-dialkyl-O-chloromethylchloroformimino phosphates interaction with neuropathy target esterase and acetylcholinesterase. Neurotoxicology. 1998;19:623–628. [PubMed] [Google Scholar]
- Makhaeva GF, Sigolaeva LV, Zhuravleva LV, Eremenko AV, Kurochkin IN, Malygin VV, Richardson RJ. Biosensor detection of neuropathy target esterase in whole blood as a biomarker of exposure to neuropathic organophosphorus compounds. J Toxicol Environ Health A. 2003;66:599–610. doi: 10.1080/152839030906375. [DOI] [PubMed] [Google Scholar]
- Makhaeva GF, Serebryakova OG, Boltneva NP, Galenko TG, Aksinenko AY, Sokolov VB, Martynov IV. Esterase profile and analysis of structure-inhibitor selectivity relationships for homologous phosphorylated 1-hydroperfluoroisopropanols. Dokl Biochem Biophys. 2008;423:352–357. doi: 10.1134/S1607672908060094. [DOI] [PubMed] [Google Scholar]
- Makhaeva GF, Aksinenko AY, Sokolov VB, Serebryakova OG, Richardson RJ. Synthesis of organophosphates with fluorine-containing leaving groups as serine esterase inhibitors with potential for Alzheimer disease therapeutics. Bioorg Med Chem Lett. 2009a;19:5528–5530. doi: 10.1016/j.bmcl.2009.08.065. [DOI] [PubMed] [Google Scholar]
- Makhaeva GF, Rudakova EV, Boltneva NP, Sigolaeva LV, Eremenko AV, Kurochkin IN, Richardson RJ. Blood esterases as a complex biomarker for exposure to organophosphorus compounds. In: Dishovsky C, Pivovarov A, editors. Counteraction to Chemical and Biological Terrorism in the East Europe Countries. Springer; Dordrecht, The Netherlands: 2009b. pp. 177–194. [DOI] [Google Scholar]
- Makhaeva GF, Aksinenko AY, Sokolov VB, Baskin II, Palyulin VA, Zefirov NS, Hein ND, Kampf JW, Wijeyesakere SJ, Richardson RJ. Kinetics and mechanism of inhibition of serine esterases by fluorinated aminophosphonates. Chem Biol Interact. 2010;187:177–184. doi: 10.1016/j.cbi.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makhaeva GF, Radchenko EV, Palyulin VA, Rudakova EV, Aksinenko AY, Sokolov VB, Zefirov NS, Richardson RJ. Organophosphorus compound esterase profiles as predictors of therapeutic and toxic effects. Chem Biol Interact. 2013;203:231–237. doi: 10.1016/j.cbi.2012.10.012. [DOI] [PubMed] [Google Scholar]
- Malygin VV, Sokolov VB, Richardson RJ, Makhaeva GF. Quantitative structure-activity relationships predict the delayed neurotoxicity potential of a series of O-alkyl-O-methylchloroformimino phenylphosphonates. J Toxicol Environ Health A. 2003;66:611–625. doi: 10.1080/15287390309353770. [DOI] [PubMed] [Google Scholar]
- Moretto A. Experimental and clinical toxicology of anticholinesterase agents. Toxicol Lett. 1998;102–103:509–513. doi: 10.1016/S03784274(98)00245-8. [DOI] [PubMed] [Google Scholar]
- Moretto A, Lotti M. The relationship between isofenphos cholinergic toxicity and the development of polyneuropathy in hens and humans. Arch Toxicol. 2002;76:367–375. doi: 10.1007/s00204-002-0352-8. [DOI] [PubMed] [Google Scholar]
- Mortensen SR, Brimijoin S, Hooper MJ, Padilla S. Comparison of the in vitro sensitivity of rat acetylcholinesterase to chlorpyrifos-oxon: what do tissue IC50 values represent? Toxicol Appl Pharmacol. 1998;148:46–49. doi: 10.1006/taap.1997.8287. [DOI] [PubMed] [Google Scholar]
- Pomeroy-Black MJ, Jortner BS, Ehrich MF. Early effects of neuropathy-inducing organophosphates on in vivo concentrations of three neurotrophins. Neurotox Res. 2007;11:85–91. doi: 10.1007/BF03033387. [DOI] [PubMed] [Google Scholar]
- Quistad GB, Sparks SE, Casida JE. Fatty acid amide hydrolase inhibition by neurotoxic organophosphorus pesticides. Toxicol Appl Pharmacol. 2001;173:48–55. doi: 10.1006/taap.2001.9175. [DOI] [PubMed] [Google Scholar]
- Quistad GB, Sparks SE, Segall Y, Nomura DK, Casida JE. Selective inhibitors of fatty acid amide hydrolase relative to neuropathy target esterase and acetylcholinesterase: Toxicological implications. Toxicol Appl Pharmacol. 2002;179:57–63. doi: 10.1006/taap.2001.9342. [DOI] [PubMed] [Google Scholar]
- Read DJ, Yong L, Chao MV, Cavanagh JB, Glynn P. Organophosphates induce distal axonal damage, but not brain oedema, by inactivating neuropathy target esterase. Toxicol Appl Pharmacol. 2010;245:108–115. doi: 10.1016/j.taap.2010.02.010. [DOI] [PubMed] [Google Scholar]
- Richardson RJ, Davis CS, Johnson MK. Subcellular distribution of marker enzymes and of neurotoxic esterase in adult hen brain. J Neurochem. 1979;32:607–615. doi: 10.1111/j.1471-4159.1979.tb00391.x. [DOI] [PubMed] [Google Scholar]
- Richardson RJ. Interaction of organophosphorus compounds with neurotoxic esterase. In: Chambers JE, Levi PE, editors. Organophosphates: Chemistry, fate, and effects. Academic Press; San Diego: 1992. pp. 299–323. [Google Scholar]
- Richardson RJ. Assessment of the neurotoxic potential of chlorpyrifos: A critical review of the literature. J Toxicol Environ Health. 1995;44:135–165. doi: 10.1080/15287399509531952. [DOI] [PubMed] [Google Scholar]
- Richardson RJ. Organophosphate Poisoning, Delayed Neurotoxicity. In: Wesler P, editor. Encyclopedia of Toxicology. 2. Vol. 4. Vol. 3. Elsevier Ltd; Oxford: 2005. pp. 302–306. [Google Scholar]
- Richardson RJ. Anticholinesterase insecticides. In: McQueen CA, editor. Comprehensive Toxicology. Vol. 13. Academic Press; Oxford: 2010. pp. 433–444. [Google Scholar]
- Richardson RJ, Moore TB, Kayyali US, Fowke JH, Randall JC. Inhibition of hen brain acetylcholinesterase and neurotoxic esterase by chlorpyrifos in vivo and kinetics of inhibition by chlorpyrifos oxon in vitro: application to assessment of neuropathic risk. Fundam Appl Toxicol. 1993;20:273–279. doi: 10.1006/faat.1993.1036. [DOI] [PubMed] [Google Scholar]
- Richardson RJ, Hein ND, Wijeyesakere SJ, Fink JK, Makhaeva GF. Neuropathy target esterase (NTE): overview and future. Chem Biol Interact. 2013;203:238–244. doi: 10.1016/j.cbi.2012.10.024. [DOI] [PubMed] [Google Scholar]
- Shaked I, Meerson A, Wolf Y, Avni R, Greenberg D, Gilboa-Geffen A, Soreq H. MicroRNA-132 potentiates cholinergic anti-inflammatory signaling by targeting acetylcholinesterase. Immunity. 2009;31:965–973. doi: 10.1016/j.immuni.2009.09.019. [DOI] [PubMed] [Google Scholar]
- Singer MA, Vernino SA, Wolfe GI. Idiopathic neuropathy: new paradigms, new promise. J Peripher Nerv Syst. 2012;17 (Suppl):43–49. doi: 10.1111/j.1529-8027.2012.00395.x. [DOI] [PubMed] [Google Scholar]
- Sogorb MA, Gonzales-Gonzales I, Pamies D, Vilanova E. An alternative in vitro method for detecting neuropathic compounds based on acetylcholinesterase inhibition and on inhibition and aging of neuropathy target esterase. Toxicol In Vitro. 2010;24:942–952. doi: 10.1016/j.tiv.2010.01.008. [DOI] [PubMed] [Google Scholar]
- Van Tienhoven M, Atkins J, Li Y, Glynn P. Human neuropathy target esterase catalyzes hydrolysis of membrane lipids. J Biol Chem. 2002;277:20942–20948. doi: 10.1074/jbc.M200330200. [DOI] [PubMed] [Google Scholar]
- Veronesi B, Padilla S, Blackmon K, Pope C. A murine model of OPIDN: neuropathic and biochemical description. Toxicol Appl Pharmacol. 1991;107:311–324. doi: 10.1016/0041-008x(91)90211-v. [DOI] [PubMed] [Google Scholar]
- Winrow CJ, Hemming ML, Allen DM, Quistad GB, Casida JE, Barlow C. Loss of neuropathy target esterase links organophosphate exposure to hyperactivity. Nat Genet. 33:477–485. doi: 10.1038/ng1131. 203. [DOI] [PubMed] [Google Scholar]
- Wu SY, Casida JE. Ethyl octylphosphonofluoridate and analogs: optimized inhibitors of neuropathy target esterase. Chem Res Toxicol. 1995;8:1070–1075. doi: 10.1021/tx00050a011. [DOI] [PubMed] [Google Scholar]
- Wu SY, Casida JE. Subacute neurotoxicity induced in mice by potent organophosphorus neuropathy target esterase inhibitors. Toxicol Appl Pharmacol. 1996;139:195–202. doi: 10.1006/taap.1996.0158. [DOI] [PubMed] [Google Scholar]