

Abstract
Objective
To identify the causative gene in SCA22, an autosomal dominant cerebellar ataxia mapped to chromosome 1p21-q23.
Subjects and Methods
We previously characterized a large Chinese family with progressive ataxia designated SCA22, which overlaps with the locus of SCA19. The disease locus in a French family and an Ashkenazi Jewish American family was also mapped to this region. Members from all three families were enrolled. Whole exome sequencing was performed to identify candidate mutations, which were narrowed by linkage analysis and confirmed by Sanger sequencing and co-segregation analyses. Mutational analyses were also performed in 105 Chinese and 55 Japanese families with cerebellar ataxia. Mutant gene products were examined in a heterologous expression system to address the changes in protein localization and electrophysiological functions.
Results
We identified heterozygous mutations in the voltage-gated potassium channel Kv4.3-encoding gene KCND3: an in-frame three-nucleotide deletion c.679_681delTTC p.F227del in both the Chinese and French pedigrees, and a missense mutation c.1034G>T p.G345V in the Ashkenazi Jewish family. Direct sequencing of KCND3 further identified three mutations, c.1034G>T p.G345V, c.1013T>C p.V338E and c.1130C>T p.T377M, in three Japanese kindreds. Immunofluorescence analyses revealed that the mutant p.F227del Kv4.3 subunits were retained in the cytoplasm, consistent with the lack of A-type K+ channel conductance in whole-cell patch-clamp recordings.
Interpretation
Our data identify the cause of SCA19/22 in patients of diverse ethnic origins as mutations in KCND3. These findings further emphasize the important role of ion channels as key regulators of neuronal excitability in the pathogenesis of cerebellar degeneration.
Keywords: exome sequencing, next generation sequencing, spinocerebellar ataxia type 22, voltage-gated potassium channel, Kv4.3, KCND3
Introduction
Spinocerebellar ataxia (SCA) is a clinically, pathologically, and genetically heterogeneous group of dominantly inherited neurodegenerative disorders characterized by progressive cerebellar ataxia variably associated with pyramidal, extrapyramidal, bulbar, spinal, and peripheral nervous system involvement. Thirty-two dominant SCAs (labeled SCA1 through SCA36) have been chromosomally mapped, and the genes causing 20 of these disorders have so far been identified.1, 2 The genetic etiologies of many SCAs have yet to be elucidated. 3, 4
Previously, we characterized a large Chinese pedigree with an autosomal dominant ataxia spanning four generations.5 The disease locus was mapped to chromosome 1p21-q23 and was designated SCA22.5 The locus of SCA22 overlaps with that of SCA19 on 1p21-q21, previously identified in a Dutch family.6, 7 SCA19 and SCA22 were therefore proposed to be allelic with a worldwide distribution.7 Here, we report mutations in KCND3 in the original SCA22 family as well as in five other SCA families of French, Ashkenazi Jewish and Japanese origin with dominant ataxia.
Subjects and Methods
Subjects
In Family A of Han Chinese origin (Fig 1A), the original SCA22 family, we enrolled 31 members, including 13 affected, 6 unaffected, 6 at-risk and 6 married-in individuals. The age at onset of ataxia in this pedigree ranged from 13 to 46 years. Clinical severity of ataxia was evaluated longitudinally using the 40-point (0 being normal) validated Scale for the Assessment and Rating of Ataxia (SARA).8,9
Figure 1.
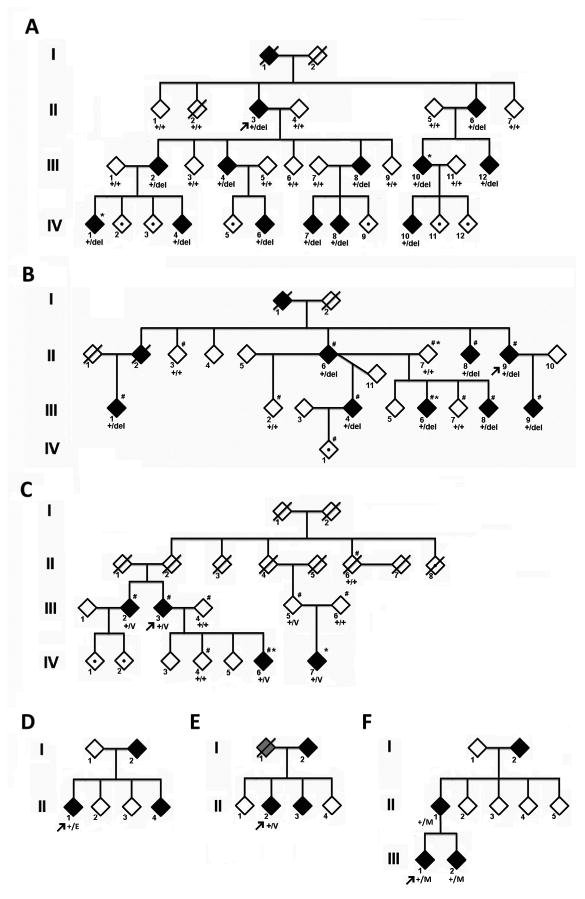
Pedigree charts of the Families A, B, C, D, E and F. The gender of the family members is obscured for privacy. The proband is denoted by an arrow. Filled diamonds represent affected members, grayed diamonds represent members with information suggesting spinocerebellar ataxia but not confirmed, open diamonds indicate unaffected individuals, and those with a dot within an open diamond denote at-risk individuals. / = deceased; *= members who underwent whole exome sequencing, # = individuals included in linkage analysis. + = wildtype allele, del = allele with F227del, V= allele with G345V, E= allele with V338E, M= allele with T377M.
In Family B of French origin (Fig 1B), 8 affected individuals, 4 at-risk relatives and 1 spouse participated in the study. Age at onset ranged from 24 to 51 years. Pathological nucleotide expansions were excluded in SCA1, 2, 3, 6, 7, 10, 12, 17, 31, 36, dentatorubral pallidoluysian atrophy (DRPLA), as were mutations in SCA5, 11, 13, 14, 23, and 28.
Family C of Ashkenazi Jewish American origin (Fig 1C) was ascertained through a proband (III-3) with ataxia with onset in her 50s. Samples from her and an affected relative were negative by commercial DNA testing, excluding SCA1, SCA2, SCA3, SCA6, SCA5, 7, 8, 10, 13, 14, 17, and DRPLA (Athena Diagnostics, MA). Four affected and 3 unaffected subjects (including 1 apparently unaffected obligate carrier), along with 2 spouses participated in the study.
In addition, we screened for mutations in the candidate gene in DNA from the index patients of 105 Chinese and 55 unrelated Japanese families with cerebellar ataxia, in whom mutations in SCA1, SCA2, MJD/SCA3, SCA6, SCA7, SCA8, SCA12, SCA17, SCA31 and DRPLA genes had been excluded.
Written informed consent was obtained from all subjects according to study protocols approved by the Institutional Review Boards of Taipei Veterans General Hospital, the Paris-Necker Ethics Committee, University of Michigan and University of Tokyo. Genomic DNA was isolated from peripheral blood leukocytes following a standard protocol.10
Genetic Studies
Linkage Analysis
Linkage analysis in family A to 1p21-q23 has been previously reported.5 In families B and C, genome scans were performed using Illumina LINKAGE_12 microarrays (6090 SNP markers). Genotypes were determined using Beadstudio (Illumina) and analyzed with MERLIN 1.0. 11
In family B, linkage analysis was run under a 0.85 penetrance model with equal allele frequencies, similar recombination fractions between males and females, and a disease frequency of 0.0005.
In family C, linkage analysis (run with 0.85 penetrance due to the presence of an unaffected obligate carrier female in the pedigree) identified ∼200 Mb regions on 8 different chromosomes with LOD scores between 0 and 2.0, reaching maximal LOD score of 1.97 on chromosome 1. To further narrow the regions, DNA of the most distantly related affected subjects (IV-6 and IV-7) (Fig 1C) was hybridized to Illumina Human660W-Quad high density SNP BeadChips. PLINK12 was used to identify chromosomal regions with large (>1000 kb) shared haplotypes.
Exome Sequencing
Exomes were captured and enriched using either the Agilent SureSelect Human All Exon 50 Mb kit (Agilent, CA, families A and B) or with the Nimblegen SeqCap EZ v1 (Roche, family C). The enriched samples were sequenced on the Illumina HiSeq2000 (Illumina, CA) platform.
In families A and C, two affected subjects were sequenced, whereas in family B, DNA from one affected (III-6), the married-in parent (II-7) and one unaffected control subject were sequenced (Fig 1). Only variants in the linked region and shared by both affected subjects sequenced (families A and C) or absent from the two controls (family B) were considered. Variants present in dbSNP, the 1000 Genomes Project,13 the exome variant server (http://evs.gs.washington.edu/EVS/), or in previously sequenced individuals without SCA were excluded. Variants were further filtered for those predicted to be functionally damaging, i.e. nonsynonymous and splice variants. The details of exome sequencing, variants filtering and analyses are available in the Supporting information.
Molecular analyses
Mutational analysis of exons and flanking introns of KCND3 was conducted by PCR followed by direct nucleotide sequence analysis as previously described.14
Expression plasmids
A human Kv4.3 expression clone pE-11.GFPIre.hKv4.3L.WT was a generous gift of Dr. Jeanne M. Nerbonne (Washington University, St. Louis, MO).15, 16 The coding region of KCND3 was subcloned into the pFLAG-CMV-5a vector (Sigma-Aldrich, St. Louis, MO). The mutation c.679_681delTTC was introduced by site-directed mutagenesis using Quick-Change method (Stratagene, Santa Clara, CA) and verified by bi-directional sequencing. The human KChIP2 expression clone was purchased from Open Biosystems (Thermo Scientific, Lafayette, CO). We constructed a plasmid expressing the integral membrane protein myelin protein zero (P0) fused to DsRed to mark the cell surface as previously described.17 The endoplasmic reticulum marker pDesRed-ER was purchased from Clontech (Mountain View, CA). All constructs were verified by sequencing.
Cell culture and transfection
Human embryonic kidney (HEK)-293T cells were maintained in high glucose Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) in a humidified incubator at 37°C under 5% CO2. Cells were grown on glass coverslips in 6-well multiwell plates. Transient transfection was performed using the calcium phosphate precipitation method.18 HEK293T cells were transiently transfected with plasmids (in 1:1:1 ratio, 200 ng each) expressing Kv4.3 (either wild-type or the F227del mutant), KChIP2, along with either P0-DsRed (to mark the cell surface) or DsRed-ER (to mark the endoplasmic reticulum).
Immunofluorescence staining
Forty eight hours after transfection, cells were fixed in 4% paraformaldehyde for 30 minutes, permeablized with 0.2% Tween-20 for 30 minutes and were blocked with 1% BSA for 30 minutes before incubation in primary antibody mouse anti-Kv4.3 at a dilution of 1:1000 (ab99045, Abcam, Cambridge, UK) overnight at 4°C. Bound primary antibodies were detected using Alexa 488-conjugated goat anti-mouse IgG (a11001, Invitrogen, Grand Island, NY).
Confocal imaging
Images were captured with a Zeiss LSM 5 Pascal Laser Scanning Confocal system mounted on an Axiovert 200M inverted fluorescence microscope with a 63X oil immersion objective. Confocal imaging was performed on expressions studies from six independent transfections. All images were processed and analyzed using ImageJ (National Institutes of Health, MD).
The midlevel optical section was selected for quantification of the surface expression of Kv4.3, with P0 staining to define the plasma membrane. A blank region was selected to calculate the average pixel intensity as the background which was subtracted from all images. In the channel of the cell surface marker P0 signal, 20% of the maximum pixel intensity was set as the threshold to define the cell contour. Surface expression of the Kv4.3 was calculated as the amount of Kv4.3 signal in the plasma membrane region normalized by the total signal intensity inside the cell contour.
Electrophysiological recordings
HEK-293T cells were transfected with either wild-type or p.F227del Kv4.3 cloned into a pGFP Ire vector, together with KChIP2 in a 1:1 ratio. Cells transfected with the vectors alone plus KChIP2 were used as controls. GFP-positive HEK-293T cells were used for electrophysiological recordings 48 hours after transfection. HEK cells were transferred to bath solution containing (in mM) NaCl 150, KCl 5, MgCl2 1, CaCl2 2.2, HEPES 10 and glucose 5; pH was adjusted to 7.3 with HCl. Transfected cells were visually selected by green fluorescence expression for recording under bright-field optics (BX51WI, Olympus). Patch pipettes (2-5 MΩ) were pulled from borosilicate glass capillaries (outer diameter 1.5 mm and inner diameter 0.86 mm; Harvard apparatus, Holliston, MA), heat-polished and then filled with internal solution containing (in mM) K-gluconate 120, KCl 24, EGTA 0.2 and HEPES 10; pH was adjusted to 7.3 with KOH. Using a Multiclamp 700B amplifier (Molecular Devices, Union City, CA), whole-cell patch recordings were made at 22-24 °C. Pipette capacitance was compensated in cell-attached configuration and patched cells were held in the voltage-clamp configuration at -90 mV with series resistance (RS) compensation (∼80-90%, lag ∼0.5 ms; RS before compensation 10-25 MΩ). Potassium currents were evoked by voltage pulses (-80 to +70 mV, 500 ms; 10 mV increments). Leakage and capacitive currents were subtracted using a ‘P over -4’ procedure. Membrane capacitances were determined from readout values (6-60 pF) of membrane capacitance compensation on the patch amplifier. Potassium equilibrium potential (-86 mV) was determined by the Nernst equation. Signals were low-pass filtered at 4 kHz (four-pole Bessel) and sampled at 10 kHz using the Digidata 1440 interface (Molecular Devices). Data acquisition was performed using the pClamp 10.2 software (Molecular Devices).
Results
Linkage identifies SCA19/22 on 1p21-q23 as a common dominant ataxia locus
Families B and C with dominant adult onset ataxia were ascertained in France and in USA in efforts to identify novel ataxia loci. Genome-wide low density SNP chips followed by linkage analysis were used to identify candidate chromosomal regions. In family B, putative or uninformative linkage to 11 candidate regions was considered, including 6 regions reached with the maximal expected value of LOD score of 2.8 according to the pedigree structure (chromosome 1, 2, 8, 9 and 14). In family C, high density SNP chips and PLINK were used to identify shared haplotypes between the most distant relatives. Only two large shared haplotypes matched the linkage peaks, one 62 Mb long haplotype on chromosome 1 and a 33 Mb haplotype on chromosome 15. The shared haplotype with the highest LOD score on chromosome 1 overlapped the SCA22 region.
Exome Sequencing Identifies KCND3 Mutations in Families with Dominant Ataxia
In family A, after analysis and filtering, 11 heterozygous coding variants that mapped to 1p21-q23 were shared by both affected subjects and were not present in thousands of control samples in various databases (Supplementary Table 1). Sanger sequencing of these variants in the remaining 23 family members identified only one variant, c.679_681delTTC in KCND3 that completely segregated with the disease phenotype (Fig 2A). This mutation was not found in 500 normal Taiwanese-Chinese controls.
Figure 2.
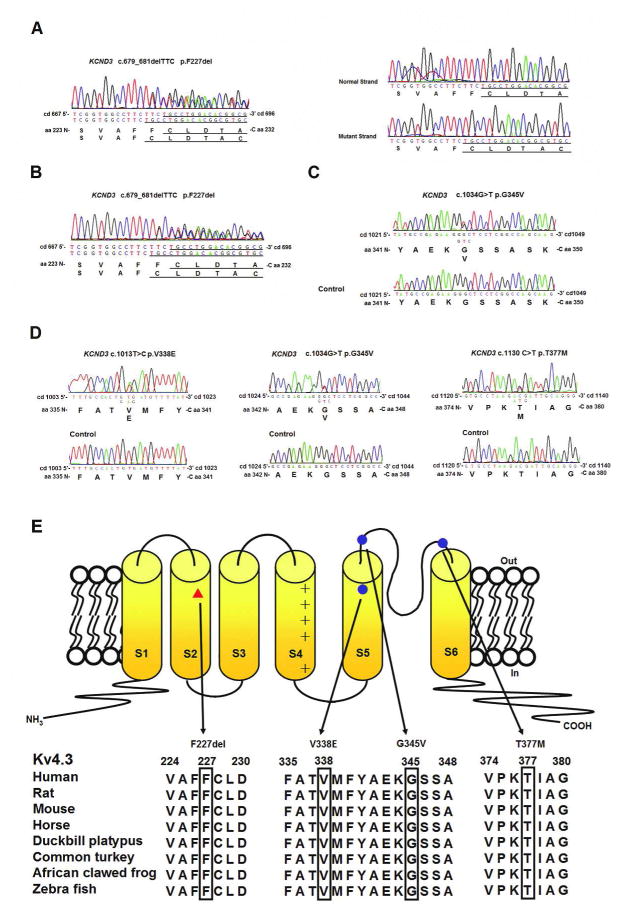
KCND3 mutations and Kv4.3 membrane topology. (A-D) The electrophoregrams of KCND3 heterozygous mutations. (E) Predicted Kv4.3 topology and locations of four mutations: red triangle- c.679_681delTTC (p.F227del), blue circles- c.1013T>C p.V338E, c.1034G>T p. G345V and c.1130C>T p.T377M. The mutated residues are evolutionarily conserved, as shown by aligning protein sequences of Kv4.3 orthologs in various organisms.
In Family B, of 1,648 nucleotide variants present in the affected subject but absent in her married-in parent and the control, only 3 variants were heterozygous, absent or rare in the Exome Variant Server and predicted to be damaging (Supplementary Table 2). Among these variants, c.679_681delTTC p.F227del in KCND3 (Fig 2B) was the strongest candidate, since mutations in the two other genes were known to have non-neurological phenotypes (detailed in the Supporting information). This mutation co-segregated with the disease in the family and was absent from 152 French control chromosomes.
In family C, exome sequencing identified a large number of shared variants between the two affected individuals. After filtering by linkage and shared haplotypes, variants were predicted to be damaging, and only one passed all filters, c.1304G>T p.G345V in KCND3 (Fig 2C) (Supplementary Table 3). Conventional sequencing confirmed segregation in the family.
Mutation Screening in Chinese and Japanese families with hereditary spinocerebellar ataxias
No mutation in KCND3 was found from any index patient of 105 Chinese families with hereditary ataxia.
Three missense mutations in KCND3 were identified in three families among 55 Japanese families with ataxia. (Fig 1D): c.1013T>C p. V338E in exon 1 was found in family D, c.1034G>T p. G345V in exon 1 in Family E, and c.1130C>T p. T377M in exon 2 in Family F. None of these mutations were present in 96 Japanese controls or public databases.
SCA22-associated mutations alter highly conserved amino acid residues
All of the ataxia-associated mutations affect amino acids in Kv4.3 that are highly conserved across a wide variety of species, from zebrafish, frog, platypus and mouse to humans (Fig 2E). In silico analysis predicted deleterious consequences from the residue changes (Supplementary Table 4).
Clinical features of patients with KCND3 mutations
Characteristically, the patients in the families with SCA 19/22 all have a very slowly progressive cerebellar ataxia. In Family A (Supplementary Table 5), II-3 has had difficulty walking for 35 years with an onset at 46 years and still manages to ride a 3-wheel motorcycle. III-2, at the age of 57 (25 years after the onset of disease), has had a very slow progression of ataxia, with an average deterioration of only 0.3 SARA score point per year over the past 5 years. III-8 has been mildly ataxic for 31 years and yet only has a SARA score of 9 points at the age of 48. There has been no cognitive impairment, myoclonus, tremors, focal weakness, sensory loss, cogwheel rigidity, visual impairment, retinopathy or ophthalmoplegia in any of the affected members. The brain MRI featured mild cerebellar atrophy (Supplementary Fig 1A). Their electrocardiograms showed sinus rhythm with normal QT intervals. There was no arrhythmia on the 24-hour Holter monitor recordings. The echocardiograms were also unremarkable.
In Family B (Supplementary Table 6), age at onset ranged between 24 and 51 years and most patients have been seen at least twice to evaluate disease evolution. Progression was slow as reflected by only one wheelchair user after 43 years of disease duration. Cerebellar ataxia was associated with impaired vibration sense at the ankles (3/8), with upwards ophthalmoplegia (1/8) or with diplopia (1/8). Hyperreflexes without positive Babinksi sign was present in 3/8. Mild cogwheel rigidity was noticed in two patients at ages 61 and 77. In the absence of pyramidal involvement, urinary urgency/incontinence was seen in 5/8. Cerebellar atrophy was present in 5 patients with cerebral MRI (Supplementary Fig 1B). The index case had sensory neuropathy.
In Family C, the proband (III-3) is tripping and falling slightly more frequently now (once or twice each month), 10 years after the beginning of imbalance and slurred speech at the age of 55, which have only slightly progressed in the absence of any visual problem or dysphagia. Neurological examination revealed breakdown of the smooth pursuit with saccades, mild dysarthria, difficulty with heel-to-shin test, mildly wide-based gait, and difficulty with tandem walk. One of the four children, one of the siblings, and one of the first cousins also have had imbalance. (Supplementary Table 7) Brain MRI revealed cerebellar vermian atrophy.
In family D, E and F, the age at onset was late and the clinical progression of ataxia was also slow in the majority of the affecteds. (Supplementary Table 8)
Immunofluorescence studies
The immunofluorescence staining pattern of HEK-293T cells expressing either wild type or p.F227del mutant Kv4.3 was characterized to address the question of channel protein localization. Cells expressing wild-type Kv4.3 displayed robust cell surface Kv4.3-specific staining (Fig 3A, D-G), whereas virtually all cells expressing the mutant Kv4.3 demonstrated diffuse cytoplasmic Kv4.3-specific staining without discernible cell surface signal (Fig 3B, H-K), suggesting impaired plasma membrane targeting of the mutant F227del Kv4.3 protein which appeared to be abnormally retained in the cytoplasm and colocalized with an ER-specific marker (Fig 3L, M, N). We observed no evidence of Kv4.3 in the mock-transfected cells (Fig3C). The expression of the cell surface marker P0 enabled us to define the cell membrane and quantify the subcellular localization of Kv4.3 using an unbiased approach. The ratio of cell surface expression for the p.F227del Kv4.3 was significantly lower than that observed in the wild type Kv4.3 (n = 10; mean ± S.E.M.: 0.28 ± 0.04 vs. 0.61 ± 0.06, p = 0.0014; Fig 3O).
Figure 3.
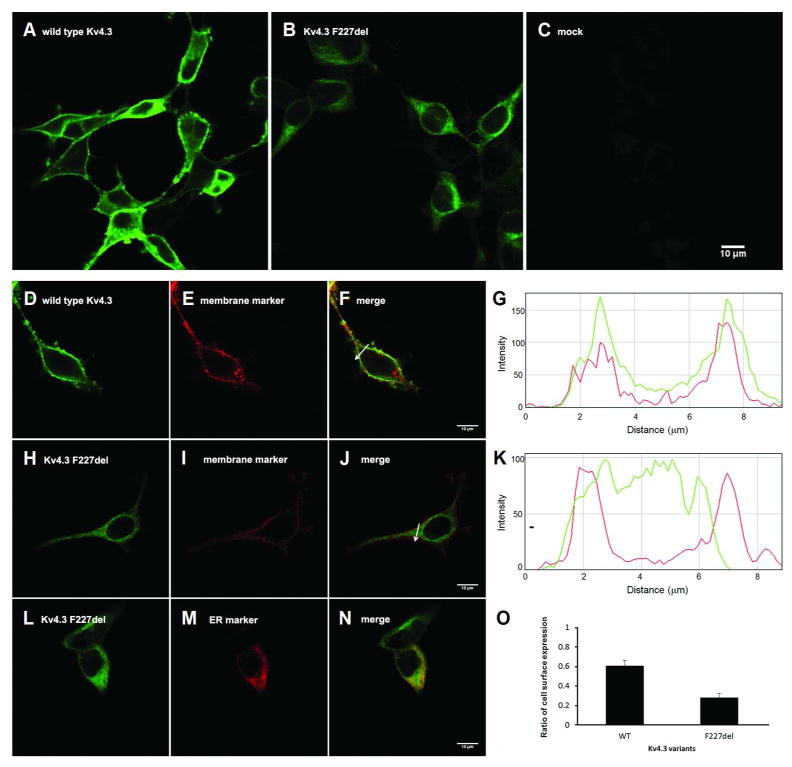
Confocal images demonstrating impaired cell surface expression and cytoplasmic retention of the mutant F227del Kv4.3. HEK-293T cells were transiently transfected with wild-type (A), p.F227del mutant (B) human Kv4.3 or empty vector (C) and immunostained with Kv4.3-specific antibody and green fluorophore-labeled secondary antibodies. Green fluorophore-labeled wild-type Kv4.3 (D) was expressed in the plasma membrane (E), colocalizing with DsRed-labeled membrane protein P0 (F). The spatial profile of fluorescent intensity of wild-type (wt) Kv4.3 (green) and P0 (red) along the arrow imposed on the images is shown in (G). The x axis displays the distance relative to the start point of the arrow and the y axis displays the fluorescence intensity. F227del Kv4.3 (H) was deficient in targeting to the plasma membrane (I, J). The spatial profile of fluorescent intensity of F227del Kv4.3 (green) and P0 (red) along the arrow imposed on the images is shown in (K). Instead, F227del Kv4.3 was retained in the cytoplasm (L) and colocalized with an ER-specific marker (M, N). The ratio of cell surface expression for the p.F227del Kv4.3 was significantly lower (O) than that observed in the wild type Kv4.3 (mean ± s.e.m.; p.F227del: 0.28 ± 0.04, n = 10; wild type Kv4.3: 0.61 ± 0.06, n = 10; p = 0.0014, Paired t test). Scale bar: 10 μm.
Electrophysiological recordings
In whole-cell voltage-clamp recording on transfected HEK-293T cells, potassium currents were evoked by voltage pulses. In the control cells (KChIP2 + GFP), HEK-293T cells exhibited endogenous non-inactivating outward currents (Fig 4A). In contrast, large transient outward currents were observed in the WT Kv4.3-transfected HEK293T cells (WT Kv4.3 + KChIP2 + GFP) (Fig 4B). The outward currents recorded from p.F227del Kv4.3-transfected HEK293T cells (p.F227del + KChIP2 + GFP) were comparable to the endogenous potassium currents recorded from mock-transfected cells (Fig 4C). After normalizing the current amplitude for cell size, WT Kv4.3-transfected cells had a significantly higher current density compared to p.F227del-transfected cells (WT: 0.52 ± 0.14 nA/pF, n= 8; p.F227del: 0.09 ± 0.02 nA/pF, n= 7, P< 0.0005, Mann-Whitney test; Fig 4D). The difference in current densities between p.F227del-transfected and control cells (p.F227del: 0.09 ± 0.02 nA/pF, n= 17; Ctrl: 0.09 ± 0.01 nA/pF, n= 3, P= 0.67, Mann-Whitney test; Fig 4D) was not statistically significant. Given that the cell surface expression level of p.F227del Kv4.3 was minimal, the finding of low current densities by electrophysiological recordings is consistent with a notion of a defect in channel expression.
Figure 4.
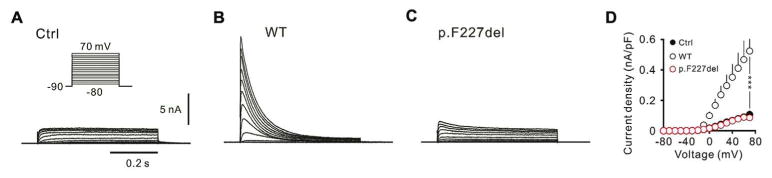
Electrophysiological recordings of the Kv4.3 channels. Whole-cell patch-clamp recordings revealed endogenous currents in un-transfected cells (A), large rapidly inactivating A-type potassium current in wild-type Kv4.3-transfected cells (B), and reduced current amplitudes in p.F227del Kv4.3-transfected cells (C). WT (black circles) had a significantly larger current density than p.F227del (red circles) (***P < 0.0005) and the current density of p.F227del was similar to that of Ctrl (filled circles) (D).
Discussions
Using exome sequencing and mutational analyses, we identified in six unrelated families of diverse ethnic origins with autosomal dominant cerebellar ataxia heterozygous mutations in the voltage-gated potassium channel Kv4.3-encoding KCND3 gene. A three-nucleotide in-frame deletion leading to p.F227del co-segregated with ataxia in Family A of Han Chinese origin and Family B of French origin. A missense mutation leading to p.G345V was found in Family C of Ashkenazi-Jewish origin and family E of Japanese origin. Two other missense mutations leading to p.V338E and p.T377M were identified in two other Japanese families. All of these amino acids involved are evolutionarily conserved in all vertebrates. The mutations were not observed in the Exome Variant Server, 10 Chinese and 32 French non-SCA subjects, as well as 1,248 chromosomes of French, Chinese or Japanese controls. Our findings provide strong genetic support for KCND3 being the causative gene in SCA19/22.
Kv4.3 is highly expressed in the brain, in particular in the cerebellar Purkinje cells, granule cells, basket cells, stellate cells, a subset of GABAergic deep neurons, and Lugaro cells adjacent to the somata of Purkinje cells.19,20-23 Kv4.3 may play an important role in the development of cerebellum.23, 24 Of note, Kv4.3 is also expressed in the heart, and rare missense mutations in the cytoplasmic C-terminus have been implicated in Brugada syndrome,25 which is a hereditary condition with cardiac arrhythmia predisposed to sudden cardiac death. There was no mention of neurological disorders in these patients. None of the patients in this report had cardiac symptoms or signs.
KCND3 encodes Kv4.3, an alpha subunit of the Shal family of the A-type voltage-gated K+ channels which are important in membrane repolarization19 in excitable cells. Similar to other voltage-gated K+ channels, Kv4.3 forms homo- or hetero-tetramers with members of the Shal subfamily channels. Each alpha subunit has six transmembrane segments (S1-S6) and a re-entrant loop linking S5 and S6 (Figure 2D). S1 through S4 form the voltage sensing domain, while S5, S6 and the re-entrant loop form the ion selective pore. F227 resides in S2 and is conserved between Kv4 and Kv1 of the Shaker family, which, similar to Shal, also carries a voltage-dependent potassium current. F227 is equivalent to F223 in Kv1.2 and F280 in Shaker. Functionally, F227 has been suggested to be a high impact residue that indirectly coordinates the omega pathway, which is formed by S1-S3 helices and S4 movement in response to membrane voltage changes.26, 27 Thus p.F227del may lead to a shift in the downstream residues in S2 lining the ion permeation pathway to interfere with the normal movement of S4. Empirically, we were surprised to not be able to detect any channel activity or changes in the biophysical properties of the channel (Fig 4). Instead, we observed intracellular retention, suggesting that p.F227del causes a loss of channel function by interfering with proper plasma membrane targeting and incorporation into a functional tetrameric channel complex. (Fig 3)
V338 resides in S5, and both G345 and T377 are in the putative outer vestibule of the channel between transmembrane segments S5 and S6. Efforts are underway to continue to characterize the functional consequences of mutations in KCND3.
Following KCNA1 (Kv1.1, NM_000217) and KCNC3 (Kv3.3, NM_004977), KCND3 (Kv4.3, NM_004980) is the third voltage-gated potassium channel gene discovered to cause human cerebellar ataxia. Point mutations 28-32,33 in Shaker Kv1.1-encoding KCNA1 cause episodic ataxia type 1 (EA1; OMIM160120). Point mutations in Shaw-related Kv3.3-encoding KCNC3 cause spinocerebellar ataxia type 13 (SCA13; OMIM605259) with phenotypes ranging from neurodevelopmental disorders to adult-onset neurodegeneration.34, 35 Although the exact mechanism is not yet known how mutations may disrupt Kv4.3 function in regulating neuronal excitability and how this may in turn lead to neurodegeneration, the discovery of KCND3 as the causative gene in SCA19/22 adds to the growing evidence of the importance of fine tuning neuronal excitability in the health and survival of neurons and especially cerebellar neurons. Other voltage-gated K+ channels have been proposed in neurodegenerative diseases.36-38
Patients with p.F227del in Kv4.3 all have a protracted clinical course with slowly progressive cerebellar ataxia. As proposed, the phenotypes of dominant cerebellar ataxias due to mutations other than polyglutamine expansions are slow and less complicated despite earlier age at onset.4 There are clinical differences between family A and B. The Chinese family presents with relatively pure cerebellar signs, which are consistent with the clinical classification of autosomal dominant cerebellar ataxia (ADCA) type III,39 similar to SCA6, while the French family presents with signs consistent with ADCA type I.39 Indeed, the clinical progression of SCA22 appears even slower than that of SCA6, as evaluated with the clinical rating scale of ataxia SARA.9 For individuals carrying the p.F227del mutation, the onset of ataxia was earlier (15-30 years) in the younger generation than in the older (30-50 years), which is also true for those with the p.G345V mutation, although the age at onset was later (50s in the older generation, compared to 30s-40s in the younger generation), suggesting that the G345V-associated phenotype may be milder. Of note, the disease is almost fully penetrant in the six families described in this report. It is our hope and expectation that additional patients and families will be identified with SCA19/22 to further define the clinical features.
Supplementary Material
Supplementary Table 1. Bioinformatic analysis of exome sequencing in Family A
Supplementary Table 2. Bioinformatic analysis of exome sequencing in Family B.
Supplementary Table 3. Bioinformatic analysis of exome sequencing in Family C
Supplementary Table 4. Deleterious consequences from residue changes predicted by in silico analysis
Supplementary Table 5. Clinical features of Family A
Supplementary Table 6. Clinical features of Family B
Supplementary Table 7. Clinical features of Family C
Supplementary Table 8. Clinical features of Family D, E, F
Supplementary FIGURE 1. MR imaging in SCA19/22. Sagittal T1-weighted scan of patient (A) III-2 (47 years of age) of family A and (B) III-9 (36 years of age) of family B, showing cerebellar volume loss and normal brainstem after 15 and 12 years, respectively, of disease duration, correlating with mild functional impairment.
Acknowledgments
We thank all patients for participating in this study. This work was supported by funds from Taiwan Ataxia Association, Hsu Tsung Pei Medical Research Fund, research grants from Taipei Veterans General Hospital (V99-C1-023, V99-C1-052, V100C-036 and V101C-045), the National Science Council, Taiwan, ROC (NSC95-2320-B-010-056-MY3, NSC96-2314-B-010-036-MY3, NSC98-2320-B-010-029-MY3 and NSC99-2314-B-010-013-MY3), the Ministry of Education, Aim for the Top University Plan (V100-E6-006 and V101E7-005), Taiwan, the European Union (6th PCRD call, to the EUROSCA consortium), the VERUM foundation (to A Brice), the Association Connaitre les Syndromes Cérébelleux (to G Stevanin and to the SPATAX Network), and National Institute of Health grant 3R21DC010074 (KM, JZL, and MB). The sequencing and analytical work was supported by the High-throughput Genome Analysis Core Facility of National Core Facility Program for Biotechnology, Taiwan (NSC-100-2319-B-010-001), National Core Facility Program for Biotechnology (Taiwan Bioinformatics Consortium of Taiwan, NSC 100-2319-B-010-002) and the University of Michigan DNA Core. Fluorescence micro-imaging analysis (confocal microscopy or live cell microscopy) were performed by the Public instrumental service center at Department of Medical Research & Education, Taipei Veterans General Hospital, Taipei, Taiwan, R.O.C. We thank Dr. Sylvie Forlani from the DNA and Cell bank of CRICM for DNA preparation. We appreciate constructive suggestions from Professors Mei-ling Tsaur, Tsung-Sheng Su, Ueng-cheng Yang and Dr. Wen-chung Yu. We also thank James Dell' Orco, Linda Gates and Weiping Peng for assistance with specific experiments, and Jishu Xu for bioinformatics.
Footnotes
Potential Conflict of Interest: Nothing to report
References
- 1.Klockgether T. Update on degenerative ataxias. Curr Opin Neurol. 2011;24:339–345. doi: 10.1097/WCO.0b013e32834875ba. [DOI] [PubMed] [Google Scholar]
- 2.Soong Bw, Paulson HL. Spinocerebellar ataxias: an update. Curr Opin Neurol. 2007;20:438–446. doi: 10.1097/WCO.0b013e3281fbd3dd. [DOI] [PubMed] [Google Scholar]
- 3.Soong BW, LU YC, Choo KB, Lee HY. Frequency analysis of autosomal dominant ataxias in Taiwanese patients and clinical and molecular characterization of spinocerebellar ataxia type 6. Arch Neurol. 2001;58:1105–1109. doi: 10.1001/archneur.58.7.1105. [DOI] [PubMed] [Google Scholar]
- 4.Durr A. Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol. 2010;9:885–894. doi: 10.1016/S1474-4422(10)70183-6. [DOI] [PubMed] [Google Scholar]
- 5.Chung MY, Lu YC, Cheng NC, Soong BW. A novel autosomal dominant spinocerebellar ataxia (SCA22) linked to chromosome 1p21-q23. Brain. 2003;126:1293–1299. doi: 10.1093/brain/awg130. [DOI] [PubMed] [Google Scholar]
- 6.Verbeek DS, Schelhaas JH, Ippel EF, et al. Identification of a novel SCA locus ( SCA19) in a Dutch autosomal dominant cerebellar ataxia family on chromosome region 1p21-q21. Hum Genet. 2002;111:388–393. doi: 10.1007/s00439-002-0782-7. [DOI] [PubMed] [Google Scholar]
- 7.Schelhaas HJ, Verbeek DS, Van de Warrenburg BP, Sinke RJ. SCA19 and SCA22: evidence for one locus with a worldwide distribution. Brain. 2004;127:E6. doi: 10.1093/brain/awh036. author reply E7. [DOI] [PubMed] [Google Scholar]
- 8.Schmitz-Hubsch T, du Montcel ST, Baliko L, et al. Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology. 2006;66:1717–1720. doi: 10.1212/01.wnl.0000219042.60538.92. [DOI] [PubMed] [Google Scholar]
- 9.Lee YC, Liao YC, Wang PS, et al. Comparison of cerebellar ataxias: A three-year prospective longitudinal assessment. Mov Disord. 2011;26:2081–2087. doi: 10.1002/mds.23809. [DOI] [PubMed] [Google Scholar]
- 10.Sambrook J, Russell DW. Molecular cloning: a laboratory manual. 3rd. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- 11.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin--rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 12.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Durbin RM, Altshuler D, Abecasis GR, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Postma AV, Bezzina CR, de Vries JF, et al. Genomic organisation and chromosomal localisation of two members of the KCND ion channel family, KCND2 and KCND3. Hum Genet. 2000;106:614–619. doi: 10.1007/s004390000308. [DOI] [PubMed] [Google Scholar]
- 15.Norris AJ, Nerbonne JM. Molecular dissection of I(A) in cortical pyramidal neurons reveals three distinct components encoded by Kv4.2, Kv4.3, and Kv1.4 alpha-subunits. J Neurosci. 30:5092–5101. doi: 10.1523/JNEUROSCI.5890-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Niwa N, Nerbonne JM. Molecular determinants of cardiac transient outward potassium current (I(to)) expression and regulation. J Mol Cell Cardiol. 48:12–25. doi: 10.1016/j.yjmcc.2009.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee YC, Yu CT, Lin KP, et al. MPZ mutation G123S characterization: evidence for a complex pathogenesis in CMT disease. Neurology. 2008;70:273–277. doi: 10.1212/01.wnl.0000296828.66915.bf. [DOI] [PubMed] [Google Scholar]
- 18.Shi G, Kleinklaus AK, Marrion NV, Trimmer JS. Properties of Kv2.1 K+ channels expressed in transfected mammalian cells. J Biol Chem. 1994;269:23204–23211. [PubMed] [Google Scholar]
- 19.Tsaur ML, Chou CC, Shih YH, Wang HL. Cloning, expression and CNS distribution of Kv4.3, an A-type K+ channel alpha subunit. FEBS Lett. 1997;400:215–220. doi: 10.1016/s0014-5793(96)01388-9. [DOI] [PubMed] [Google Scholar]
- 20.Kong W, Po S, Yamagishi T, et al. Isolation and characterization of the human gene encoding Ito: further diversity by alternative mRNA splicing. Am J Physiol. 1998;275:H1963–1970. doi: 10.1152/ajpheart.1998.275.6.H1963. [DOI] [PubMed] [Google Scholar]
- 21.Dilks D, Ling HP, Cockett M, et al. Cloning and expression of the human kv4.3 potassium channel. J Neurophysiol. 1999;81:1974–1977. doi: 10.1152/jn.1999.81.4.1974. [DOI] [PubMed] [Google Scholar]
- 22.Isbrandt D, Leicher T, Waldschutz R, et al. Gene structures and expression profiles of three human KCND (Kv4) potassium channels mediating A-type currents I(TO) and I(SA) Genomics. 2000;64:144–154. doi: 10.1006/geno.2000.6117. [DOI] [PubMed] [Google Scholar]
- 23.Hsu YH, Huang HY, Tsaur ML. Contrasting expression of Kv4.3, an A-type K+ channel, in migrating Purkinje cells and other post-migratory cerebellar neurons. Eur J Neurosci. 2003;18:601–612. doi: 10.1046/j.1460-9568.2003.02786.x. [DOI] [PubMed] [Google Scholar]
- 24.Oberdick J, Baader SL, Schilling K. From zebra stripes to postal zones: deciphering patterns of gene expression in the cerebellum. Trends Neurosci. 1998;21:383–390. doi: 10.1016/s0166-2236(98)01325-3. [DOI] [PubMed] [Google Scholar]
- 25.Giudicessi JR, Ye D, Tester DJ, et al. Transient outward current (I(to)) gain-of-function mutations in the KCND3-encoded Kv4.3 potassium channel and Brugada syndrome. Heart Rhythm. 2011;8:1024–1032. doi: 10.1016/j.hrthm.2011.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tombola F, Pathak MM, Gorostiza P, Isacoff EY. The twisted ion-permeation pathway of a resting voltage-sensing domain. Nature. 2007;445:546–549. doi: 10.1038/nature05396. [DOI] [PubMed] [Google Scholar]
- 27.Khalili-Araghi F, Tajkhorshid E, Roux B, Schulten K. Molecular dynamics investigation of the omega-current in the Kv1.2 voltage sensor domains. Biophys J. 2012;102:258–267. doi: 10.1016/j.bpj.2011.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.D'Adamo MC, Liu Z, Adelman JP, et al. Episodic ataxia type-1 mutations in the hKv1.1 cytoplasmic pore region alter the gating properties of the channel. EMBO J. 1998;17:1200–1207. doi: 10.1093/emboj/17.5.1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.D'Adamo MC, Imbrici P, Sponcichetti F, Pessia M. Mutations in the KCNA1 gene associated with episodic ataxia type-1 syndrome impair heteromeric voltage-gated K(+) channel function. FASEB J. 1999;13:1335–1345. doi: 10.1096/fasebj.13.11.1335. [DOI] [PubMed] [Google Scholar]
- 30.Eunson LH, Rea R, Zuberi SM, et al. Clinical, genetic, and expression studies of mutations in the potassium channel gene KCNA1 reveal new phenotypic variability. Ann Neurol. 2000;48:647–656. [PubMed] [Google Scholar]
- 31.Rea R, Spauschus A, Eunson LH, et al. Variable K(+) channel subunit dysfunction in inherited mutations of KCNA1. J Physiol. 2002;538:5–23. doi: 10.1113/jphysiol.2001.013242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Imbrici P, D'Adamo MC, Kullmann DM, Pessia M. Episodic ataxia type 1 mutations in the KCNA1 gene impair the fast inactivation properties of the human potassium channels Kv1.4-1.1/Kvbeta1.1 and Kv1.4-1.1/Kvbeta1.2. Eur J Neurosci. 2006;24:3073–3083. doi: 10.1111/j.1460-9568.2006.05186.x. [DOI] [PubMed] [Google Scholar]
- 33.Shook SJ, Mamsa H, Jen JC, et al. Novel mutation in KCNA1 causes episodic ataxia with paroxysmal dyspnea. Muscle Nerve. 2008;37:399–402. doi: 10.1002/mus.20904. [DOI] [PubMed] [Google Scholar]
- 34.Waters MF, Minassian NA, Stevanin G, et al. Mutations in voltage-gated potassium channel KCNC3 cause degenerative and developmental central nervous system phenotypes. Nat Genet. 2006;38:447–451. doi: 10.1038/ng1758. [DOI] [PubMed] [Google Scholar]
- 35.Figueroa KP, Waters MF, Garibyan V, et al. Frequency of KCNC3 DNA variants as causes of spinocerebellar ataxia 13 (SCA13) PLoS One. 2011;6:e17811. doi: 10.1371/journal.pone.0017811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ariano MA, Cepeda C, Calvert CR, et al. Striatal potassium channel dysfunction in Huntington's disease transgenic mice. J Neurophysiol. 2005;93:2565–2574. doi: 10.1152/jn.00791.2004. [DOI] [PubMed] [Google Scholar]
- 37.Angulo E, Noe V, Casado V, et al. Up-regulation of the Kv3.4 potassium channel subunit in early stages of Alzheimer's disease. J Neurochem. 2004;91:547–557. doi: 10.1111/j.1471-4159.2004.02771.x. [DOI] [PubMed] [Google Scholar]
- 38.Baranauskas G, Tkatch T, Surmeier DJ. Delayed rectifier currents in rat globus pallidus neurons are attributable to Kv2.1 and Kv3.1/3.2 K(+) channels. J Neurosci. 1999;19:6394–6404. doi: 10.1523/JNEUROSCI.19-15-06394.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harding AE. Clinical features and classification of inherited ataxias. Adv Neurol. 1993;61:1–14. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Bioinformatic analysis of exome sequencing in Family A
Supplementary Table 2. Bioinformatic analysis of exome sequencing in Family B.
Supplementary Table 3. Bioinformatic analysis of exome sequencing in Family C
Supplementary Table 4. Deleterious consequences from residue changes predicted by in silico analysis
Supplementary Table 5. Clinical features of Family A
Supplementary Table 6. Clinical features of Family B
Supplementary Table 7. Clinical features of Family C
Supplementary Table 8. Clinical features of Family D, E, F
Supplementary FIGURE 1. MR imaging in SCA19/22. Sagittal T1-weighted scan of patient (A) III-2 (47 years of age) of family A and (B) III-9 (36 years of age) of family B, showing cerebellar volume loss and normal brainstem after 15 and 12 years, respectively, of disease duration, correlating with mild functional impairment.