

Abstract
Inflammatory bowel disease (IBD) is a complex disease that involves unpredictable and destructive inflammation in the gastrointestinal tract resulting in gastrointestinal symptoms, infection, and tissue destruction, and which can be associated with an increased risk of colon cancer. The underlying cause of IBD involves disruption of the innate and adaptive immune mechanisms that maintain homeostasis between the gut mucosa and its environment. Elucidating how the homeostatic mechanisms controlling gut mucosal immunity and inflammation are disrupted in IBD represents the first steps to identifying novel therapeutic targets. Sphingosine-1-phosphate (S1P) is a bioactive sphingolipid that is enriched in the blood and lymph, and functions in innate and adaptive immunity. S1P signaling regulates inflammation via its impact on the trafficking, differentiation, and effector functions of bone marrow-derived immune cells. S1P also activates nuclear factor kappa B and signal transducer and activator of transcription 3 inflammatory pathways. S1P is generated by the ubiquitously expressed lipid kinase, sphingosine kinase (SphK)1 and its tissue-restricted homolog, SphK2. S1P is irreversibly degraded by S1P lyase, which is highly expressed in enterocytes. Recent studies targeting S1P metabolism and signaling have shown promise in preclinical models of IBD and have shed light on the mechanisms by which S1P signaling impacts IBD. The evidence suggests that targeting S1P signaling and metabolism may represent a novel strategy in treating IBD and it may reduce colon cancer risk by interrupting the progression from inflammation to carcinogenesis.
Keywords: sphingolipids, sphingadienes, sphingosine phosphate lyase, sphingosine kinase, STAT3, S1PR
Introduction
Inflammatory bowel disease (IBD) is a complex disease comprised of Crohn’s Disease or Crohn Disease (CD) and ulcerative colitis (UC), two conditions that involve unpredictable and destructive inflammatory processes resulting in weight loss, diarrhea, abdominal pain, and fever.1–3 UC is restricted to the colon, whereas CD is characterized by skip lesions and can involve any site in the gastrointestinal tract.4 Symptoms include abdominal pain, ulcers, bloody or nonbloody diarrhea, cramping, weight loss, anorexia, and fatigue. Affected children can also exhibit stunted growth and delayed puberty.5 Pathological features include ulceration, abscesses, inflammatory infiltrates, edema, mucin depletion, and loss of the normal crypt architecture. IBD leads to fistulas, perforations, infection, abscesses, and increases the risk of intestinal dysplasia and colon cancer in proportion to its duration.6,7
IBD is caused by dysregulation of the immune mechanisms that maintain homeostasis between the intestinal epithelium and commensal and pathogenic gut flora.3,8–11 Genetic analyses have implicated the human genes ATG16L, NOD2/CARD15, IBD5, CTLA4, TNFSF15, JAK2, STAT3, IL23R, and ORMDL3 as risk factors in IBD.12–19 These clues, combined with basic research, have revealed that antimicrobial peptides, autophagy, endoplasmic reticulum stress, innate and adaptive immune cell function, T-helper (Th)17 cells, regulatory T-cells, and cytokines (tumor necrosis factor [TNF]-α, interleukin [IL]-17, IL-23/IL-12, IL-22, and IL-6) are contributing factors in IBD.20–23 These mediators initiate signaling pathways that activate key inflammatory transcription factors including nuclear factor kappa B (NFκB) and signal transducer and activator of transcription (STAT)3, which integrate and amplify signals from a wide range of intrinsic and environmental stimuli.24–26 Many cell compartments of the gut including enterocytes, Paneth cells, T-cells, mature and immature myeloid cells, and vascular cells contribute to the regulation of NFκB, STAT3, and the inflammatory milieu.27,28 Elucidating the complex interactions between intestinal cells, secreted proteins, and transcription factors, their modulation by factors in the gut mucosa and its environment, and how these interactions are disrupted in IBD are the necessary first steps to identifying new targets and curing IBD. Targeted therapy and dietary chemoprevention strategies hold the promise of reducing the toxicities and risks associated with global immunosuppressive regimens that are currently being employed to treat IBD.
Sphingosine-1-phosphate (S1P) is a signaling lipid found in the circulation and in most tissues.29,30 S1P is derived from the recycling of endogenous human sphingolipids and the metabolism of sphingolipids found in dietary animal products that, like human tissues, contain sphingolipids, which are built upon a sphingosine structural backbone.29 S1P has many functions in angiogenesis, development, innate and adaptive immunity, and is a regulator of lymphocyte trafficking.31 A majority of S1P’s biological functions have been linked to its ability to activate a family of five G protein-coupled receptors, S1P receptors 1–5 (S1PR1–5).29 However, S1P exerts some actions that have not yet been definitively or completely attributed to S1PRs. For example, S1P serves as a major activator of the IL-6/STAT3 pathway implicated in the pathophysiology and genetic basis of IBD, as well as the pathogenesis of colon cancer.24,32–38 In fact, S1P production appears to be oncogenic in colon cancer.39,40 S1P is also the cofactor for the TNF receptor associated factor 2 E3 ubiquitin ligase required for activation of NFκB downstream of TNF-α and nucleotide-binding oligomerization domain-containing protein 2.16,41 Nuclear actions of S1P have also not been linked to S1PR functions.42
S1P is generated from sphingosine through the actions of sphingosine kinase (SphK) enzymes, as shown in Figure 1. There are two isoforms of SK: the ubiquitously expressed major SK, SphK1; and the more tissue-restricted isoform, SphK2. S1P can be dephosphorylated by specific and nonspecific lipid phosphatases.43 However, the irreversible degradation of S1P to ethanolamine phosphate and hexadecenal is catalyzed by the conserved endoplasmic reticulum enzyme, sphingosine phosphate lyase (SPL), which is expressed in differentiated enterocytes of the small and large intestine, Paneth cells, and inflammatory cells44,45 (Saba, unpublished data, 2014). SPL is downregulated in colon cancer, leading to S1P accumulation in neoplastic intestinal tissues, thereby implicating SPL in colon carcinogenesis.46,47
Figure 1.
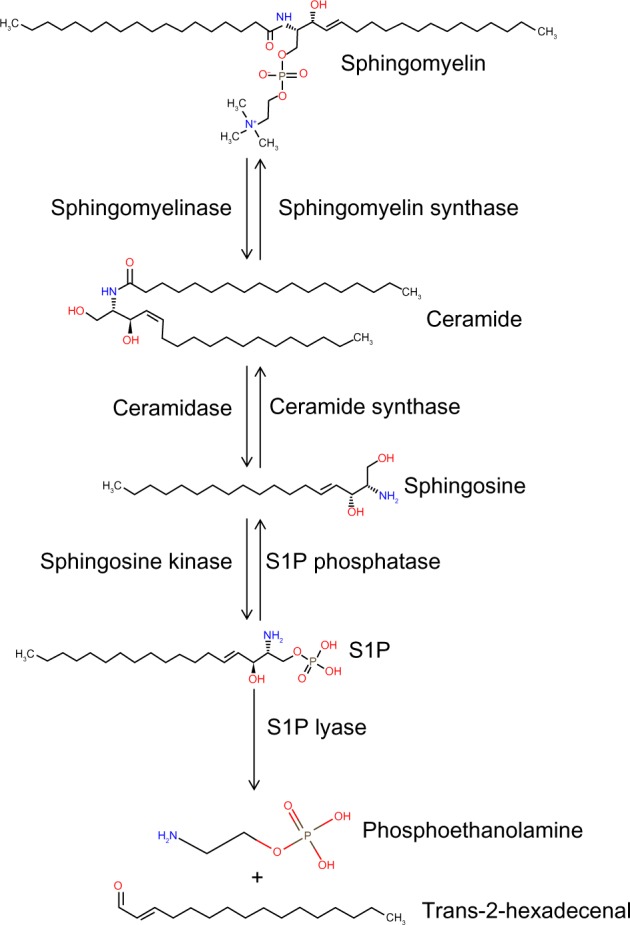
The sphingolipid metabolic pathway.
Notes: S1P is generated by the catabolism of ceramide, which is the central molecule of the sphingolipid metabolic pathway. Sphingomyelin is hydrolyzed by sphingomyelinase, yielding phosphorylcholine and ceramide; the latter is further metabolized to form a free fatty acid and sphingosine. Sphingosine can be phosphorylated by sphingosine kinase resulting in S1P. S1P can be dephosphorylated back to sphingosine by S1P phosphatase (or nonspecific lipid phosphatases), or irreversibly cleaved by S1P lyase into phosphoethanolamine and trans-2-hexadecenal.
Abbreviation: S1P, sphingosine-1-phosphate.
Sphingolipids are implicated in the regulation of immune functions and key inflammatory pathways involving STAT3 and NFκB.32,48 Further, there is high expression of the genes involved in sphingolipid metabolism in the small and large intestine, where they function in the metabolism of dietary sphingolipids.49 Based on these findings, there has been interest in exploring the possible role of S1P signaling in IBD. The relevance of sphingolipids in the pathophysiology of IBD is heightened by the recent finding that polymorphisms of orosomucoid (ORM)1-like 3, a homologue of the sphingolipid regulatory protein ORM1, are genetically linked to IBD.19,50 This review will summarize the scientific evidence implicating S1P signaling as a mediator and potential target in IBD.
S1P signaling as a target in autoimmune disease
In 2004 and 2005, several publications from one laboratory51–53 demonstrated that S1P signaling through S1PR1 and metabolism by SPL control lymphocyte egress from the thymus into the circulatory system. These studies showed that the pharmacological inhibition of SPL (which disrupts the S1P gradient between the blood and tissues) or treatment with an S1PR1 modulator (which induces downregulation and attenuates signaling through this receptor) induced lymphopenia in mice. The role of S1P signaling and metabolism in the regulation of lymphocyte trafficking has been further confirmed through genetic approaches.54,55 Most exciting was the fact that targeting the S1P signaling pathway blocked lymphocyte trafficking without necessarily reducing global immune functions as other immunosuppressive agents do. The strategy of targeting S1PR1 to induce lymphopenia was later shown to be therapeutically effective in the treatment of the human autoimmune disorder, multiple sclerosis.56–59
Other lines of evidence demonstrate that S1P signaling is linked to inflammation, autoimmune disease, and cancer.32 In 2010, the research group of Lee et al32 demonstrated that the STAT3 signaling pathway and the S1P/S1PR1 pathway coactivate one another in an inflammatory loop that promotes carcinogenesis. In an atherosclerosis mouse model, S1PR2 activation increased NFκB activity in macrophages, one of the cell types implicated in the development of atherosclerosis.60 In another study, S1PR1 was found to directly activate the STAT3 pathway via IL-6 in a mouse model of autoimmune encephalomyelitis.61 These studies implicate the S1P axis in the pathophysiology of autoimmune disease and highlight the need to explore S1P targeting approaches in other autoimmune and inflammatory conditions.
S1P signaling as a target in IBD
The results from preclinical studies employing various mouse models of colitis suggest that targeting S1P signaling and other sphingolipids may also be useful in the treatment of IBD.62–65 FTY720, KRP-203, and W-061 are S1PR agonist/functional antagonists that have a high affinity for S1PR1, causing its internalization and downmodulation, as shown in Figure 2A. These agents have been shown to reduce murine models of colitis produced by IL-10 deficiency, dextran sodium sulfate (DSS) treatment, and adoptive T-cell transfer.66–69 In contrast to CD, which is characterized by a Th1 cytokine profile, UC is defined by a Th2 cytokine profile.70 Th2 cytokines are necessary for the differentiation of cluster of differentiation (CD)4+ T-cells into natural killer T (NKT) cells. NKT cells produce IL-13 following activation by glycolipid antigens that are found on the bacteria of the gut or in epithelial cells infected by bacteria.71 The lamina propria of UC patients is characterized by high expression of IL-13 and IL-13-producing CD4+ T-cells.72 NKT cells have been shown in vitro to cause cytotoxicity via IL-13 and to negatively affect the epithelial barrier function of a monolayer of HT29.72 In the oxazolone mouse model of UC, an ex vivo culture of CD4+ T-cells isolated from FTY720-treated mice showed reduced expression of proinflammatory cytokines IL-13, IL-5, and IL-4, produced by Th2 cells.73 This suggests that FTY720 may have the potential to correct fundamental immune imbalances in the UC microenvironment. In a Th1-driven 2,4,6-trinitrobenzenesulfonic acid (TNBS) mouse model of colitis, FTY720 also showed efficacy in the reduction of colitis.74 Treg cells are thought to play a key role in IBD by preventing the proliferation and activation of T lymphocytes. In this study, FTY720 was found to directly affect the functional activity of CD4+CD25+ regulatory T-cells. Administration of FTY720 to TNBS-treated mice resulted in the decreased expression of Th1 cytokines, namely IL-12p70 and TNF-α, and an upregulation of the immunosuppresive Treg cytokines IL-10 and transforming growth factor-β. This was accompanied by an increase in the expression of cytotoxic T-lymphocyte antigen 4 and FoxP3, both of which are markers of Treg cells. The ability of FTY720 to attenuate colitis in both Th1- and Th2-driven models of colitis makes it an attractive candidate for the treatment of IBD patients.
Figure 2.

S1P axis modulators and their effects on S1P signaling.
Notes: (A) S1PR1 is activated by its natural ligand, S1P, resulting in the activation of its G-coupled protein Gαi. FTY720, KRP-203, and W-061 are all superagonists of S1PR1 with functional antagonist activity leading to the desensitization and internalization of S1PR1, thus attenuating S1P-associated signaling through this receptor. Note that FTY720 also acts on S1PR3–5, KRP-203 has a slight agonist activity on S1PR4, and W-061 also acts on S1PR4–5. (B) Sphingosine kinases produce S1P from sphingosine. ABC747080, ABC294640, and FTY720 are sphingosine kinase inhibitors that either block SK activity or, in the case of FTY720, cause proteasomal degradation of the kinases. ABC747080 and FTY720 are SphK1 inhibitors and ABC294640 inhibits Sphk2.
Abbreviations: S1P, sphingosine-1-phosphate; S1PR, sphingosine-1-phosphate receptor; ER, endoplasmic reticulum; SphK, sphingosine kinase.
In addition to the effects on colitis afforded by agents targeting S1PRs, it was observed that mice lacking SphK1 were less susceptible than littermate controls to colitis induced by DSS.75 This suggests that SphK1 could be targeted to reduce S1P signaling (Figure 2B). Compared to control mice, SphK1 null mice showed a similar expression of TNF-α, but decreased expression of cyclooxygenase (COX)-2 and no systemic inflammatory response. These findings pinpoint SphK1’s action downstream of TNF-α, but upstream of COX-2. The evidence suggests that targeting SphK1 with a specific inhibitor could attenuate inflammatory responses in IBD patients. In support of this notion, two inhibitors of SK, ABC747080 and ABC294640, prevented TNF-α activation of NFκB, prostaglandin E2 production, vascular cell adhesion molecule and intercellular adhesion molecule expression, and reduced clinical and histological signs of DSS-induced colitis in C57Bl/6 mice.76 The effects of the S1P axis on adaptive immunity are shown in Figure 3.
Figure 3.

The role of the S1P axis in adaptive immunity.
Notes: Dietary or bacterial S1P from the intestinal lumen bind to S1PRs and activate the STAT3 pathway in intestinal epithelial cells. Mucosal NFκB is activated by intracellular S1P produced by the phosphorylation of sphingosine by SphK. These two transcription factors transactivate numerous proinflammatory cytokine genes, thereby contributing to the recruitment of immune cells to the inflamed site. Moreover, STAT3 can amplify S1P/S1PR signaling by increasing the expression of S1PR1. Adaptive immune cells, namely Treg, Th1, and Th2 cells, produce a variety of cytokines in response to intestinal inflammation. Most notably, in response to NFκB and STAT3 pathway activation, Th1 and Th2 cells can produce an array of proinflammatory cytokines, including IL-1β, TNF-α, IL-5, and so on. The S1PR1 superagonist FTY720 increases Treg production of IL-10 and TGF-β, but inhibits proinflammatory cytokine production by Th1 and Th2 cells, possibly by acting through S1PRs.
Abbreviations: S1P, sphingosine-1-phosphate; S1PR, sphingosine-1-phosphate receptor; SphK, sphingosine kinase; NFκB, nuclear factor kappa-light-chain-enhancer of activated B cells; STAT3, signal transducer and activator of transcription 3; Treg, regulatory T-cells; IL, interleukin; Th, T-helper; iNOS, inducible nitric oxide synthase; TNF, tumor necrosis factor; TGF, transforming growth factor.
SPL is highly expressed throughout the intestine, especially in the differentiated epithelial cells of the duodenum and jejunum and, to a lesser degree, in the ileum and colon.77,78 In a recent study,79 an inverse correlation between the colonic concentrations of S1P and SPL’s cofactor, pyridoxal-5-phosphate was found. Of note, plasma pyridoxal-5-phosphate concentration was inversely linked with the concentration of inflammatory markers.79 These results suggest that SPL expression, as well as its cofactor availability, may be important in preventing or reducing inflammation. Patients suffering from IBD are at a higher risk of developing colon cancer the longer they suffer from unresolved inflammation.2 Interestingly, we found that SPL expression and activity were downregulated in adenomatous lesions of ApcMin/+ mice.46 Moreover, human colon cancer tissues expressed less SPL than the normal adjacent tissue. It was demonstrated that SPL expression is required for p38- and p53-induced cell death via caspase 2 activation.46 The results of this study raise the possibility that changes in SPL expression or enzyme activity could contribute to the initiation of intestinal tumorigenesis.
S1P in colitis-associated cancer
Liang et al48 have provided new evidence supporting a role for SphK2 in the pathogenesis of colitis and colitis-associated cancer (CAC). In their study, the azoxymethane (AOM)/DSS mouse model of CAC was implemented in both wild type and SphK2 knockout (KO) mice. They found that the tumor number, size, and load were higher in SphK2 KO mice compared to wild type mice.48 The SphK2 KO mice also exhibited elevated disease activity, suffering from severe diarrhea, as well as blood and weight loss. In addition, the KO mice exhibited shorter colons (indicating inflammation-induced stricture and fibrosis), extensive colon architectural damage, and immune cell infiltration.48
In performing biochemical analyses of the two groups, the authors made a surprising discovery. They found increased circulating and colonic S1P levels in SphK2 KO mice compared to controls. Moreover, SphK1 protein and messenger ribonucleic acid levels were higher in the colons of SphK2 KO mice.48 The authors suggested that the upregulation of SphK1 could be the result of reduced nuclear SphK2. SphK2 in the nucleus produces a source of S1P that inhibits histone deacetylase 1/2. Importantly, one of the transcriptional regulators of SphK1 expression, c-Jun, is regulated by histone acetylation.42,80,81 Therefore, in the colons of SphK2 KO mice, the authors suggest that increased histone deacetylase activity results in increased activation of c-Jun, thereby driving increased SphK1 expression.48
S1P activates NFκB, a transcription factor involved in the expression of many proinflammatory cytokines that contribute to the pathogenesis of IBD and CAC.41,82,83 IL-6, which is a NFκB target gene, can activate STAT3, a regulator of S1PR1 expression.32,84 Accordingly, NFκB activation, IL-6 expression and S1PR1 expression were all increased in the colons of SphK2 KO mice following DSS treatment to a greater extent than in controls.48 To determine the cell type responsible for IL-6 secretion, the authors generated bone marrow chimeras by transferring bone marrow cells of either wild type or SphK2 KO mice to an irradiated recipient. SphK2 disruption in bone marrow cells alone was sufficient to recapitulate the SphK2 global KO mouse phenotype following DSS treatment.48 Flow cytometry analysis identified macrophages and dendritic cells as the major producers of IL-6.48 However, intestinal epithelial cells were at least in part responsible for the increased colonic expression of IL-6 in global SphK2 KO mice, as shown by immunohistochemistry.48
The authors proposed a model involving “a feed-forward amplification loop” in which the SK/S1P/S1PR1 axis leads to the activation of NFκB and persistent STAT3 activation in CAC, as shown in Figure 4.48 To disrupt the feed-forward cycle, the authors administered FTY720, an S1PR1 functional antagonist that can also inhibit SphK1 by causing its proteasomal degradation (Figure 2B).85–87 Administration of FTY720 in SphK2 global KO mice treated with DSS reduced disease activity, SphK1, IL-6, and S1PR1 expression, and NFκB and STAT3 activity.48 FTY720 could prevent CAC in SphK2 KO mice if administered at the beginning of the AOM/DSS regimen, but not when it was administered at a later time point. This was true even though FTY720 administration could still reduce STAT3 activation and S1PR1 expression.48 The authors concluded that FTY720 should be explored as a drug to treat CAC in IBD patients, since it reduces both the expression of S1PR1 and SphK1, thereby disrupting the S1P/SphK1/S1PR1 signaling loop.
Figure 4.
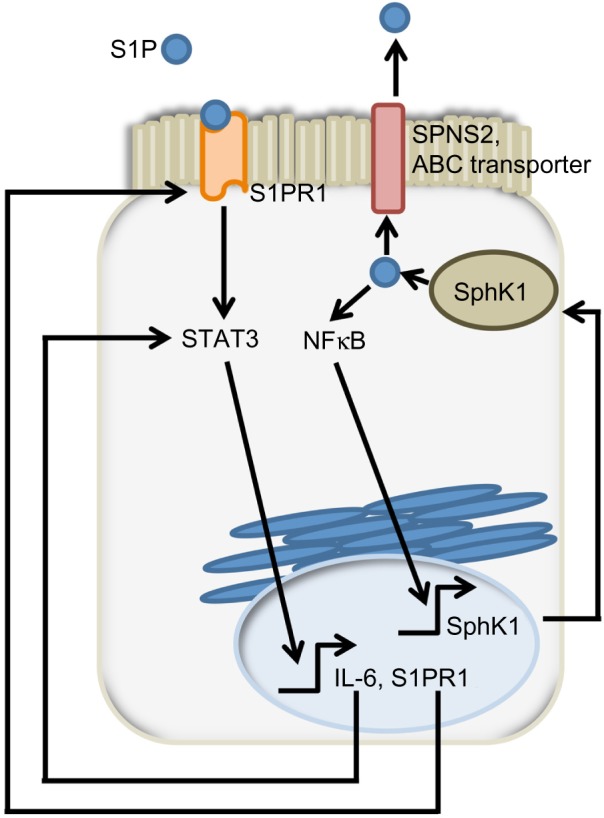
SphK1/S1P/S1PR1 feed-forward loop sustain signals promoting colon tumorigenesis.
Notes: SphK1 produces S1P, which is kept in the cell and/or is exported in the extracellular milieu. Extracellular S1P activates S1PR1 which, in turn, activates the JAK2/STAT3 pathway. STAT3 is one of the transcription factors involved in the regulation of IL-6 and S1PR1 expression. Intracellular S1P can activate NFκB, a transcriptional regulator of SphK1. All these pathways are interrelated in an amplification loop, allowing for the sustained activation of STAT3, which is involved in cell transformation and tumor progression. Bent arrows indicate transcriptional regulation.
Abbreviations: SphK, sphingosine kinase; S1P, sphingosine-1-phosphate; S1PR, sphingosine-1-phosphate receptor; STAT3, signal transducer and activator of transcription 3; IL, interleukin; NFκB, nuclear factor kappa-light-chain-enhancer of activated B cells; JAK2, Janus kinase 2; SPNS2, spinster homolog 2; ABC, ATP Binding Cassette; ATP, adenosine triphosphate.
Dietary sphingolipids
Each day, we consume between 0.3–0.4 g of sphingolipids though our diet.88 In the intestinal tract, these sphingolipids are digested to form sphingoid bases and ceramides.49 In the past three decades, the potential of sphingolipids as dietary bioactive molecules has been studied intensively in different mouse models of IBD and colon cancer. 1,2 dimethylhydrazine (DMH) is a carcinogenic compound used in mouse and rat models of CAC.89,90 DMH promotes colon tumorigenesis by causing oxidative stress and inflammation.91 One report of a study employing the DMH mouse model found that dietary sphingomyelin can reduce the number of premalignant lesions and tumors by 20%.92 In another study, the plant sphingolipid soy glucosylceramide added to the diet of ApcMin/+ or DMH-treated mice decreased the number of adenomas and aberrant crypt foci arising in these two respective models of colon cancer.93 The authors confirmed that the expression levels of two transcription factors involved in tumorigenesis, hypoxia-inducible factor 1α and transcription factor 4, were decreased in response to sphingolipid treatment.93
Mazzei et al94 explored the effects of dietary sphingomyelin in the AOM/DSS mouse model of CAC. They found that adding 1 g/kg of sphingomyelin in the mouse diet decreased both the disease activity index and the tumor number.94 They found that sphingomyelin attenuated the expression of proinflammatory cytokines (interferon-γ, IL-17, and IL-23), but induced antiinflammatory cytokines and receptors implicated in Th2 and Treg cell differentiation (IL-4, IL-3, IL-13ra2, and IL-10rb).94 In contrast, in an acute DSS colitis model, gavage of 4 mg or 8 mg of sphingomyelin/day exacerbated the disease activity by causing increased apoptosis of the intestinal epithelial cells.95 This finding was associated with an increased activity of cathepsin D that mediated caspase-3- and caspase-9-induced apoptosis.95 Using the IL-10 KO mouse model of spontaneous colitis, the authors showed that sphingomyelin aggravated mucosal inflammation.95 These findings – using a variety of mouse models, different sphingomyelin doses, and measuring different disease-related endpoints – raise the possibility that dietary sphingolipids may have untoward effects depending on the state of the target tissue, and possibly the source and structure of the sphingolipid. They also suggest that there may be a tight range of sphingomyelin concentrations that will afford therapeutic benefit in CAC.
Sphingadienes are orally available sphingolipids containing a sphingolipid backbone harboring conjugated trans double bonds. They are found in soy products and other natural sources.96 Sphingadienes containing double bonds at C4,5 and C8,9 were shown to induce apoptosis in HCT116, DLD1, and SW480 colon cancer cell lines and to decrease tumor development in an ApcMin/+ mouse model of colon cancer.96 Sphingadienes act, at least in part, by blocking the translocation of Akt from the cytoplasm to the membrane, thereby inactivating Akt. This effect of sphingadienes leads to the induction of apoptosis and autophagy, processes that are repressed by Akt.96 Sphingadiene treatment also inactivated GSK-3β and, as a consequence, downregulated growth-activating Wnt signaling.97 Sphingadienes are being explored for their potential efficacy as antiinflammatory agents in IBD and in their ability to prevent the development of CAC (Degagne, unpublished data, 2014).29 Importantly, unlike mammalian sphingolipids, sphingadienes cannot be metabolized to S1P. Further, since sphingadienes are not produced in appreciable amounts in humans, their metabolism by the enzymes present in human colonic epithelial cells is limited, their uptake into the bloodstream is minimal, and their action is local, making them appealing candidates for the long-term chemoprevention of CAC.
Risks, controversies, and remaining questions
The studies described above highlight the possibility that strategies involving S1PR1 downregulation or inhibition that have proven useful in other autoimmune diseases may afford promise in the context of IBD. However, chemically targeting S1PRs may be complicated by their diverse functions. For example, S1PR1 activity could prove critical in maintaining vascular integrity in the colonic mucosa. This concern is illustrated by results from preclinical studies testing S1PR1 disruption in mice suffering from experimental colitis.98 However, treatment of C57BL6/J mice with FTY720 did not cause more bleeding than the control group in an experimental colitis model.98 Of additional concern, administration of FTY720 to mice in an experimental colitis model impaired mucosal immunity such that the mice were then unable to eliminate an enteric bacterial infection.99 FTY720 treatment was associated with a shift in the immune cell populations present in the colon during the infection, decreasing the number of T and B lymphocytes and macrophages, and resulting in the decreased expression of Th1 cytokines (Il-1β, inducible nitric oxide synthase [iNOS], IL-17a, IL-6, and TNF-α). Even when the treatment was discontinued prior to infection, the mice were still unable to clear the bacterial infection.99 These findings suggest that the safety of strategies that modulate S1PR signaling in IBD patients must be investigated further. Specifically, it will be critical to understand how S1PR modulation affects the innate immune response to a wide range of bacteria and viruses to which IBD patients are particularly susceptible.
The recent article by Liang et al48 has established the importance of SphK2 in inflammatory signaling through STAT3. Interestingly, others have shown that CD4+ T-cell SphK2 is required for suppressing IL-2 responses that aggravate IBD via a STAT5-dependent mechanism.100 Sphk2 null mouse CD4+ T-cells have a hyperactive phenotype, increased proliferation, and cytokine secretions in response to IL-2 signaling. Moreover, these Sphk2 null mouse CD4+ T-cells were not responsive to Treg cell-mediated suppression. This study raises concerns regarding the use of SK inhibitors in treating IBD patients, as they could have adverse effects on immune cells by directly increasing cytokine secretions through their effects on CD4+ T-cells and also by counteracting the immunomodulatory role of Treg cells. Thus, the S1P signaling pathway may be affecting multiple aspects of the innate and adaptive immune response relevant to IBD.
Additional questions remained unanswered. Is the decreased expression of SphK2 a characteristic of or a precursor state to the development of IBD or colon cancer? What are the key mechanisms by which S1P signaling (and downstream NFκB and STAT3 inflammatory nodes) promotes the switch from a state of inflammation to a preneoplastic state and, ultimately, to the development of CAC? At this time, the exact role of S1P in IBD remains incompletely understood. Further, the potential for S1P to be used as a biomarker of disease activity has not been explored. In that regard, there is sparse information regarding the lipid metabolic profiles of IBD colons compared to controls. How lipid metabolomics, S1P degradation, and the enzymes of sphingolipid metabolism that are enriched in the gut epithelium may influence gut immunity and IBD still remain to be determined.
Conclusion
The mechanisms that underlie the pathophysiology of IBD are complex and involve numerous signal transduction pathways that regulate innate and adaptive immunity in the gut. Of particular interest are a triumvirate of interrelated pathways centered on the sphingolipid metabolite S1P, as well as on the NFκB and STAT3 inflammatory signaling pathways with which it interacts. S1P functions as a critical regulator of lymphocyte trafficking and a cofactor in inflammatory signaling.31 Further, many S1P-related targets, including S1PRs and the enzymes involved in S1P biosynthesis or degradation, are located in the epithelium of the small and large intestine wherein IBD arises.49 As such, these targets are of interest in the treatment of IBD and in the prevention of its progression to CAC. In addition, dietary sphingolipids may represent another strategy for reducing colitis and preventing colon carcinogenesis. While some safety concerns remain, the preclinical evidence supports further investigation of these strategies in the context of IBD.
Acknowledgments
This work was supported by the Broad Medical Research Program of The Broad Foundation Grant IBD-0353, Crohn’s and Colitis Foundation of America Senior Research Award 277014, National Institutes of Health grants CA129438 and R21AT005336, AICR grant 09A041, Swim Across America Foundation support (JDS), Crohn’s and Colitis Canada, and Fonds de recherche du Québec-Santé fellowship grant 28137 (ED). We thank Nelle Cronen for her expert administrative assistance.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Odze R. Diagnostic problems and advances in inflammatory bowel disease. Mod Pathol. 2003;16(4):347–358. doi: 10.1097/01.MP.0000064746.82024.D1. [DOI] [PubMed] [Google Scholar]
- 2.Ullman TA, Itzkowitz SH. Intestinal inflammation and cancer. Gastroenterology. 2011;140(6):1807–1816. doi: 10.1053/j.gastro.2011.01.057. [DOI] [PubMed] [Google Scholar]
- 3.Bosani M, Ardizzone S, Porro GB. Biologic targeting in the treatment of inflammatory bowel diseases. Biologics. 2009;3:77–97. [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 4.Neurath MF. Cytokines in inflammatory bowel disease. Nat Rev Immunol. 2014;14(5):329–342. doi: 10.1038/nri3661. [DOI] [PubMed] [Google Scholar]
- 5.Sylvester FA, Gordon CM, Thayu M, et al. Report of the CCFA pediatric bone, growth and muscle health workshop, New York City, November 11–12, 2011, with updates. Inflamm Bowel Dis. 2013;19(13):2919–2926. doi: 10.1097/MIB.0b013e3182a5a004. [DOI] [PubMed] [Google Scholar]
- 6.Baumgart DC, Sandborn WJ. Crohn’s disease. Lancet. 2012;380(9853):1590–1605. doi: 10.1016/S0140-6736(12)60026-9. [DOI] [PubMed] [Google Scholar]
- 7.Ordás I, Eckmann L, Talamini M, Baumgart DC, Sandborn WJ. Ulcerative colitis. Lancet. 2012;380(9853):1606–1619. doi: 10.1016/S0140-6736(12)60150-0. [DOI] [PubMed] [Google Scholar]
- 8.Blumberg RS. Inflammation in the intestinal tract: pathogenesis and treatment. Dig Dis. 2009;27(4):455–464. doi: 10.1159/000235851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abraham C, Medzhitov R. Interactions between the host innate immune system and microbes in inflammatory bowel disease. Gastroenterology. 2011;140(6):1729–1737. doi: 10.1053/j.gastro.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaser A, Blumberg RS. Adaptive immunity in inflammatory bowel disease: state of the art. Curr Opin Gastroenterol. 2008;24(4):455–461. doi: 10.1097/MOG.0b013e328304d60d. [DOI] [PubMed] [Google Scholar]
- 11.Iskandar HN, Ciorba MA. Biomarkers in inflammatory bowel disease: current practices and recent advances. Transl Res. 2012;159(4):313–325. doi: 10.1016/j.trsl.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Christophi GP, Rong R, Holtzapple PG, Massa PT, Landas SK. Immune markers and differential signaling networks in ulcerative colitis and Crohn’s disease. Inflamm Bowel Dis. 2012;18(12):2342–2356. doi: 10.1002/ibd.22957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Biswas A, Petnicki-Ocwieja T, Kobayashi KS. Nod2: a key regulator linking microbiota to intestinal mucosal immunity. J Mol Med (Berl) 2012;90(1):15–24. doi: 10.1007/s00109-011-0802-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boniface K, Blom B, Liu YJ, de Waal Malefyt R. From interleukin-23 to T-helper 17 cells: human T-helper cell differentiation revisited. Immunol Rev. 2008;226:132–146. doi: 10.1111/j.1600-065X.2008.00714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dubinsky MC, Wang D, Picornell Y, et al. Western Regional Research Alliance for Pediatric IBD IL-23 receptor (IL-23R) gene protects against pediatric Crohn’s disease. Inflamm Bowel Dis. 2007;13(5):511–515. doi: 10.1002/ibd.20126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Naser SA, Arce M, Khaja A, et al. Role of ATG16L, NOD2 and IL23R in Crohn’s disease pathogenesis. World J Gastroenterol. 2012;18(5):412–424. doi: 10.3748/wjg.v18.i5.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maloy KJ, Kullberg MC. IL-23 and Th17 cytokines in intestinal homeostasis. Mucosal Immunol. 2008;1(5):339–349. doi: 10.1038/mi.2008.28. [DOI] [PubMed] [Google Scholar]
- 18.Noble CL, Abbas AR, Lees CW, et al. Characterization of intestinal gene expression profiles in Crohn’s disease by genome-wide microarray analysis. Inflamm Bowel Dis. 2010;16(10):1717–1728. doi: 10.1002/ibd.21263. [DOI] [PubMed] [Google Scholar]
- 19.McGovern DP, Gardet A, Törkvist L, et al. NIDDK IBD Genetics Consortium Genome-wide association identifies multiple ulcerative colitis susceptibility loci. Nat Genet. 2010;42(4):332–337. doi: 10.1038/ng.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grivennikov S, Karin E, Terzic J, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15(2):103–113. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Festen EA, Szperl AM, Weersma RK, Wijmenga C, Wapenaar MC. Inflammatory bowel disease and celiac disease: overlaps in the pathology and genetics, and their potential drug targets. Endocr Metab Immune Disord Drug Targets. 2009;9(2):199–218. doi: 10.2174/187153009788452426. [DOI] [PubMed] [Google Scholar]
- 22.Erdman SE, Poutahidis T. Roles for inflammation and regulatory T cells in colon cancer. Toxicol Pathol. 2010;38(1):76–87. doi: 10.1177/0192623309354110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hayakawa Y, Maeda S, Nakagawa H, et al. Effectiveness of IkappaB kinase inhibitors in murine colitis-associated tumorigenesis. J Gastroenterol. 2009;44(9):935–943. doi: 10.1007/s00535-009-0098-7. [DOI] [PubMed] [Google Scholar]
- 24.Bromberg J, Wang TC. Inflammation and cancer: IL-6 and STAT3 complete the link. Cancer Cell. 2009;15(2):79–80. doi: 10.1016/j.ccr.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aaronson DS, Horvath CM. A road map for those who don’t know JAK-STAT. Science. 2002;296(5573):1653–1655. doi: 10.1126/science.1071545. [DOI] [PubMed] [Google Scholar]
- 26.Grivennikov SI, Karin M. Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010;21(1):11–19. doi: 10.1016/j.cytogfr.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scaldaferri F, Correale C, Gasbarrini A, Danese S. Molecular signaling blockade as a new approach to inhibit leukocyte-endothelial interactions for inflammatory bowel disease treatment. Cell Adh Migr. 2009;3(3):296–299. doi: 10.4161/cam.3.3.9152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Danese S. Role of the vascular and lymphatic endothelium in the pathogenesis of inflammatory bowel disease: ‘brothers in arms’. Gut. 2011;60(7):998–1008. doi: 10.1136/gut.2010.207480. [DOI] [PubMed] [Google Scholar]
- 29.Fyrst H, Saba JD. An update on sphingosine-1-phosphate and other sphingolipid mediators. Nat Chem Biol. 2010;6(7):489–497. doi: 10.1038/nchembio.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spiegel S, Milstien S. The outs and ins of sphingosine-1-phosphate in immunity. Nat Rev Immunol. 2011;11(6):403–415. doi: 10.1038/nri2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rivera J, Proia RL, Olivera A. The alliance of sphingosine-1-phosphate and its receptors in immunity. Nat Rev Immunol. 2008;8(10):753–763. doi: 10.1038/nri2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee H, Deng J, Kujawski M, et al. STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat Med. 2010;16(12):1421–1428. doi: 10.1038/nm.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Slattery ML, Lundgreen A, Kadlubar SA, Bondurant KL, Wolff RK. JAK/STAT/SOCS-signaling pathway and colon and rectal cancer. Mol Carcinog. 2013;52(2):155–166. doi: 10.1002/mc.21841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Corvinus FM, Orth C, Moriggl R, et al. Persistent STAT3 activation in colon cancer is associated with enhanced cell proliferation and tumor growth. Neoplasia. 2005;7(6):545–555. doi: 10.1593/neo.04571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin Q, Lai R, Chirieac LR, et al. Constitutive activation of JAK3/STAT3 in colon carcinoma tumors and cell lines: inhibition of JAK3/STAT3 signaling induces apoptosis and cell cycle arrest of colon carcinoma cells. Am J Pathol. 2005;167(4):969–980. doi: 10.1016/S0002-9440(10)61187-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Santandreu FM, Valle A, Oliver J, Roca P. Resveratrol potentiates the cytotoxic oxidative stress induced by chemotherapy in human colon cancer cells. Cell Physiol Biochem. 2011;28(2):219–228. doi: 10.1159/000331733. [DOI] [PubMed] [Google Scholar]
- 37.Wang Z, Jin H, Xu R, Mei Q, Fan D. Triptolide downregulates Rac1 and the JAK/STAT3 pathway and inhibits colitis-related colon cancer progression. Exp Mol Med. 2009;41(10):717–727. doi: 10.3858/emm.2009.41.10.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li Y, de Haar C, Chen M, et al. Disease-related expression of the IL6/STAT3/SOCS3 signalling pathway in ulcerative colitis and ulcerative colitis-related carcinogenesis. Gut. 2010;59(2):227–235. doi: 10.1136/gut.2009.184176. [DOI] [PubMed] [Google Scholar]
- 39.Oskouian B, Saba J. Sphingosine-1-phosphate metabolism and intestinal tumorigenesis: lipid signaling strikes again. Cell Cycle. 2007;6(5):522–527. doi: 10.4161/cc.6.5.3903. [DOI] [PubMed] [Google Scholar]
- 40.Furuya H, Shimizu Y, Kawamori T. Sphingolipids in cancer. Cancer Metastasis Rev. 2011;30(3–4):567–576. doi: 10.1007/s10555-011-9304-1. [DOI] [PubMed] [Google Scholar]
- 41.Alvarez SE, Harikumar KB, Hait NC, et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature. 2010;465(7301):1084–1088. doi: 10.1038/nature09128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hait NC, Allegood J, Maceyka M, et al. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science. 2009;325(5945):1254–1257. doi: 10.1126/science.1176709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mandala SM. Sphingosine-1-phosphate phosphatases. Prostaglandins. 2001;64(1–4):143–156. doi: 10.1016/s0090-6980(01)00111-3. [DOI] [PubMed] [Google Scholar]
- 44.Aguilar A, Saba JD. Truth and consequences of sphingosine-1-phosphate lyase. Adv Biol Regul. 2012;52(1):17–30. doi: 10.1016/j.advenzreg.2011.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saba JD, Nara F, Bielawska A, Garrett S, Hannun YA. The BST1 gene of Saccharomyces cerevisiae is the sphingosine-1-phosphate lyase. J Biol Chem. 1997;272(42):26087–26090. doi: 10.1074/jbc.272.42.26087. [DOI] [PubMed] [Google Scholar]
- 46.Oskouian B, Sooriyakumaran P, Borowsky AD, et al. Sphingosine-1-phosphate lyase potentiates apoptosis via p53- and p38-dependent pathways and is down-regulated in colon cancer. Proc Natl Acad Sci U S A. 2006;103(46):17384–17389. doi: 10.1073/pnas.0600050103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kohno M, Momoi M, Oo ML, et al. Intracellular role for sphingosine kinase 1 in intestinal adenoma cell proliferation. Mol Cell Biol. 2006;26(19):7211–7223. doi: 10.1128/MCB.02341-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liang J, Nagahashi M, Kim EY, et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell. 2013;23(1):107–120. doi: 10.1016/j.ccr.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Duan RD. Physiological functions and clinical implications of sphin-golipids in the gut. J Dig Dis. 2011;12(2):60–70. doi: 10.1111/j.1751-2980.2011.00481.x. [DOI] [PubMed] [Google Scholar]
- 50.Skieceviciene J, Kiudelis G, Ellinghaus E, et al. Replication study of ulcerative colitis risk loci in a Lithuanian-Latvian case-control sample. Inflamm Bowel Dis. 2013;19(11):2349–2355. doi: 10.1097/MIB.0b013e3182a3eaeb. [DOI] [PubMed] [Google Scholar]
- 51.Matloubian M, Lo CG, Cinamon G, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427(6972):355–360. doi: 10.1038/nature02284. [DOI] [PubMed] [Google Scholar]
- 52.Lo CG, Xu Y, Proia RL, Cyster JG. Cyclical modulation of sphingosine-1-phosphate receptor 1 surface expression during lymphocyte recirculation and relationship to lymphoid organ transit. J Exp Med. 2005;201(2):291–301. doi: 10.1084/jem.20041509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schwab SR, Pereira JP, Matloubian M, Xu Y, Huang Y, Cyster JG. Lymphocyte sequestration through S1P lyase inhibition and disruption of S1P gradients. Science. 2005;309(5741):1735–1739. doi: 10.1126/science.1113640. [DOI] [PubMed] [Google Scholar]
- 54.Vogel P, Donoviel MS, Read R, et al. Incomplete inhibition of sphingosine 1-phosphate lyase modulates immune system function yet prevents early lethality and non-lymphoid lesions. PLoS One. 2009;4(1):e4112. doi: 10.1371/journal.pone.0004112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bréart B, Ramos-Perez WD, Mendoza A, et al. Lipid phosphate phosphatase 3 enables efficient thymic egress. J Exp Med. 2011;208(6):1267–1278. doi: 10.1084/jem.20102551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kappos L, Antel J, Comi G, et al. FTY720 D2201 Study Group Oral fingolimod (FTY720) for relapsing multiple sclerosis. N Engl J Med. 2006;355(11):1124–1140. doi: 10.1056/NEJMoa052643. [DOI] [PubMed] [Google Scholar]
- 57.Kappos L, Radue EW, O’Connor P, et al. FREEDOMS Study Group A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362(5):387–401. doi: 10.1056/NEJMoa0909494. [DOI] [PubMed] [Google Scholar]
- 58.Cohen JA, Barkhof F, Comi G, et al. TRANSFORMS Study Group Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362(5):402–415. doi: 10.1056/NEJMoa0907839. [DOI] [PubMed] [Google Scholar]
- 59.Comi G, O’Connor P, Montalban X, et al. FTY720D2201 Study Group Phase II study of oral fingolimod (FTY720) in multiple sclerosis: 3-year results. Mult Scler. 2010;16(2):197–207. doi: 10.1177/1352458509357065. [DOI] [PubMed] [Google Scholar]
- 60.Wang F, Okamoto Y, Inoki I, et al. Sphingosine-1-phosphate receptor-2 deficiency leads to inhibition of macrophage proinflammatory activities and atherosclerosis in apoE-deficient mice. J Clin Invest. 2010;120(11):3979–3995. doi: 10.1172/JCI42315. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 61.Garris CS, Wu L, Acharya S, et al. Defective sphingosine 1-phosphate receptor 1 (S1P1) phosphorylation exacerbates TH17-mediated autoimmune neuroinflammation. Nat Immunol. 2013;14(11):1166–1172. doi: 10.1038/ni.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nixon GF. Sphingolipids in inflammation: pathological implications and potential therapeutic targets. Br J Pharmacol. 2009;158(4):982–993. doi: 10.1111/j.1476-5381.2009.00281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Duan RD, Nilsson A. Metabolism of sphingolipids in the gut and its relation to inflammation and cancer development. Prog Lipid Res. 2009;48(1):62–72. doi: 10.1016/j.plipres.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 64.Soo I, Madsen KL, Tejpar Q, et al. VSL#3 probiotic upregulates intestinal mucosal alkaline sphingomyelinase and reduces inflammation. Can J Gastroenterol. 2008;22(3):237–242. doi: 10.1155/2008/520383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mazzei JC, Zhou H, Brayfield BP, Hontecillas R, Bassaganya-Riera J, Schmelz EM. Suppression of intestinal inflammation and inflammation-driven colon cancer in mice by dietary sphingomyelin: importance of peroxisome proliferator-activated receptor γ expression. J Nutr Biochem. 2011;22(12):1160–1171. doi: 10.1016/j.jnutbio.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Deguchi Y, Andoh A, Yagi Y, et al. The S1P receptor modulator FTY720 prevents the development of experimental colitis in mice. Oncol Rep. 2006;16(4):699–703. [PubMed] [Google Scholar]
- 67.Sanada Y, Mizushima T, Kai Y, et al. Therapeutic effects of novel sphingosine-1-phosphate receptor agonist W-061 in murine DSS colitis. PLoS One. 2011;6(9):e23933. doi: 10.1371/journal.pone.0023933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mizushima T, Ito T, Kishi D, et al. Therapeutic effects of a new lymphocyte homing reagent FTY720 in interleukin-10 gene-deficient mice with colitis. Inflamm Bowel Dis. 2004;10(3):182–192. doi: 10.1097/00054725-200405000-00002. [DOI] [PubMed] [Google Scholar]
- 69.Song J, Matsuda C, Kai Y, et al. A novel sphingosine 1-phosphate receptor agonist, 2-amino-2-propanediol hydrochloride (KRP-203), regulates chronic colitis in interleukin-10 gene-deficient mice. J Pharmacol Exp Ther. 2008;324(1):276–283. doi: 10.1124/jpet.106.119172. [DOI] [PubMed] [Google Scholar]
- 70.Fuss IJ, Neurath M, Boirivant M, et al. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn’s disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J Immunol. 1996;157(3):1261–1270. [PubMed] [Google Scholar]
- 71.Strober W, Fuss IJ. Proinflammatory cytokines in the pathogenesis of inflammatory bowel diseases. Gastroenterology. 2011;140(6):1756–1767. doi: 10.1053/j.gastro.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fuss IJ, Heller F, Boirivant M, et al. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. J Clin Invest. 2004;113(10):1490–1497. doi: 10.1172/JCI19836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Daniel C, Sartory NA, Zahn N, et al. FTY720 ameliorates oxazolone colitis in mice by directly affecting T helper type 2 functions. Mol Immunol. 2007;44(13):3305–3316. doi: 10.1016/j.molimm.2007.02.026. [DOI] [PubMed] [Google Scholar]
- 74.Daniel C, Sartory N, Zahn N, Geisslinger G, Radeke HH, Stein JM. FTY720 ameliorates Th1-mediated colitis in mice by directly affecting the functional activity of CD4+CD25+ regulatory T cells. J Immunol. 2007;178(4):2458–2468. doi: 10.4049/jimmunol.178.4.2458. [DOI] [PubMed] [Google Scholar]
- 75.Snider AJ, Kawamori T, Bradshaw SG, et al. A role for sphingosine kinase 1 in dextran sulfate sodium-induced colitis. FASEB J. 2009;23(1):143–152. doi: 10.1096/fj.08-118109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Maines LW, Fitzpatrick LR, French KJ, et al. Suppression of ulcerative colitis in mice by orally available inhibitors of sphingosine kinase. Dig Dis Sci. 2008;53(4):997–1012. doi: 10.1007/s10620-007-0133-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Borowsky AD, Bandhuvula P, Kumar A, et al. Sphingosine-1-phosphate lyase expression in embryonic and adult murine tissues. Natl Acad Sci USA. 2006;103(46):17384–17389. doi: 10.1073/pnas.0600050103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Oskouian B1, Sooriyakumaran P, Borowsky AD, et al. Sphingosine-1-phosphate lyase potentiates apoptosis via p53- and p38-dependent pathways and is down-regulated in colon cancer. J Lipid Res. 2012;53(9):1920–1931. doi: 10.1073/pnas.0600050103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Selhub J, Byun A, Liu Z, Mason JB, Bronson RT, Crott JW. Dietary vitamin B6 intake modulates colonic inflammation in the IL10−/− model of inflammatory bowel disease. J Nutr Biochem. 2013;24(12):2138–2143. doi: 10.1016/j.jnutbio.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Paugh BS, Bryan L, Paugh SW, et al. Interleukin-1 regulates the expression of sphingosine kinase 1 in glioblastoma cells. J Biol Chem. 2009;284(6):3408–3417. doi: 10.1074/jbc.M807170200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yamaguchi K, Lantowski A, Dannenberg AJ, Subbaramaiah K. Histone deacetylase inhibitors suppress the induction of c-Jun and its target genes including COX-2. J Biol Chem. 2005;280(38):32569–32577. doi: 10.1074/jbc.M503201200. [DOI] [PubMed] [Google Scholar]
- 82.Atreya I, Atreya R, Neurath MF. NF-kappaB in inflammatory bowel disease. J Intern Med. 2008;263(6):591–596. doi: 10.1111/j.1365-2796.2008.01953.x. [DOI] [PubMed] [Google Scholar]
- 83.Karin M. NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb Perspect Biol. 2009;1(5):a000141. doi: 10.1101/cshperspect.a000141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xiao W, Hodge DR, Wang L, Yang X, Zhang X, Farrar WL. NF-kappaB activates IL-6 expression through cooperation with c-Jun and IL6-AP1 site, but is independent of its IL6-NFkappaB regulatory site in autocrine human multiple myeloma cells. Cancer Biol Ther. 2004;3(10):1007–1017. doi: 10.4161/cbt.3.10.1141. [DOI] [PubMed] [Google Scholar]
- 85.Brinkmann V. FTY720 (fingolimod) in multiple sclerosis: therapeutic effects in the immune and the central nervous system. Br J Pharmacol. 2009;158(5):1173–1182. doi: 10.1111/j.1476-5381.2009.00451.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vessey DA, Kelley M, Zhang J, Li L, Tao R, Karliner JS. Dimethylsphingosine and FTY720 inhibit the SK1 form but activate the SK2 form of sphingosine kinase from rat heart. J Biochem Mol Toxicol. 2007;21(5):273–279. doi: 10.1002/jbt.20193. [DOI] [PubMed] [Google Scholar]
- 87.Lim KG, Tonelli F, Li Z, et al. FTY720 analogues as sphingosine kinase 1 inhibitors: enzyme inhibition kinetics, allosterism, proteasomal degradation, and actin rearrangement in MCF-7 breast cancer cells. J Biol Chem. 2011;286(21):18633–18640. doi: 10.1074/jbc.M111.220756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vesper H, Schmelz EM, Nikolova-Karakashian MN, Dillehay DL, Lynch DV, Merrill AH., Jr Sphingolipids in food and the emerging importance of sphingolipids to nutrition. J Nutr. 1999;129(7):1239–1250. doi: 10.1093/jn/129.7.1239. [DOI] [PubMed] [Google Scholar]
- 89.Thaker AI, Shaker A, Rao MS, Ciorba MA. Modeling colitis-associated cancer with azoxymethane (AOM) and dextran sulfate sodium (DSS) J Vis Exp. 2012;(67) doi: 10.3791/4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Santiago C, Pagán B, Isidro AA, Appleyard CB. Prolonged chronic inflammation progresses to dysplasia in a novel rat model of colitis-associated colon cancer. Cancer Res. 2007;67(22):10766–10773. doi: 10.1158/0008-5472.CAN-07-1418. [DOI] [PubMed] [Google Scholar]
- 91.Hamiza OO, Rehman MU, Tahir M, et al. Amelioration of 1,2 dimethylhydrazine (DMH) induced colon oxidative stress, inflammation and tumor promotion response by tannic acid in Wistar rats. Asian Pac J Cancer Prev. 2012;13(9):4393–4402. doi: 10.7314/apjcp.2012.13.9.4393. [DOI] [PubMed] [Google Scholar]
- 92.Dillehay DL, Webb SK, Schmelz EM, Merrill AH., Jr Dietary sphingomyelin inhibits 1,2-dimethylhydrazine-induced colon cancer in CF1 mice. J Nutr. 1994;124(5):615–620. doi: 10.1093/jn/124.5.615. [DOI] [PubMed] [Google Scholar]
- 93.Symolon H, Schmelz EM, Dillehay DL, Merrill AH., Jr Dietary soy sphingolipids suppress tumorigenesis and gene expression in 1,2-dimethylhydrazine-treated CF1 mice and ApcMin/+ mice. J Nutr. 2004;134(5):1157–1161. doi: 10.1093/jn/134.5.1157. [DOI] [PubMed] [Google Scholar]
- 94.Mazzei JC, Zhou H, Brayfield BP, Hontecillas R, Bassaganya-Riera J, Schmelz EM. Suppression of intestinal inflammation and inflammation-driven colon cancer in mice by dietary sphingomyelin: importance of peroxisome proliferator-activated receptor γ expression. J Nutr Biochem. 2011;22(12):1160–1171. doi: 10.1016/j.jnutbio.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fischbeck A, Leucht K, Frey-Wagner I, et al. Sphingomyelin induces cathepsin D-mediated apoptosis in intestinal epithelial cells and increases inflammation in DSS colitis. Gut. 2011;60(1):55–65. doi: 10.1136/gut.2009.201988. [DOI] [PubMed] [Google Scholar]
- 96.Fyrst H, Oskouian B, Bandhuvula P, et al. Natural sphingadienes inhibit Akt-dependent signaling and prevent intestinal tumorigenesis. Cancer Res. 2009;69(24):9457–9464. doi: 10.1158/0008-5472.CAN-09-2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kumar A, Pandurangan AK, Lu F, et al. Chemopreventive sphingadienes downregulate Wnt signaling via a PP2A/Akt/GSK3β pathway in colon cancer. Carcinogenesis. 2012;33(9):1726–1735. doi: 10.1093/carcin/bgs174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Montrose DC, Scherl EJ, Bosworth BP, et al. S1P. localizes to the colonic vasculature in ulcerative colitis and maintains blood vessel integrity. J Lipid Res. 2013;54(3):843–851. doi: 10.1194/jlr.M034108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Murphy CT, Hall LJ, Hurley G, et al. The sphingosine-1-phosphate analogue FTY720 impairs mucosal immunity and clearance of the enteric pathogen Citrobacter rodentium. Infect Immun. 2012;80(8):2712–2723. doi: 10.1128/IAI.06319-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Samy ET, Meyer CA, Caplazi P, et al. Cutting edge: Modulation of intestinal autoimmunity and IL-2 signaling by sphingosine kinase 2 independent of sphingosine 1-phosphate. J Immunol. 2007;179(9):5644–5648. doi: 10.4049/jimmunol.179.9.5644. [DOI] [PubMed] [Google Scholar]